
Abstract
Chemoresistance is one of the major hurdles to overcome for the successful treatment of breast cancer. At present, there are several mechanisms proposed to explain drug resistance to chemotherapeutic agents, including decreased intracellular drug concentrations, mediated by drug transporters and metabolic enzymes; impaired cellular responses that affect cell cycle arrest, apoptosis, and DNA repair; the induction of signaling pathways that promote the progression of cancer cell populations; perturbations in DNA methylation and histone modifications; and alterations in the availability of drug targets. Both genetic and epigenetic theories have been put forward to explain the mechanisms of drug resistance. Recently, a small non-coding class of RNAs, known as microRNAs, has been identified as master regulators of key genes implicated in mechanisms of chemoresistance. This article reviews the role of microRNAs in regulating chemoresistance and highlights potential therapeutic targets for reversing miRNA-mediated drug resistance. In the future, microRNA-based treatments, in combination with traditional chemotherapy, may be a new strategy for the clinical management of drug-resistant breast cancers.
Similar content being viewed by others

Introduction
Breast cancer is the most common malignancy in women that affected an estimated three million women worldwide in 2008 alone (Jemal et al. 2011). Current treatment strategies combine surgery with adjuvant therapy, such as cytotoxic anticancer drugs, hormonal therapy, targeted drugs, or a combination thereof. It is estimated that one of two breast cancer patients will fail to respond to initial treatments or will rapidly acquire resistance to chemotherapeutic agents (O’Driscoll and Clynes 2006). Moreover, the majority of cancer patients, even if they show an initial response to treatment, will develop aggressive malignancies, which exhibit up to 90% resistance to one or more drugs (Ellis and Hicklin 2009; Sorrentino et al. 2008). This clearly suggests that drug resistance, whether intrinsic or acquired over time, constitutes a major hurdle to overcome for the successful treatment of breast cancer.
The underlying mechanisms of the acquisition of resistance to chemotherapeutic agents are still poorly understood. Presently, two main hypotheses, genetic and epigenetic, have been proposed to explain the basis of cancer drug resistance (Baker and El-Osta 2003; Glasspool et al. 2006; Iwasa et al. 2006; Lee et al. 2011). The term “genetic” is commonly used to define a heritable change in the DNA sequence, and according to this model, drug-induced mutational events select for drug-resistant cell populations (Iwasa et al. 2006). Conversely, the “epigenetic” hypothesis refers to heritable changes in gene expression that occur without altering the sequence of DNA, through processes such as DNA methylation and histone modifications (Egger et al. 2004), and suggests that changes in the epigenome facilitates resistance to cytotoxic drugs (Baker and El-Osta 2003; Glasspool et al. 2006).
Although evidence regarding genetic changes following chemotherapeutic treatment is limited, numerous studies have demonstrated substantial epigenetic alterations in drug-resistant cancer cells (Baker et al. 2005; Roberti et al. 2006). It is well documented that epigenetic dysregulation of genes, whose products modulate the intracellular concentration of anticancer agents, such as the multidrug-resistant 1 (MDR1) gene, breast cancer resistance protein (BCRP) gene, and glutathione S-transferase π (GSTπ) gene, influences drug efficacy (Baker et al. 2005; Chekhun et al. 2006). Additionally, alterations in DNA repair proteins, including O6-methylguanine DNA methyltransferase, and increased expression of proteins involved in cell signaling pathways that promote cancer cell survival have been associated with cytosine methylation and histone modifications, clearly indicating the importance of epigenetic mechanisms in drug resistance (Chekhun et al. 2006, 2007; Starlard-Davenport et al. 2010). Furthermore, acquisition of resistance has been associated with profound dysregulation of the epigenomic landscape, suggesting that epigenetic changes may be a driving force in acquiring resistance to anticancer agents (Baylin 2011; Glasspool et al. 2006).
In addition to these well-studied mechanisms of cancer drug resistance, recent studies have linked the acquisition of cancer drug resistance to altered expression of microRNAs (miRNAs; Kovalchuk et al. 2008; Liang et al. 2010; Pogribny et al. 2010; Zhao et al. 2008). Evidence of miRNA-mediated drug resistance is accumulating, and much attention is now being focused on targeting miRNAs as a novel strategy for therapeutic intervention. Future studies will be necessary to address the clinical significance and feasibility of implementing miRNA-based approaches to modulate sensitivity to chemotherapeutic agents.
