
Abstract
Whereas the physiologic wound healing (WH) successfully proceeds through the clearly defined sequence of the individual phases of wound healing, chronic non-healing wounds/ulcers fail to complete the individual stages and the entire healing process. There are many risk factors both modifiable (such as stress, smoking, inappropriate alcohol consumption, malnutrition, obesity, diabetes, cardio-vascular disease, etc.) and non-modifiable (such as genetic diseases and ageing) strongly contributing to the impaired WH. Current statistics demonstrate that both categories are increasingly presented in the populations, which causes dramatic socio-economic burden to the healthcare sector and society at large. Consequently, innovative concepts by predictive, preventive and personalised medicine are crucial to be implemented in the area. Individual risk factors, causality, functional interrelationships, molecular signature, predictive diagnosis, and primary and secondary prevention are thoroughly analysed followed by the expert recommendations in this paper.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A physiologic wound healing is a highly orchestrated process initiated by the tissue injury and resolved by the restoration of tissue integrity. It involves several overlapping phases: hemostasis, inflammation, proliferation, and remodeling [1]. Immediately after injury, the hemostasis phase is triggered, accompanied by immediate vascular contraction, platelet aggregation, and fibrin clot formation, which altogether initiate the next phase, namely the inflammatory one. During this phase, the platelets get activated, and the injured tissue releases a well-controlled panel of growth factors, cytokines, and chemoattractants which, in turn, do attract neutrophils, macrophages, and lymphocytes to the wound site. The locally involved extracellular matrix (ECM) and the entire wound area get enriched by the recruited platelets, macrophages, and bone marrow-derived stem cells which altogether release the core of growth factors promoting fibroblast activation and initiating the next phase - the proliferative one. Fibroblasts migrate into the wound area and proliferate almost simultaneously with endothelial cells triggering revascularisation by de novo capillary growth within the wound area. The fibroblasts secrete the essential molecular repertoire used to build up new ECM including collagen, glycosaminoglycans, and proteoglycans and launch the final remodelling phase of the wound healing. During this phase, an extensive qualitative and quantitative remodelling of ECM and local vascular system occurs by strictly regulated matrix metalloproteinases (MMPs)/ tissue inhibitor of metalloproteinase (TIMPs) complexes. This phase is a long-term process persisting over months and even years depending on the wound characteristics and individual health condition of the patient [1,2,3].
Whereas the physiologic wound healing successfully proceeds through the clearly defined sequence of the individual phases described above, chronic (non-healing) wounds/ulcers fail to complete some individual stages and the entire healing process, and stagnate usually at the early inflammatory stage. Chronic wounds are defined as those which do not follow the normal healing process and show no signs of effective healing within 3 months after the tissue injury [4]. The features characteristic for the chronic wounds are prolonged or excessive inflammatory phase [5], overabundant neutrophil infiltration [6], persistent infections [7], and frequent formation of tissue/organ atypical biofilms [8, 9].
Substantial socio-economical consequences caused by complex medical services dedicated specifically to the care needed for the chronic wounds negatively impact healthcare systems worldwide. In the USA alone, over 25 billion US $ are spent annually for the treatment of chronic wounds affecting around 6.5 million patients [10]. Problematic wound healing and chronic wounds may result from a broad spectrum of sub-optimal health conditions, severe pathologies and comorbidities predisposing the affected individuals and corresponding patient cohorts to hindered wound healing and consequent pathologic developments such as chronic inflammation, persistent infections, “open wounds”, cancerogenous wound transformation, etc. This article provides deep analysis of the risk factors, causal interrelations, and consequences linked to impaired wound healing, in order to motivate multi-professional considerations and development of innovative medical and technological approaches focused on prediction, prevention, and personalised treatments in the field.
Risk factors contributing to non-physiologic and impaired wound healing
There is a great number of risk factors which individually and combined may predispose to impaired wound healing. Here, we have categorised them as non-modifiable (unpreventable) risk factors (part A) and modifiable (preventable) (part B) ones as presented below. Further, comorbidities (part C) and risks of infection (part D) known as strongly contributing to the delayed and impaired wound healing are analysed in the paper.
Non-modifiable risk factors
Genetic component as unpreventable risk factor of ineffective healing processes
As described above, wound healing is a complex process involving a number of key-pathways regulating the response of many gene panels. Therefore, the genetic component is a prominent contributor to the wound healing. Indeed, some inborn genetic diseases are known to lead to non-physiologic and impaired wound healing, e.g. in case of Down syndrome [11] and Ataxia-telangiectasia [12, 13]. Further, rare genetic diseases are known to be responsible for the occurrence of venous ulcers in about 10% of cases [14]. The associated genetic defects can involve mutations in individual genes or gene-clusters, chromosomal aberrations, etc. Those defects may lead to the clinical manifestation in disorders of the immune system, of haemoglobin synthesis, of vasculopathies, of connective tissue diseases, of progeroid syndromes, etc. [14]. Hence, chromosomal aberrations are known for “Klinefelter syndrome” characterised by the presence of an additional X-chromosome. In Klinefelter patients, the incidence of varicosis and thrombosis is significantly increased, and about 13% of these patients develop venous ulcers, due to a post-thrombotic syndrome caused by high levels of the fibrinolysis inhibitor PAI-1 and diminished fibrinolysis [14,15,16].
Further, disorders of the immune system are known risk factors of the impaired WH, due to their particular role in the inflammatory phase. Leukocyte adhesion deficiencies (LADs)—LAD-I is caused by a mutation of the gene encoding the β2 chain of the integrins. LAD II is caused by a genetic defect in the synthesis of fucosylated glycostructures resulting in the absence of functional selectin ligands. LAD III is caused by a genetic defect of the leukocyte integrin [14]. All the LADs result in decreased migration of neutrophils to the wounded tissue and lacking phagocytosis ability of neutrophils in the wound bed. These genetic defects increase the susceptibility of affected patients to infections and consequent risks of severe wound complications [17]. TAP deficiency syndrome is characterised by the reduced expression of HLA class I molecules on the cell surface. Consequently, patients demonstrate abundant bacterial infections of the respiratory tract and chronic granulomatous skin lesions possibly caused by cells bearing inhibitory HLA class I receptors, i.e. NK and γδ T cells [18].
The next group is disorders of haemoglobin synthesis. Sickle cell anaemia caused by a point mutation in the β-globin gene results in an abnormality of the haemoglobin protein responsible for sickle shaped erythrocytes [14]. The disease-characteristic sickle shape and reduced deformability of affected erythrocytes result in vascular occlusion of small vessels and subsequent necrotic and ischemic injury [19]. Consequently, any trauma happened to the lower extremities results in their ulceration in 10–70% of sickle cell anaemia patients [19, 20]. Further, in case of Thalassemia which is a quantitative disorder of haemoglobin synthesis, about 27% of the affected patients develop chronic ulcers [21] due to poor peripheral oxygenation resulting from the existing underlying haemolytic anaemia [22].
Another group is vasculopathies. About 40% of the etiologically unexplained thromboses occur due to mutations in genes involved specifically in the coagulation system [14]. MTHFR polymorphism caused by a mutation in the 5-MTHFR gene leads to a development of hyperhomocysteinaemia—well-acknowledged risk factor for arterial and venous thrombosis and venous ulcers [14]. Further, mutations in factor V gene lead to an enhanced risk of thrombosis, since an activated protein C fails to inactivate factor V [14]. Other less common coagulation defects include prothrombin mutations, protein C deficiency, protein S deficiency, and antithrombin deficiency [14].