This review focuses on the role of miRNAs in acquiring the drug-resistant phenotype and how dysregulation of the miRNAome may contribute to the cross-resistance of cancer cells to various chemotherapeutic agents.
miRNA biogenesis and function in normal and tumor cells
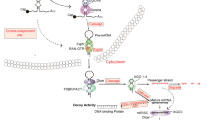
miRNAs are 16- to 29-nucleotide-long single-stranded RNA sequences that function at the post-transcriptional level to negatively regulate gene expression (Bartel 2004; Guo et al. 2010a; Zhang et al. 2009). These mature miRNAs arise from a multistep process (Fig. 1) in which they are first transcribed as long primary transcripts (pri-miRNA) by RNA polymerase II (Lee et al. 2004) or RNA polymerase III (Borchert and LanierW 2006). Nuclear cleavage of the pri-miRNA by Drosha, a RNase III-type enzyme, liberates an ∼60- to 70-nucleotide stem loop intermediate known as the miRNA precursor (pre-miRNA; Filippov et al. 2000; Snyder et al. 2009). These precursor miRNAs are then transported from the nucleus to the cytoplasm by Exportin-5 for further processing by a second RNase III enzyme, Dicer (Kim et al. 2009). The double-stranded complex is unwound to free the mature strand for incorporation into a RNA-induced silencing complex, leaving the second strand to be degraded. Through sequence-specific interactions between the mature miRNA and mRNA, the ribonucleoprotein complex is positioned on either the 3′- or 5′-untranslated region (UTR) of their targets, resulting in mRNA cleavage if complementarity is perfect or, in the case of imperfect base pairing, translational repression (Bartel 2004; Kim et al. 2009; Guo et al. 2010a; Selbach et al. 2008).

miRNA biogenesis and function. Mature miRNAs arise from a multistep process in which they are first transcribed by RNA polymerase II (RNA pol II) as a primary miRNA (pri-miRNA) transcript. After being cleaved in the nucleus by the RNAse III ribonuclease, Drosha, and its cofactor Pasha, the precursor miRNA (pre-miRNA) is exported to the cytoplasm by Exportin 5, where it is cleaved by a second RNAse III ribonuclease, Dicer. This 16- to 29-nucleotide-long miRNA duplex is then unwound to free the mature strand for incorporation into a RNA-induced silencing complex (RISC) and, based on sequence complementarity, directs translational repression or cleavage of its mRNA target by binding to either the 3′- or 5′-untranslated (UTR) regions
Currently, more than 1,200 mammalian miRNAs have been identified that can potentially target up to one third of the protein-coding genes (The miRBase Sequence Database—Release 16.0) involved in development, cell differentiation, metabolic pathways, signal transduction, proliferation, and apoptosis (Bartel 2004; Selbach et al. 2008). Given that cancer is characterized by the deregulation of these very same biological processes (Hanahan and Weinberg 2011), hypothetical conjectures, confirmed by empirical evidence, have indicated that aberrant miRNA expression may play an important role in cancer (Ruan et al. 2009). Indeed, the list of miRNAs found to be up- or down-regulated in cancer, whose targets are tumor suppressor genes or oncogenes, respectively, is rapidly expanding (Andorfer et al. 2011; Garofalo and Croce 2011), and these dysregulated miRNAs have been associated with every aspect of tumor biology, including tumor progression, invasion, metastasis, and acquisition of resistance to various chemotherapeutic agents (Aigner 2011; Blower et al. 2007; Kastl et al. 2011; Korpal et al. 2008; Ma et al. 2010; Pogribny et al. 2010).
miRNAs and mechanisms of drug resistance in breast cancer
At present, there are several mechanisms proposed to explain drug resistance to chemotherapeutic agents, including (1) decreased intracellular drug concentrations, mediated by drug transporters and metabolic enzymes; (2) impaired cellular responses that affect cell cycle arrest, apoptosis, and DNA repair; (3) the induction of signaling pathways that promote the malignant transformation and invasiveness of cell populations; (4) perturbations in DNA methylation and histone modifications; and (5) alterations in the availability of drug targets. These mechanisms have recently been shown to be targeted by miRNAs in drug-resistant breast cancer (Table 1; Kovalchuk et al. 2008; Liang et al. 2010; Pogribny et al. 2010; Xin et al. 2009).
miRNAs in defensive mechanisms
Defensive mechanisms, such as the up-regulation of drug transporters and metabolic enzymes, play an important role in regulating absorption, distribution, and clearance of chemotherapeutic agents and their metabolites (Baguley 2010; Gatti and Zunino 2005). Recent miRNA profiling studies have identified a subset of target genes involved in drug efflux and metabolism (Kovalchuk et al. 2008; Pan et al. 2009; Pogribny et al. 2010; Xin et al. 2009).
Chemotherapeutic failure is often attributed to increases in energy-dependent drug efflux, mediated via ATP-binding cassette (ABC) drug transporters (Gottesman and Ling 2006). The classic representative of this family is P-glycoprotein (P-gp) encoded by the MDR1 gene (Gottesman and Ling 2006) whose overexpression has been associated with cellular resistance to a wide variety of anticancer agents, including anthracycline antibiotics, plant alkaloids, taxanes, and platinum-based drugs (Allen et al. 2003; Zunino et al. 1999). One hypothesis to explain P-gp-mediated resistance is a marked decrease of miR-451, which was directly correlated to elevated levels of its target, P-gp, in MCF-7 cells resistant to doxorubicin (Kovalchuk et al. 2008). Furthermore, MCF-7 cells transfected with miR-451 exhibited enhanced sensitivity to doxorubicin (Kovalchuk et al. 2008). Similarly, BCRP, which shares drug substrates with P-gp (Allen et al. 2003), is also a target for miRNA regulation, specifically miR-328 (Pan et al. 2009; Xin et al. 2009).
Another subfamily of the ABC transporter family, the multidrug resistance-associated proteins (MRPs), acts on a number of drugs, including glutathione (GSH)-conjugated derivatives (Jedlitschky et al. 1996; Konig et al. 1999). Indeed, resistance to cisplatin has been correlated with MRP2-mediated efflux of cisplatin–GSH complexes (Konig et al. 1999; Siddik 2002, 2003). Currently, miR-489 is being explored as a putative regulator of MRP2 whose down-regulation is associated with resistance to cisplatin and doxorubicin (Kovalchuk et al. 2008; Pogribny et al. 2010). Studies in our lab have further demonstrated the importance of miRNA-mediated regulation of MRP in which miR-7 and miR-345 were found to target directly the 3′-UTR of the MRP1 transcript (Pogribny et al. 2010). Decreased levels of these miRNAs conferred resistance to cisplatin in MCF-7 cells (Pogribny et al. 2010). Additionally, loss of miR-326 in MCF-7/VP multidrug-resistant cell lines contributed to the overexpression of MRP1, whereas enforced levels of this miRNA sensitized MCF-7/VP to VP-16 and doxorubicin (Liang et al. 2010). Similar associations were observed in a panel of advanced breast cancers in which decreased levels of miR-326 consistently exhibited an inverse correlation with MRP1 expression levels (Liang et al. 2010).
Sensitivity to chemotherapeutic agents is also modulated by the rate of metabolic clearance. Increased drug metabolism, mediated by the induction of phase I and phase II enzymes, has been correlated with decreased responses to breast cancer treatment (Ingelman-Sundberg and Rodriguez-Antona 2005). Our findings link the up-regulation of cytochrome P450 enzymes, CYP1B1 and CYP3A4, with the down-regulation of miR-27 and a subsequent increase in doxorubicin metabolism (Kovalchuk et al. 2008).
Reversal of drug resistance is of great importance for establishing effective chemotherapeutic practices. In light of recent research indicating a prominent role of miRNAs in reversing resistance to a wide range of drugs (Zhang et al. 2010; Zhao et al. 2010a), miRNA-mediated regulation of drug transporters and metabolic enzymes may prove to be a promising strategy to regulate the absorption, distribution, and clearance of chemotherapeutic agents.
miRNAs in cellular responses to chemotherapeutic agents
Cytotoxic drugs, such as doxorubicin, paclitaxel, and cisplatin, primarily function by damaging the genetic material of cells, thereby interfering with the ability of cancer cells to divide. Several distinct cellular responses are activated to cope with genotoxic damage, including the induction of cell cycle arrest and, if the damage exceeds the cellular capacity for repair, the induction of apoptosis. The fine tuning of pro- and anti-apoptotic programs, which are carefully balanced by miRNAs in normal cells, is shifted toward cell survival in cancerous cells.