Inborn genetic disorders causing a connective tissue disease such as the Ehlers-Danlos syndrome lead to impairments in the remodelling phase of wound healing [23]. Furthermore, Progeroid syndromes such as Werner syndrome tend to generate skin ulcerations [24].
In addition to clinically manifested genetic diseases, polymorphisms in the WH relevant genes may also have a vital role in predisposing affected individuals to impaired wound healing and ulceration. For example, in ECM regulation, which is highly relevant in case of WH, a polymorphism in MMP and/or fibrinolytic system genes can lead to delayed and even impaired healing [25]. HFE and FPN1 gene functions are associated with increased iron efflux from macrophages and depending on the gene polymorphism can strongly affect the efficiency of the fibrinolytic system. The C282Y variant HFE gene polymorphism has been demonstrated to increase the risk of developing venous leg ulcers by almost 7 times [26]. The FPN1 polymorphism 8CG also increases leg ulcer susceptibility [27]. The MMP-12 gene polymorphism -82AG has been proposed as a prognostic marker for venous leg ulceration progression [27]. Different FXIII polymorphisms have also been shown to modulate and even have protective effects against ulcers progression [25]. Similarly, the SNP-1562C/T detected in the MMP-9 gene, which downregulates MMP-9 expression, has been shown to have a protective effect against pressure ulcers [28].
Autoimmune diseases deteriorate the physiologic wound healing processes
Patients suffering from immune diseases have significantly larger wounds and their time to heal is much more prolonged compared to the general population [29]. Leg ulcerations have been monitored in several autoimmune diseases, especially in those linked to connective tissue pathologies. The highest rates of ulceration are recorded for rheumatoid arthritis and systemic lupus erythematosus. However, ulcerations were also seen in primary antiphospholipid syndrome and other autoimmune diseases [30].
Patients with rheumatoid arthritis are predisposed to develop chronic leg ulcers [31]. Foot ulcerations in rheumatoid arthritis are frequently recurrent, and the significantly extended time needed for them to heal, further, increases the risk of infections [32]. The aetiology of the ulcers was found to be multifactorial with the most common factors being venous insufficiency, trauma or pressure, arterial insufficiency, and vasculitis [33].
Modelling systemic lupus erythematosus by lupus prone mice revealed impairments in the inflammation phase and accelerated angiogenesis, which deeply impact the overall wound healing [34]. Further studies have demonstrated that specifically the presence of antiphospholipid antibodies associated with thrombosis development strongly promotes the leg ulceration observed in autoimmune diseases. These antibodies were reported to be involved in the pathology of primary antiphospholipid syndrome, in systemic lupus erythematosus, and in rheumatoid arthritis [30].
Accelerate and advanced ageing is a risk factor for healing
Aged subpopulations frequently demonstrate chronic non-healing wounds, and their impaired WH is a major medical care issue [35]. Individuals experiencing accelerated ageing, for example, in the case of Down syndrome or progeroid syndromes are also at great risk for slowed down and impaired wound healing [11, 24]. There are evident alterations at any stage of the healing process in aged individuals. Hence, they demonstrate an altered inflammatory response characterised by sustained elevation of proinflammatory cytokines such as IL-6 and TNFα and by declined levels of growth factors. This combination leads to high levels of TGF-β that may play a role in transforming wounds from acute to chronic ones by inhibiting the reepithelialisation [35]. Accelerated and advanced ageing is also associated with slowed macrophage and T cell infiltration to the wound area and reduced macrophage function [36, 37]. Contextually, aged mice demonstrate severe neutrophils depletion shown to delay wound closure [38].
Disturbed microcirculation and hypoperfusion characteristic for ageing skin contributes to the impaired inflammatory response and hinders the physiologic angiogenic phase in the overall WH [39].
Another characteristic of the ageing skin is a strongly reduced ECM production and overexpressed MMPs, especially MMP-2 that collectively leads to impairments in the remodelling phase [40, 41].
Sex hormones have a role in the physiologic wound healing. Further, there are gender-dependent particularities in WH of aged individuals: healing of acute wounds in aged males is significantly slower compared to this in aged females, due to positive regulatory effects of oestrogen in the WH [42].
Lifestyle related modifiable risk factors
Psychological stress modulates healing processes
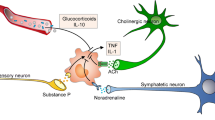
Psychological stress demonstrates strong modulating effects towards WH by influencing mood, behaviour, and health condition of the affected individual. Issue-dedicated studies demonstrated its adverse effects on wound healing [43, 44]. Stress reduces the levels of the pro-inflammatory cytokines IL-1β, IL-6, and TNFα at the wound site. It also reduces expression levels of cytokine IL-1α and chemoattractant IL-8 and, consequently, interferes with the well-regulated inflammatory phase of the physiologic wound healing [45,46,47,48]. Some of the detrimental effects of stress on WH may be due to upregulated glucocorticoids suppressing immune cell proliferation and decreasing production of IL-1α, IL-1β, and TNFα cytokines at the wound site [49, 50]. Further, stress leads to dysregulation of MMP-9 and MMP-2 levels at the wound site [51,52,53]. Although indirectly, stress is frequently associated with harmful habits such as cigarette smoking, inappropriate alcohol consumption and imbalanced nutrition—each of them adversely affects physiologic wound healing as demonstrated in detail below [54, 55].
Smoking strongly impairs wound healing
Smoking demonstrates detrimental effects on physiologic wound healing. Amongst over 4000 substances detected in tobacco smoke, several ones negatively impact healing processes [56]. To this end, nicotine strongly promotes vasoconstriction leading to disturbed microcirculation that negatively impacts WH [56, 57]. Further, smoking attenuates the inflammation phase by impairing white blood cell migration, reducing neutrophil bactericidal activity, and depressing IL-1 production [56, 58]. The proliferative phase is impaired by the reduced fibroblast migration and proliferation in addition to the downregulated collagen synthesis and deposition in smokers [56, 58]. Additionally, smoking disrupts epithelial regeneration and normal angiogenesis and decreases ECM production [56]. Overall, smokers show delayed wound healing, increased frequency of wound healing complications and wound dehiscence compared to non-smokers [56, 59, 60].
Inappropriate alcohol consumption is linked to non-physiologic healing processes
Moderate alcohol consumption demonstrates some protective effects against cardiovascular disease [61,62,63] which might be beneficial for WH as a whole. In contrast, inappropriate alcohol consumption may have strong adverse effects on an individual response towards an acute and chronic injury. Alcohol has been shown to play a role of immunomodulatory agent: acute alcohol consumption has inhibitory effects on the release of pro-inflammatory cytokines, while chronic alcohol consumption lead to a significantly prolonged response of inflammatory cells [64]. In experimental models, binge drinking before trauma resulted in attenuated levels of TNFα, IL-1, and IL-6 [65]. In human alcoholics, the levels of the immunosuppressive cytokine IL-10 were significantly increased over those in non-alcoholic individuals after major surgical intervention [66]. Alcohol consumption is also linked to the reduced T cell proliferation capacity important for the physiologic WH [67, 68]. Relevant murine models demonstrate that acute alcohol consumption decreases the expression of VEGF receptors and reduces nuclear expression of HIF-1α in endothelial cells, thereby affecting angiogenesis and the proliferative phase of wound healing [69, 70]. There are also studies showing that inappropriate alcohol consumption has an adverse effect on the physiologic reepithelialisation and collagen production [69,70,71,72].