Cell cycle arrest
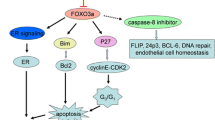
DNA damage is detected by sensor molecules, such as p53, which then initiate checkpoint responses that block the activity of cyclin-dependent kinases (CDKs) and consequently halt cell cycle progression. Loss of p53 is associated with decreased sensitivity to DNA damage and drug resistance. One mechanism to explain this decreased sensitivity is the up-regulation of miR-125, which binds to the 3′-UTR of the p53 gene to block translation (Le et al. 2009) in cell lines resistant to paclitaxel and doxorubicin (Kovalchuk et al. 2008; Zhao et al. 2008). The downstream effects of this response are numerous because p53 activates the expression of several important cell regulatory genes, including cyclin-dependent kinase inhibitor p21 (CDKN1A). Moreover, miR-20, which targets p21 (CDKN1A; Trompeter et al. 2011) is up-regulated in drug-resistant cell lines (Kovalchuk et al. 2008), further decreasing the levels of this negative cell cycle regulator. Loss of p21, which normally binds to CDK2 to prevent cells from entering the S-phase, may be one of the mechanisms enabling cancer cells to progress through the cell cycle with DNA damage. The G1/S checkpoint is also targeted by several other miRNAs dysregulated in drug-resistant breast cancers, including miR-221/222 and miR-214 (Kovalchuk et al. 2008; Pogribny et al. 2010; Zhao et al. 2008), both of which target PTEN (Garofalo et al. 2009; Li et al. 2011), a CDK inhibitor, and the miR-16 family, miR-29, miR-34, and miR-107 (Liang et al. 2010; Kovalchuk et al. 2008; Pogribny et al. 2010), which target CDK6 (Feng et al. 2011; Liu et al. 2008; Sun et al. 2008; Zhao et al. 2010b).
CDKs, when bound to their cyclin counterparts, actively phosphorylate substrates to regulate the entry of the cell into the cell cycle. Phosphorylation of RB releases E2F, which enters the nucleus to activate the transcription of cell cycle-related genes to promote the cell’s entry into the S-phase. Both RB and E2F are targets for miRNA repression, including miR-17, miR-20, miR-34, miR-93, miR-106, miR-205, and miR-331 (Dar et al. 2011; Guo et al. 2010b; Tazawa et al. 2007; Trompeter et al. 2011) whose expression is dysregulated in genotoxic drug resistance (Kovalchuk et al. 2008; Pogribny et al. 2010). The inhibitory phosphate group can be removed by cell division control proteins (CDC), which are also a target for miRNAs, including miR-29 and miR-224 (Park et al. 2009; Zhu et al. 2010a), both of which are up-regulated in drug-resistant cells (Kovalchuk et al. 2008; Liang et al. 2010; Pogribny et al. 2010).
To add further to the complexity, there are many miRNAs dysregulated in chemoresistant cell lines (Kovalchuk et al. 2008; Liang et al. 2010; Xin et al. 2009) that target cyclins directly, including let-7, miR-16, miR-17, and miR-34 (Liu et al. 2008; Schultz et al. 2008; Sun et al. 2008; Yu et al. 2008). Clearly, miRNAs play a key role in regulating cell cycle progression in drug-resistant cell lines by targeting mRNA at several different stages.
Apoptosis
The ability to evade programmed cell death is one of the hallmarks of cancer cells (Hanahan and Weinberg 2011). Cytotoxic drugs, such as cisplatin, are designed to reactivate apoptotic pathways by inducing stress pathways, such as via p38, or by suppressing MAPK/AKT signaling (Winograd-Katz and Levitzki 2006). Activation of MAPK/AKT through the up-regulation of several oncogenic miRNAs that target PTEN (e.g., miR-22, miR-205, miR-214, and miR221/222; Bar and Dikstein 2010; Garofalo et al. 2009; Greene et al. 2010; Li et al. 2011), a negative regulator of AKT, and the down-regulation of tumor suppressor miR-375, which targets 14-3-3zeta (Tsukamoto et al. 2010), an integral component of the AKT pathway, has been associated with resistance to DNA-damaging agents (Chen et al. 2010; Kovalchuk et al. 2008; Pogribny et al. 2010; Xin et al. 2009). It has been demonstrated that enforced down-regulation of 14-3-3zeta, which is overexpressed in 40% of breast cancer cases, sensitized drug-resistant cell populations to stress-induced apoptosis (Neal et al. 2009; Niemantsverdriet et al. 2008).