Imbalanced diet is critical for the physiologic wound healing
An optimal setup of nutrients is extremely important for the regulation of all individual phases of wound healing and for the entire capacity of the body to perform the wound healing process successfully. Both deviations from the physiologic body mass index (BMI), namely too high as well as too low body BMI may predispose an individual to delayed and even impaired wound healing. The detailed analysis of the issue is provided below.
Malnutrition
The need for cell proliferation and protein synthesis during the wound healing process increases the body’s nutritional needs [73, 74]. Consequently, deficiencies or depletions in carbohydrates, protein, fatty acids, vitamins, or micronutrients may lead to impaired wound healing [75]. In particular, carbohydrates are needed to supply energy for the healing process and have been shown to be the key factor for activating several enzymatic complexes essential for the wound repair [76,77,78,79]. Certain amino acids, specifically leucine, glutamine, and arginine, possess anabolic activity required for the healing process. Moreover, protein deficiencies decrease leukocyte phagocytosis and increase susceptibility to infection [80, 81]. Fatty acids are required to provide additional sources for the highly required energy; they act as signalling molecules and contribute to the inflammatory process and cell proliferation [80]. Micronutrients such as zinc and vitamins B and C act as essential cofactors for energy production and protein synthesis, and demonstrate antioxidant properties. Regarding the aged individuals who are at higher risk for impaired WH as described above, they require 50% more protein compared to young individuals and thus are more prone to suffer from protein deficiencies [80].
Obesity
In 2014, 39% of adults aged 18+ worldwide were recorded as being overweight and 13% as being obese. Thus, worldwide nearly two billion adults are overweight and, of these, more than half a billion are obese [82]. Obese individuals have been demonstrated as being predisposed to several severe pathologies including impaired wound healing [76], which might be explained by hypoperfusion and ischemic effects that occurs in subcutaneous adipose tissue. Thus, if the tissue in the vicinity of the wound is inadequately oxygenated, the oxygen strongly dependent cellular repair processes do not take place adequately [83, 84]. Hypovascularity frequently observed in obese individuals, further contributes to poor perfusion and increases the risk of infections, due to a decreased infiltration of immune cells to the wound area [83, 85]. In addition, obese individuals frequently demonstrate increased tension on the wound edges contributing to the wound dehiscence [76, 86]. Consequently, the pathogenic bacteria which thrive in the moist environment of skin folds have an ideal environment for invasion and tissue breakdown. Lastly, skin-on-skin contact causes friction that can lead to ulceration that are particularly frequent in obese individuals [76, 86, 87].
Pathologies/comorbidities
Diabetes mellitus and cascaded comorbidities: major issues in medical care of wounds
The global prevalence of diabetes has reached an epidemic scale by almost half of billion patients worldwide [82]; the prediction for the next decades is highly pessimistic. This reflects a dramatic increase in associated risk factors and severe comorbidities frequently linked to disturbed WH. Diabetic individuals demonstrate disturbances in all individual processes and healing stages that collectively lead to overall impaired healing of acute wounds and are prone to chronic non-healing wounds such as the diabetic foot ulcers. Rates of lower limb amputation in populations with diabetic history are up to 20 times higher compared to those in non-diabetic populations [82].
Diabetic patients demonstrate deficient neutrophil chemotaxis, phagocytic, and microbicidal activities contributing to the high susceptibility to infections [88,89,90]. Aberrant cellular infiltration [91], insufficient macrophage activation [92], decreased release of TNFα, IL-1β and VEGF from macrophages [93], and impaired leukocyte function [94] have been shown to negatively impact wound healing in diabetic individuals.
Fibroblasts from diabetic foot ulcers are characterised by strongly decreased proliferative response to growth factors [95] and impaired signalling resulting in a diminished formation of granulation tissue [96, 97].
Between 30 to 50% of diabetic patients suffer from peripheral neuropathy, which increases the risk of ulcer development and predisposes them to delayed cutaneous tissue repair [98,99,100]. In the diabetic condition, the entire core of neuropeptides is strongly dysregulated such as downregulated SP, NPY, CGRP and upregulated CRF, α-MSH and NT. These neuropeptides play a key role in several stages of WH acting as chemoattractants, modulating permeability of blood vessels, improving adhesion of leucocytes, regulating expression of cytokines, stimulating endothelial cell proliferation, and enhancing VEGF release. The overall altered neuropeptide expression leads to the dysregulation of the downstream cytokines in the skin that results in the impaired wound healing [101].
Excessive activation of MMP-2 and MMP-9 combined with persistent nitrosative and oxidative stress and with excessive formation of advanced glycation end-products leads to ECM instability and to the breakdown of essential matrix proteins and growth factors [102,103,104,105]. Diabetic individuals show delayed reepithelialisation [106] and altered sensitivity to VEGF resulting in decreased angiogenesis [107].
Lastly, diabetic patients are strongly predisposed to severe comorbidities each of them individually and collectively altogether are functionally linked to impaired wound healing as analysed below in detail.
Cardio-vascular disease is critical for WH and follow-up cascade of pathologies
In general, vascular diseases resulting in local and/or systemic ischemic effects strongly affect healing processes, due to undersupplied oxygen and diminished levels of essential nutrients delivered to the tissue. Chronic non-healing wounds developed in the lower limbs is the typical complication of advanced diabetes as described above. Cardiac component plays a role as well. Hence, it has been shown that heart failure is predictive for the delayed healing of diabetic foot ulcers being strongly associated with poor prognosis [108, 109]. Further, heart failure is an independent risk factor for venous leg ulcers [110].
From a statistical viewpoint, venous ulcers alone affect up to 2.5 million patients annually in the USA for example, severely diminishing the patients’ quality of life and negatively impacting medical care as a whole [111]. However, early/predictive diagnosis followed by timely prevention may help corresponding patient cohorts to avoid severe complications linked to impaired WH. For example, critical limb ischemia (CLI) is characterised by non-healing ulcerations [112]. However, well-targeted preventive procedures such as endovascular revascularisation capable of restoring the blood supply to the tissue have been shown to be effective for improving the wound healing quality [113].
Another example of impaired wound healing is provided by venous ulceration. The exact mechanism underlying this pathology is not well understood yet; however, venous hypertension resulting from venous reflux is assumed to be the main cause of the disease and its complications [111]. These wounds, if become persistent, have been reported as leading to malignant transformation of the chronic leg ulcers providing, therefore, the clear functional link for the cascade of pathologies developed in a clear sequence, namely, untreated persistent vascular disease resulting in the chronic non-healing wounds finishing with aggressive cancer development at the wound site [114,115,116].
Cancer and impaired wound healing: multi-faceted interrelationships
Wound healing on one side and tumour pathologies on the other side are two areas which are characterised by multi-faceted interrelationships with each other as well as with other highly relevant medical areas already mentioned above. Herewith we provide some examples for the multi-functional links strongly supported by independent studies, available data and literature sources:
-
1.
Chronic non-healing wounds—high risk of cancerous transformation of the affected tissue [114,115,116,117,118,119,120,121,122,123]
-
2.
Relevant genetic diseases (e.g. Down syndrome)—increased stress (excessive production of SOD2)—insufficient repair processes—strong predisposition to impaired wound healing and cancer [11, 124, 125]
-
3.
Malnutrition—non-physiologically low BMI—impaired wound healing and poor prognosis in metastatic disease [80, 126]
-
4.
Obesity—risk of diabetes—risk of chronic CVD—strong predisposition to chronic non-healing wounds and cancer [76, 127, 128]
-
5.
Systemic hypoxia—impaired wound healing and strong predisposition to aggressive metastatic disease [75, 129]
-
6.
Chronic inflammatory processes—chronic non-healing wounds stagnating at the early inflammatory phase but not progressing into the later phases of healing—increased risks of cancer by chronic inflammation [130]
-
7.
Autoimmune diseases—strong predisposition to impaired wound healing and cancer [29, 131]
-
8.
At the molecular level, non-physiologically upregulated activities of metalloproteinases (in particular MMP-2 and MMP-9) and dysregulated enzymatic complexes MMPs/TIMPs are characteristic for both impaired wound healing and aggressive tumour promotion and metastatic disease [132, 133]
All the cascades listed above demand broad attention at the levels of fundamental research and complex healthcare approaches.
The causality between the WH and cancer has been demonstrated to be both sided:
-
Chronic non-healing wounds may lead to the cancer development [114,115,116,117,118,119,120,121,122,123]; some authors characterise tumours as “wounds which do not heal” [134].
-
Cancer patients frequently demonstrate delayed and impaired wound healing; these impairments diminish treatment success and contribute to aggressive metastatic disease [130, 135]
-
Wound healing and cancer development share common cellular and molecular mechanisms [136]
Contextually, independent studies indicate that the wound healing environment provides an opportunistic matrix for tumour growth [137, 138]. For instance, human basal cell carcinoma has been observed in areas of wound healing, including sites of vaccination [117, 118], surgery [119, 120], burns [121], and trauma [122, 123]. Modelling of breast cancer in mice has demonstrated that wounding next to the tumour significantly increases tumour size, and injecting the wound fluid closely to the tumour site results in strongly promoted tumour growth [139]. Additionally, acute inflammation triggered by a biopsy in mammary mouse model was shown to enhance the risk of developing peripheral metastases. This is probably due the inflammation in the primary tumours and in targeted organs, favouring the seeding of released tumour cells [140].
The presence of tumor appears to inhibit wound healing in cutaneous wounds [141]. In consensus, a small sample size study registered higher rates of non-healing wounds in patients with cancer [142]. Another study examined the cellular and molecular alterations of the dermal wound healing process in rats bearing oral carcinoma. On a macroscopic level, reduced rates of wound closure have been demonstrated compared to the tumour-free controls. On a microscopic level, enhanced numbers of immature macrophages in the wound area have been detected demonstrating adverse effects on the healing. In the tumour bearing mice reduced maturation of those macrophage have been seen, which negatively impacts the inflammatory processes of wound healing. Tumours reduce expression levels of the immunomodulatory genes Tlr4, IL-1β, Ccl2, IL-10, Ccl3, and Cxcl1, which are essential for physiologic wound healing. Taken the above summarised data together, it is evident that, in particular, the physiologic recruitment of the immune cells and the initiation and resolution of the inflammatory response are suppressed in the presence of a tumour contributing to the impairments of wound healing [143].
In addition to the molecular and cellular mechanisms linked to the primary tumour development, impaired wound healing in cancer patients may secondary result from the systemic toxic effects of anti-cancer treatments such as irradiation and chemotherapy [126]. Finally, in most studies approximately 40 to 80% of patients with cancer are presented as malnourished, which increases their susceptibility to infection and overall tendency to delayed wound healing [126]. To this end, please see the subchapter “Infection” provided below.
Infection impairs healing processes of the host
Infection to the wound is an extrinsic factor strongly retarding healing processes. Live bacteria and bacterial toxins—both lead to a strong upregulation and prolonged activity of pro-inflammatory cytokines, excessive inflammatory responses and damage to the affected tissue. In turn, the recruited inflammatory cells as well as invaded bacteria themselves contribute to the overexpression of matrix metalloproteases degrading the ECM and growth factors overloading the wound bed [75, 144]. Some pathologies such as diabetes mellitus are known to increase risks of chronic infections, due to synergic effects of ineffective immune response on one side and on the other side systemic oxygen undersupply—both dramatically increasing the risks of infection [145, 146].
Pathogenic bacteria colonising chronic wounds often form biofilms consisting of the aggregated bacteria embedded in a self-secreted extracellular polysaccharide matrix. Those biofilms provide the hosted bacteria with highly protective environment making them more resistant against any antibiotic treatments [75, 144]. Formation of the bacterial films within the wound impairs key healing processes such as the inflammatory immune response, granulation tissue formation, and reepithelialisation of the host’s injured tissue [147].
Concluding remarks, working hypotheses and expert recommendations
As mentioned above, predictive diagnostics and targeted prevention of pathologies negatively impacting wound healing might be the clue to the most effective approach against impaired wound healing in stratified patient groups e.g. within the primary and secondary prevention in diabetes care. Further, well-controlled physiologic wound healing is effective in protecting affected individuals against potential pathologies cascaded by chronic non-healing wounds such as a malignant transformation of the affected tissue at the wound site.
Additionally to both above summarised approaches, herewith, we would like to propose some new concepts for multi-professional considerations. The first one strongly involves expertise and daily practice of general practitioners who might be in duty to put a particular attention to any delayed wound healing in their patient pools within the broad population. Well elaborated issue-dedicated questionnaires would be of great help for this group of professionals, in order to estimate potentially undiagnosed pathologies linked to impaired healing processes and even individual predisposition of the affected individual which can be diagnosed well in time. This approach is highly promising for an optimistic scenario by reconsidering currently applied reactive medical care and may lead to a great success by innovative screening programmes, if appropriately applied to the broad population.
Another innovative concept comprises a development of new research areas of great clinical utility of the knowledge accumulated regarding individuals in suboptimal health conditions who demonstrate some symptoms potentially relevant for impaired repair and healing processes. This kind of stratification may be of particular value for predictive diagnosis and targeted prevention within the young subpopulations. Herewith, we provide an example by so-called “Flammer Syndrome” for innovative research in the area.
Potential relevance of “Flammer syndrome” for altered wound healing: facts and hypotheses
Flammer syndrome (FS) describes a highly specific phenotype characterised by strongly pronounced sensitivity towards stress stimuli and primary vascular dysregulation accompanied by a cluster of the syndrome-typical symptoms and signs such as altered gene regulation, frequently decreased blood pressure and low BMI amongst others; FS is more prevalent in young women and academic professions [148]. FS-individuals are considered to comprise otherwise healthy subpopulations which, however, may be strongly predisposed to some severe pathologies [148]. In turn, patients being already affected by severe pathology may be strongly predisposed to particularly poor outcomes, if demonstrating symptoms of FS [149].
In FS individuals, plasma levels of endothelin-1 (ET-1) are moderately till strongly increased [148] resulting in insufficient vasodilatation or inappropriate vasoconstriction clinically established as systemic hypoxia with ischemic lesions described for several organs including life-important ones [150]. Systemic hypoxia and local ischemic lesions play an important regulatory role in repair processes leading to impairments in wound healing as discussed above in several subchapters. To this end, it is notable that the quality of repair processes in general and, in particular, this of wound healing has not been investigated in FS-affected individuals. However, there is a large number of direct and indirect indications which motivates the healing relevant research to be dedicated specifically to the FS affected individuals, namely
-
Elevated ET-1 and systemic hypoxia typical for FS and detrimental for WH [75, 151]
-
Retinal vein occlusion is frequently demonstrated by FS individuals [150]
-
At the molecular level, repair processes are diminished and both MMP-2 and MMP-9 are highly activated in FS individuals as well as in FS-affected patients suffering from normal-tension glaucoma; consequently tissues remodelling is altered in this patient cohort [152, 153]
-
Finally, FS may predispose cancer patients to both impaired wound healing and aggressive metastatic disease, due to systemic hypoxic effects and upregulation of MMPs as proposed by dedicated research [149,155,, 154–156].
References
Mathieu D, Linke JC, Wattel F. Non-healing wounds. In: Mathieu D, editor. Handbook on hyperbaric medicine. Dordrecht: Springer Netherlands; 2006. p. 401–28.
Lorenz HP, Longaker MT. Wounds: biology, pathology, and management. In: Norton JA, Barie PS, Bollinger RR, Chang AE, Lowry SF, Mulvihill SJ, editors. Surgery: basic science and clinical evidence. New York: Springer New York; 2008. p. 191–208.
Stolzenburg-Veeser L, Golubnitschaja O. Mini-encyclopaedia of the wound healing repertoire for multiprofessional considerations. J Proteomics. 2017;Submitted.
Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. Dis Model Mech. 2014;7(11):1205–13.
Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127(3):514–25.
Martin P, Nunan R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br J Dermatol. 2015;173(2):370–8.
Edwards R, Harding KG. Bacteria and wound healing. Curr Opin Infect Dis. 2004;17(2):91–6.
Wolcott RD, Rhoads DD, Dowd SE. Biofilms and chronic wound inflammation. J Wound Care. 2008;17(8):333–41.
Thomson CH. Biofilms: do they affect wound healing? Int Wound J. 2011;8(1):63–7.
Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, et al. Human skin wounds: a major and snowballing threat to public health and economy. Wound Repair Regen. 2009;17(6):763–71.
Mik G, Gholve PA, Scher DM, Widmann RF, Green DW. Down syndrome: orthopedic issues. Curr Opin Pediatr. 2008;20(1):30–6.
Götz A, Eckert F, Landthaler M. Ataxia-telangiectasia (Louis-Bar syndrome) associated with ulcerating necrobiosis lipoidica. J Am Acad Dermatol. 1994;31(1):124–6.
Paller AS, Massey RB, Curtis MA, Pelachyk JM, Dombrowski HC, Leickly FE, et al. Cutaneous granulomatous lesions in patients with ataxia-telangiectasia. J Pediatr. 1991;119(6):917–22.
Elsharkawi-Welt K, Hepp J, Scharffetter-Kochanek K. Genetic causes of impaired wound healing. Rare differential diagnosis of the non-healing wound. Hautarzt. 2008;59(11):893–903.
Higgins EJ, Tidman MJ, Savidge GF, Beard J, MacDonald DM. Platelet hyperaggregability in two patients with Klinefelter’s syndrome complicated by leg ulcers. Br J Dermatol. 1989;120(2):322.
Zollner TM, Veraart JCJM, Wolter M, Hesse S, Villemur B, Wenke A, et al. Leg ulcers in Klinefelter’s syndrome—further evidence for an involvement of plasminogen activator inhibitor-1. Br J Dermatol. 1997;136(3):341–4.
Roos D, Law SK. Hematologically important mutations: leukocyte adhesion deficiency. Blood Cells Mol Dis. 2001;27(6):1000–4.
Gadola SD, Moins-Teisserenc HT, Trowsdale J, Gross WL, Cerundolo V. TAP deficiency syndrome. Clin Exp Immunol. 2000;121(2):173–8.
Cackovic M, Chung C, Bolton LL, Kerstein MD. Leg ulceration in the sickle cell patient. J Am Coll Surg. 1998;187(3):307–9.
Ballas SK. Sickle cell anaemia: progress in pathogenesis and treatment. Drugs. 2002;62(8):1143–72.
Stevens DM, Shupack JL, Javid J, Silber R. Ulcers of the leg in thalassemia. Arch Dermatol. 1977;113(11):1558–60.
Sawhney H, Weedon J, Gillette P, Solomon W, Braverman A. Predilection of hemolytic anemia-associated leg ulcers for the medial malleolus. Vasa. 2002;31(3):191–3.
Shirley ED, Demaio M, Bodurtha J. Ehlers-danlos syndrome in orthopaedics: etiology, diagnosis, and treatment implications. Sports Health. 2012;4(5):394–403.
Muftuoglu M, Oshima J, von Kobbe C, Cheng W-H, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008;124(4):369–77.
Singh AV, Subhashree L, Milani P, Gemmati D, Zamboni P. Interplay of iron metallobiology, metalloproteinases, and FXIII, and role of their gene variants in venous leg ulcer. Int J Low Extrem Wounds. 2010;9:166–79.
Zamboni P, Tognazzo S, Izzo M, Pancaldi F, Scapoli GL, Liboni A, et al. Hemochromatosis C282Y gene mutation increases the risk of venous leg ulceration. J Vasc Surg. 2005;42(2):309–14.
Gemmati D, Federici F, Catozzi L, Gianesini S, Tacconi G, Scapoli GL, et al. DNA-array of gene variants in venous leg ulcers: detection of prognostic indicators. J Vasc Surg. 2009;50(6):1444–51.
Latifa K, Sondess S, Hajer G, Manel B-H-M, Souhir K, Nadia B, et al. Evaluation of physiological risk factors, oxidant-antioxidant imbalance, proteolytic and genetic variations of matrix metalloproteinase-9 in patients with pressure ulcer. Sci Rep. 2016;6:29371.
Shanmugam VK, Schilling A, Germinario A, Mete M, Kim P, Steinberg J, et al. Prevalence of immune disease in patients with wounds presenting to a tertiary wound healing center. Int Wound J. 2012;9(4):403–11.
Shanmugam VK, Steen VD, Cupps TR. Lower extremity ulcers in connective tissue disease. Isr Med Assoc J. 2008;10(7):534–6.
Öien RF, Håkansson A, Hansen BU. Leg ulcers in patients with rheumatoid arthritis—a prospective study of aetiology, wound healing and pain reduction after pinch grafting. Rheumatology (Oxford). 2001;40(7):816–20.
Siddle HJ, Firth J, Waxman R, Nelson EA, Helliwell PS. A case series to describe the clinical characteristics of foot ulceration in patients with rheumatoid arthritis. Clin Rheumatol. 2012;31(3):541–5.
Pun YL, Barraclough DR, Muirden KD. Leg ulcers in rheumatoid arthritis. Med J Aust. 1990;153(10):585–7.
Campos PP, Bakhle YS, Andrade SP. Mechanisms of wound healing responses in lupus-prone New Zealand White mouse strain. Wound Repair Regen. 2008;16(3):416–24.
Gould L, Abadir P, Brem H, Carter M, Conner-Kerr T, Davidson J, et al. Chronic wound repair and healing in older adults: current status and future research. J Am Geriatr Soc. 2015;63(3):427–38.
Brubaker AL, Rendon JL, Ramirez L, Choudhry MA, Kovacs EJ. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol. 2013;190(4):1746–57.
Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol. 2001;117(5):1027–35.
Nishio N, Okawa Y, Sakurai H, Isobe KI. Neutrophil depletion delays wound repair in aged mice. Age (Omaha). 2008;30(1):11–9.
Bentov I, Reed MJ. Anesthesia, microcirculation, and wound repair in aging. Anesthesiology. 2014;120(3):760–72.
Ashcroft G, Mills S, Ashworth J. Ageing and wound healing. Biogerontology. 2002;3(6):337–45.
Ballas CB, Davidson JM. Delayed wound healing in aged rats is associated with increased collagen gel remodeling and contraction by skin fibroblasts, not with differences in apoptotic or myofibroblast cell populations. Wound Repair Regen. 2001;9(3):223–37.
Gilliver SC, Ashworth JJ, Ashcroft GS. The hormonal regulation of cutaneous wound healing. Clin Dermatol. 2007;25(1):56–62.
Kiecolt-Glaser JK, Marucha PT, Mercado AM, Malarkey WB, Glaser R. Slowing of wound healing by psychological stress. Lancet. 1995;346(8984):1194–6.
Marucha PT, Kiecolt-Glaser JK, Favagehi M. Mucosal wound healing is impaired by examination stress. Psychosom Med. 1998;60(3):362–5.
Glaser R, Kiecolt-Glaser JK, Marucha PT, MacCallum RC, Laskowski BF, Malarkey WB. Stress-related changes in proinflammatory cytokine production in wounds. Arch Gen Psychiatry. 1999;56(5):450–6.
Kiecolt-Glaser JK, Loving TJ, Stowell JR, Malarkey WB, Lemeshow S, Dickinson SL, et al. Hostile marital interactions, proinflammatory cytokine production, and wound healing. Arch Gen Psychiatry. 2005;62:1377–84.
Godbout JP, Glaser R. Stress-induced immune dysregulation: implications for wound healing, infectious disease and cancer. J Neuroimmune Pharmacol. 2006;1(4):421–7.
Boyapati L, Wang H-L. The role of stress in periodontal disease and wound healing. Periodontol 2000. 2007;44:195–210.
Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6(4):318–28.
Hübner G, Brauchle M, Smola H, Madlener M, Fässler R, Werner S. Differential regulation of pro-inflammatory cytokines during wound healing in normal and glucocorticoid-treated mice. Cytokine. 1996;8(7):548–56.
Broadbent E, Petrie J, Alley P, Booth R. Psychological stress impairs early wound repair following surgery. Psychosom Med. 2003;65(5):865–9.
Yang EV, Bane CM, MacCallum RC, Kiecolt-Glaser JK, Malarkey WB, Glaser R. Stress-related modulation of matrix metalloproteinase expression. J Neuroimmunol. 2002;133(1–2):144–50.
Romana-Souza B, Otranto M, Vieira AM, Filgueiras CC, Fierro IM, Monte-Alto-Costa A. Rotational stress-induced increase in epinephrine levels delays cutaneous wound healing in mice. Brain Behav Immun. 2010;24(3):427–37.
Steptoe A, Wardle J, Pollard TM, Canaan L, Davies GJ. Stress, social support and health-related behavior: a study of smoking, alcohol consumption and physical exercise. J Psychosom Res. 1996;41(2):171–80.
Vitaliano PP, Scanlan JM, Zhang J, Savage MV, Hirsch IB, Siegler IC. A path model of chronic stress, the metabolic syndrome, and coronary heart disease. Psychosom Med. 2002;64(3):418–35.
Ahn C, Mulligan P, Salcido RS. Smoking-the bane of wound healing: biomedical interventions and social influences. Adv Skin Wound Care. 2008;21(5):227–36. quiz 237-8.
Sørensen LT, Jørgensen S, Petersen LJ, Hemmingsen U, Bülow J, Loft S, et al. Acute effects of nicotine and smoking on blood flow, tissue oxygen, and aerobe metabolism of the skin and subcutis. J Surg Res. 2009;152(2):224–30.
Sørensen LT, Toft B, Rygaard J, Ladelund S, Teisner B, Gottrup F. Smoking attenuates wound inflammation and proliferation while smoking cessation restores inflammation but not proliferation. Wound Repair Regen. 2010;18(2):186–92.
Chan LKW, Withey S, Butler PE. Smoking and wound healing problems in reduction mammaplasty: is the introduction of urine nicotine testing justified? Ann Plast Surg. 2006;56(2):111–5.
Manassa EH, Hertl CH, Olbrisch RR. Wound healing problems in smokers and nonsmokers after 132 abdominoplasties. Plast Reconstr Surg. 2003;111(6):2082–9.
O’Keefe JH, Bybee KA, Lavie CJ. Alcohol and cardiovascular health. the razor-sharp double-edged sword. J Am Coll Cardiol. 2007;50(11):1009–14.
Jackson R, Scragg R, Beaglehole R. Alcohol consumption and risk of coronary heart disease. BMJ. 1991;303(6796):211–6.
Stampfer MJ, Colditz GA, Willett WC, Speizer FE, Hennekens CH. A prospective study of moderate alcohol consumption and the risk of coronary disease and stroke in women. N Engl J Med. 1988;319(5):267–73.
Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33(2):220–32.
Greiffenstein P, Mathis KW, Stouwe CV, Molina PE. Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcohol Clin Exp Res. 2007;31(4):704–15.
Sander M, von Heymann C, Neumann T, Braun JP, Kastrup M, Beholz S, et al. Increased interleukin-10 and cortisol in long-term alcoholics after cardiopulmonary bypass: a hint to the increased postoperative infection rate? Alcohol Clin Exp Res. 2005;29(9):1677–84.
Heinz R, Waltenbaugh C. Ethanol consumption modifies dendritic cell antigen presentation in mice. Alcohol Clin Exp Res. 2007;31(10):1759–71.
Mandrekar P, Catalano D, Dolganiuc A, Kodys K, Szabo G. Inhibition of myeloid dendritic cell accessory cell function and induction of T cell anergy by alcohol correlates with decreased IL-12 production. J Immunol. 2004;173(5):3398–407.
Radek KA, Matthies AM, Burns AL, Heinrich SA, Kovacs EJ, Dipietro LA. Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am J Physiol Heart Circ Physiol. 2005;289(3):H1084–90.
Radek KA, Kovacs EJ, Gallo RL, DiPietro L. a. Acute ethanol exposure disrupts VEGF receptor cell signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295(1):H174–84.
Radek KA, Kovacs EJ, DiPietro LA. Matrix proteolytic activity during wound healing: Modulation by acute ethanol exposure. Alcohol Clin Exp Res. 2007;31(6):1045–52.
Fitzgerald DJ, Radek KA, Chaar M, Faunce DE, DiPietro LA, Kovacs EJ. Effects of acute ethanol exposure on the early inflammatory response after excisional injury. Alcohol Clin Exp Res. 2007;31(2):317–23.
Mora S, Pessin JE. An adipocentric view of signaling and intracellular trafficking. Diabetes Metab Res Rev. 2002;18(5):345–56.
Zhang XJ, Chinkes DL, Doyle D, Wolfe RR. Metabolism of skin and muscle protein is regulated differently in response to nutrition. Am J Physiol Endocrinol Metab. 1998;274(3):E484–92.
Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010;89(3):219–29.
Wilson JA, Clark JJ. Obesity: impediment to postsurgical wound healing. Adv Skin Wound Care. 2004;17(8):426–35.
Patel GK. The role of nutrition in the management of lower extremity wounds. Int J Low Extrem Wounds. 2005;4(1):12–22.
Ireton-Jones C, Liepa GU. Carbohydrates and wound healing. In: Molnar J, editor. Nutrition and wound healing. CRC Press; 2006. pp. 15–26.
De Feo M, Gregorio R, Renzulli A, Ismeno G, Romano GP, Cotrufo M. Treatment of recurrent postoperative mediastinitis with granulated sugar. J Cardiovasc Surg (Torino). 2000;41(5):715–9.
Demling RH. Nutrition, anabolism, and the wound healing process: an overview. Eplasty. 2009;9:e9.
Gogia PP. Physiology of wound healing. In: Gogia PP, editor. Clinical wound management. Thorofare: Slack Inc.; 1995. p. 8–12.
World Health Organization. Global Report on Diabetes. Geneva: WHO; 2016. http://www.who.int/diabetes/global-report/en. Accessed 18 Jan 2017.
Goldman RJ. Hyperbaric oxygen therapy for wound healing and limb salvage: a systematic review. PM R. 2009;1(5):471–89.
Shipman AR, Millington GWM. Obesity and the skin. Br J Dermatol. 2011;165(4):743–50.
Kranke P, Bennett MH, Martyn-St James M, Schnabel A, Debus SE. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst Rev. 2012;4(1):CD004123.
Anaya DA, Dellinger EP. The obese surgical patient: a susceptible host for infection. Surg Infect (Larchmt). 2006;7(5):473–80.
Greco JA, Castaldo ET, Nanney LB, Wendel JJ, Summitt JB, Kelly KJ. The effect of weight loss surgery and body mass index on wound complications after abdominal contouring operations. Ann Plast Surg. 2008;61(3):235–42.
Alba-Loureiro TC, Hirabara SM, Mendonça JR, Curi R, Pithon-Curi TC. Diabetes causes marked changes in function and metabolism of rat neutrophils. J Endocrinol. 2006;188(2):295–303.
Mastej K, Adamiec R. Neutrophil surface expression of CD11b and CD62L in diabetic microangiopathy. Acta Diabetol. 2008;45(3):183–90.
Marhoffer W, Stein M, Schleinkofer L, Federlin K. Evidence of ex vivo and in vitro impaired neutrophil oxidative burst and phagocytic capacity in type 1 diabetes mellitus. Diabetes Res Clin Pract. 1993;19(3):183–8.
Rosner K, Ross C, Karlsmark T, Petersen AA, Gottrup F, Vejlsgaard GL. Immunohistochemical characterization of the cutaneous cellular infiltrate in different areas of chronic leg ulcers. APMIS. 1995;103(4):293–9.
Moore K, Ruge F, Hadring KG. T lymphocytes and the lack of activated macrophages in wound margin biopsies from chronic leg ulcers. Br J Dermatol. 1997;137(2):188–94.
Zykova SN, Jenssen TG, Berdal M, Olsen R, Myklebust R, Seljelid R. Altered cytokine and nitric oxide secretion in vitro by macrophages from diabetic type II-like db/db mice. Diabetes. 2000;49(9):1451–8.
Bagdade JD, Root RK, Bulger RJ. Impaired leukocyte function in patients with poorly controlled diabetes. Diabetes. 1974;23(1):9–15.
Loot MA, Kenter SB, Au FL, van Galen WJ, Middelkoop E, Bos JD, et al. Fibroblasts derived from chronic diabetic ulcers differ in their response to stimulation with EGF, IGF-I, bFGF and PDGF-AB compared to controls. Eur J Cell Biol. 2002;81:153–60.
Werner S, Breeden M, Hübner G, Greenhalgh DG, Longaker MT. Induction of keratinocyte growth factor expression is reduced and delayed during wound healing in the genetically diabetic mouse. J Invest Dermatol. 1994;103(4):469–73.
Werner S, Krieg T, Smola H. Keratinocyte-fibroblast interactions in wound healing. J Invest Dermatol. 2007;127(5):998–1008.
Adler AI, Boyko EJ, Ahroni JH, Stensel V, Forsberg RC, Smith DG. Risk factors for diabetic peripheral sensory neuropathy: results of the Seattle Prospective Diabetic Foot Study. Diabetes Care. 1997;20(7):1162–7.
Sheehan P, Jones P, Caselli A, Giurini JM, Veves A. Percent change in wound area of diabetic foot ulcers over a 4-week period is a robust predictor of complete healing in a 12-week prospective trial. Diabetes Care. 2003;26(6):1879–82.
Illigens BM, Gibbons CH. A human model of small fiber neuropathy to study wound healing. PLoS One. 2013;8(1):e54760.
Pradhan L, Nabzdyk C, Andersen ND, LoGerfo FW, Veves A. Inflammation and neuropeptides: the connection in diabetic wound healing. Expert Rev Mol Med. 2009;11:e2.
Signorelli SS, Malaponte G, Libra M, Di Pino L, Celotta G, Bevelacqua V, et al. Plasma levels and zymographic activities of matrix metalloproteinases 2 and 9 in type II diabetics with peripheral arterial disease. Vasc Med. 2005;10(1):1–6.
Soneja A, Drews M, Malinski T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol Rep. 2005;57(Suppl):108–19.
Huijberts MS, Schaper NC, Schalkwijk CG. Advanced glycation end products and diabetic foot disease. Diabetes Metab Res Rev. 2008;24 Suppl 1:S19–24.
Haines DD, Juhasz B, Tosaki A. Management of multicellular senescence and oxidative stress. J Cell Mol Med. 2013;17(8):936–57.
Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol. 1998;111(5):850–7.
Tchaikovski V, Olieslagers S, Böhmer FD, Waltenberger J. Diabetes mellitus activates signal transduction pathways resulting in vascular endothelial growth factor resistance of human monocytes. Circulation. 2009;120(2):150–9.
Xu L, Qian H, Gu J, Shi J, Gu X, Tang Z. Heart failure in hospitalized patients with diabetic foot ulcers: clinical characteristics and their relationship with prognosis. J Diabetes. 2013;5(4):429–38.
Rhou YJ, Henshaw FR, McGill MJ, Twigg SM. Congestive heart failure presence predicts delayed healing of foot ulcers in diabetes: an audit from a multidisciplinary high-risk foot clinic. J Diabetes Complic. 2015;29(4):556–62.
Augey F, Pinet A, Renaudier P. Heart failure and stasis ulcer: a significant association (prospective study of 100 cases). Ann Dermatol Venereol. 2010;137(5):353–8.
Brem H, Kirsner RS, Falanga V. Protocol for the successful treatment of venous ulcers. Am J Surg. 2004;188(Suppl 1A):1–8.
Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG. Inter-society consensus for the management of peripheral arterial disease (TASC II). J Vasc Surg. 2007;45(1 Suppl):S5–67.
Bae JI, Won JH, Han SH, Lim SH, Hong YS, Kim JY, et al. Endovascular revascularization for patients with critical limb ischemia: impact on wound healing and long term clinical results in 189 limbs. Korean J Radiol. 2013;14(3):430–8.
Baldursson BT, Beitner H, Syrjänen S. Human papillomavirus in venous ulcers with and without squamous cell carcinoma. Arch Dermatol Res. 2000;292(6):275–8.
Baldursson B, Sigurgeirsson B, Lindelof B. Venous leg ulcers and squamous cell carcinoma: a large-scale epidemiological study. Br J Dermatol. 1995;133(4):571–4.
Lehnert W, Kohl K, Riebe H, Jünger M, Ladwig A. The treatment of malignant tumors on venous leg ulcers. Case presentation and literature review. Hautarzt. 2008;59(11):912–6.
Rich JD, Shesol BF, Horne 3rd DW. Basal cell carcinoma arising in a smallpox vaccination site. J Clin Pathol. 1980;33(2):134–5.
Zelickson A. Basal cell epithelioma at site and following smallpox vaccination: report of a case. Arch Dermatol. 1968;98(1):35–6.
Ozyazgan I, Kontacs O. Basal cell carcinoma arising from surgical scars: a case and review of the literature. Dermatol Surg. 1999;25(12):965–8.
Jorquero E, Moreno JC, Díaz-Cano SJ, Rodríguez-Adrados F, Camacho F. Basal cell carcinoma arising in a surgical scar: reconstructive surgical treatment. J Dermatol Surg Oncol. 1994;20(12):846–7.
Koga Y, Sawada Y. Basal cell carcinoma developing on a burn scar. Burns. 1997;23(1):75–7.
Özyazgan İ, Kontaş O. Previous injuries or scars as risk factors for the development of basal cell carcinoma. Scand J Plast Reconstr Surg Hand Surg. 2004;38(1):11–5.
Rustin MHA, Chambers TJ, Munro DD. Post-traumatic basal cell carcinomas. Clin Exp Dermatol. 1984;9(4):379–83.
Perrone S, Lotti F, Geronzi U, Guidoni E, Longini M, Buonocore G. Oxidative stress in cancer-prone genetic diseases in pediatric age: the role of mitochondrial dysfunction. Oxid Med Cell Longev. 2016;2016:4782426.
Muramatsu H. Genetic predisposition to pediatric myeloid malignancies. Rinsho Ketsueki. 2016;57(6):730–5.
Payne WG, Naidu DK, Wheeler CK, Barkoe D, Mentis M, Salas RE, et al. Wound healing in patients with cancer. Eplasty. 2008;8:e9.
Manna P, Jain SK. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab Syndr Relat Disord. 2015;13(10):423–44.
Arnold M, Pandeya N, Byrnes G, Renehan AG, Stevens GA, Ezzati M, et al. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol. 2015;16(1):36–46.
Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29(2):285–93.
Arnold KM, Opdenaker LM, Flynn D, Sims-Mourtada J. Wound healing and cancer stem cells: inflammation as a driver of treatment resistance in breast cancer. Cancer Growth Metastasis. 2015;8:1–13.
Rahat MA, Shakya J. Parallel aspects of the microenvironment in cancer and autoimmune disease. Mediat Inflamm. 2016;2016:4375120.
Lazaro JL, Izzo V, Meaume S, Davies AH, Lobmann R, Uccioli L. Elevated levels of matrix metalloproteinases and chronic wound healing: an updated review of clinical evidence. J Wound Care. 2016;25(5):277–87.
Kerkelä E, Saarialho-Kere U. Matrix metalloproteinases in tumor progression: focus on basal and squamous cell skin cancer. Exp Dermatol. 2003;12(2):109–25.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–9.
Harréus U. Surgical errors and risks - the head and neck cancer patient. GMS Curr Top Otorhinolaryngol Head Neck Surg. 2013; 12: Doc04.
Byun JS, Gardner K. Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol. 2013;182(4):1055–64.
Abramovitch R, Marikovsky M, Meir G, Neeman M. Stimulation of tumour angiogenesis by proximal wounds: spatial and temporal analysis by MRI. Br J Cancer. 1998;77(3):440–7.
Wong SY, Reiter JF. Wounding mobilizes hair follicle stem cells to form tumors. Proc Natl Acad Sci U S A. 2011;108(10):4093–8.
Stuelten CH, Barbul A, Busch JI, Sutton E, Katz R, Sato M, et al. Acute wounds accelerate tumorigenesis by a T cell-dependent mechanism. Cancer Res. 2008;68(18):7278–82.
Hobson J, Gummadidala P, Silverstrim B, Grier D, Bunn J, James T, et al. Acute Inflammation induced by the biopsy of mouse mammary tumors promotes the development of metastasis. Breast Cancer Res Treat. 2013;139(2):391–401.
Lawrence WT, Norton JA, Harvey AK, Gorschboth CM, Talbot TL, Grotendorst GR. Wound healing in sarcoma-bearing rats: tumor effects on cutaneous and deep wounds. J Surg Oncol. 1987;35(1):7–12.
McNees P, Meneses KD. Pressure ulcers and other chronic wounds in patients with and patients without cancer: a retrospective, comparative analysis of healing patterns. Ostomy Wound Manag. 2007;53(2):70–8.
Pyter LM, Husain Y, Calero H, McKim DB, Lin HY, Godbout JP, et al. Tumors alter inflammation and impair dermal wound healing in female mice. PLoS One. 2016;11(8):e0161537.
Demidova-Rice TN, Hamblin MR, Herman IM. Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 1: normal and chronic wounds: biology, causes, and approaches to care. Adv Skin Wound Care. 2012;25(7):304–14.
Mustoe T. Understanding chronic wounds: a unifying hypothesis on their pathogenesis and implications for therapy. Am J Surg. 2004;187(Suppl 5A):65S–70.
Cebioglu M, Schild HH, Golubnitschaja O. Cancer predisposition in diabetics: risk factors considered for predictive diagnostics and targeted preventive measures. EPMA J. 2010;1(1):130–7.
Hurlow J, Couch K, Laforet K, Bolton L, Metcalf D, Bowler P. Clinical biofilms: a challenging frontier in wound care. Adv Wound Care (New Rochelle). 2015;4(5):295–301.
Konieczka K, Ritch R, Traverso CE, Kim DM, Kook MS, Gallino A, et al. Flammer syndrome. EPMA J. 2014;5(1):11.
Golubnitschaja O, Debald M, Kuhn W, Yeghiazaryan K, Bubnov RV, Goncharenko VM. lammer Syndrome and potential formation of pre-metastatic niches: a multi-centred study on phenotyping, patient stratification, prediction and potential prevention of aggressive breast cancer and metastatic disease. EPMA J. 2016;7 Suppl 1:A25.
Flammer J, Konieczka K. Retinal venous pressure: the role of endothelin. EPMA J. 2015; 1–12.
Khimji AK, Rockey DC. Endothelin and hepatic wound healing. Pharmacol Res. 2011;63(6):512–8.
Yeghiazaryan K, Flammer J, Orgül S, Wunderlich K, Golubnitschaja O. Vasospastic individuals demonstrate significant similarity to glaucoma patients as revealed by gene expression profiling in circulating leukocytes. Mol Vis. 2009;15:2339–48.
Yeghiazaryan K, Flammer J, Golubnitschaja O. Predictive molecular profiling in blood of healthy vasospastic individuals: clue to targeted prevention as personalised medicine to effective costs. EPMA J. 2010;1(2):263–72.
Golubnitschaja O, Debald M, Yeghiazaryan K, Kuhn W, Pešta M, Costigliola V, et al. Breast cancer epidemic in the early twenty-first century: evaluation of risk factors, cumulative questionnaires and recommendations for preventive measures. Tumor Biol. 2016;37(10):12941–57.
Golubnitschaja O, Yeghiazaryan K, Stricker H, Trog D, Schild HH, Berliner L. Patients with hepatic breast cancer metastases demonstrate highly specific profiles of matrix metalloproteinases MMP-2 and MMP-9 after SIRT treatment as compared to other primary and secondary liver tumours. BMC Cancer. 2016;16:357.
Golubnitschaja O, Yeghiazaryan K, Abraham JA, Schild HH, Costigliola V, Debald M, et al. Breast cancer risk assessment: a non-invasive multiparametric approach to stratify patients by MMP-9 serum activity and RhoA expression patterns in circulating leucocytes. Amino Acids. 2017;49(2):273–281.
Acknowledgements
The authors thank the Scholarship Programme of the German State of North Rhine-Westphalia for students from Israel which has supported the actual project by granting the study stay of Eden Avishai in the scientific group of Prof. Dr. Olga Golubnitschaja at the Radiological clinic, University of Bonn in Germany in the year 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Statement of informed consent
Patients have not been involved in the study
Statement of human and animal rights
No experiments have been performed including patients and/or animals.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Avishai, E., Yeghiazaryan, K. & Golubnitschaja, O. Impaired wound healing: facts and hypotheses for multi-professional considerations in predictive, preventive and personalised medicine. EPMA Journal 8, 23–33 (2017). https://doi.org/10.1007/s13167-017-0081-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13167-017-0081-y