Downstream of AKT, imbalances in the BCL2 superfamily, favoring cell survival over apoptosis, have also been associated with miRNA-mediated drug resistance. Reduced suppression of BCL2, arising from the down-regulation of miR-15, miR-16, miR-21, miR-34, and miR-181 (Cimmino et al. 2005; Ji et al. 2008; Zhu et al. 2010b), in cell lines resistant to apoptosis-inducing drugs (Chen et al. 2010; Kovalchuk et al. 2008; Zhou et al. 2010) may contribute to the enhanced tumorigenicity and metastatic potential of BCL2 overexpressing malignant cell populations (Del Bufalo et al. 1997). Moreover, the BCL2 homologous antagonist/killer (BAK1) is suppressed by overexpression of miR-125, a biomarker of paclitaxel resistance (Zhou et al. 2010), further supporting a reduced apoptotic program. Studies have shown that the loss of miR-125 may increase the sensitivity of breast cancer patients to anthracycline-based chemotherapy (Climent et al. 2007). In contrast to miR-125, loss of miR-127 is associated with increased cell survival in cell lines resistant to cisplatin and doxorubicin (Kovalchuk et al. 2008; Pogribny et al. 2010) by targeting BCL6 (Saito et al. 2006). Several BCL2 family members are also indirectly dysregulated by the overexpression of miR-155 and miR-221/222, which inhibit apoptosis by suppressing the FOXO3 transcription factor (Di Leva et al. 2010; Kong et al. 2008).
The anti-apoptotic program is further reinforced at one of the final stages of apoptosis regulation as a result of the effector caspase-3 being suppressed by let-7 (Tsang and Kwok 2008), a miRNA found up-regulated in cyclophosphamide-resistant breast cancer (Salter et al. 2008). Transfection of MDA-MB-435 breast cancer cells with anti-apoptotic miRNAs, such as miR-145 or miR-155, was also shown to block caspase-3 activity (Ovcharenko et al. 2007).
Taken together, these findings suggest that numerous miRNAs are modulated as part of a complex network establishing an anti-apoptotic program in cells resistant to apoptosis-inducing chemotherapeutic agents.
DNA repair
Up-regulation of DNA repair mechanisms, although important for maintaining genetic integrity in normal cells, has been associated with resistance to genotoxic drugs including alkylating agents, platinum compounds, and topoisomerase inhibitors (Reardon et al. 1999). Activation of the nucleotide excision repair (NER) pathway has been shown to protect cells against drug-induced DNA damage (Reardon et al. 1999), indicating that the down-regulation of miR-373 (Xin et al. 2009), which suppresses the NER repair protein, RAD23B, may facilitate DNA repair and cell survival in drug-resistant cell lines.
Alternatively, defects in repair pathways may provide a source of mutations contributing to the selective evolution of progressively advanced malignant cell populations. Tumors with defects in the mismatch repair (MMR) pathway, which removes single-base mispairs and mismatched loops incorporated as a result of drug exposure, exhibit a marked resistance to the very anticancer agents that create substrates for the MMR system (Aebi et al. 1996). Indeed, drug-resistant breast cancer cell lines exhibit dysregulated expression of miR-141 and miR-21, which target two key members of the MMR system, human DNA MutS homolog 2 (hMSH2) and 6 (hMSH6; Bandrés et al. 2006; Valeri et al. 2010).
Similarly, defects in breast cancer 1 (BRCA1), involved in the repair of double-strand breaks and interstrand cross-links, diminish the apoptotic response to chemotherapeutic agents (Thangaraju et al. 2000). Although mutations in the BRCA1 gene are well known, recent studies have shown that variants in pri-miR-17 only occurred in a subset of non-carriers of BRCA1/2 mutations (Shen et al. 2010) and that miR-17, commonly up-regulated in cancer (Kovalchuk et al. 2008; Liang et al. 2010; Salter et al. 2008), could bind the 3′-UTR of BRCA1 mRNA (Shen et al. 2010). Additionally, miR-28, miR-146, and miR-182 were also shown to target BRCA1 (Yao and Ventura 2011), and each of these miRNAs has been shown to be dysregulated in breast cancer cell lines resistant to genotoxic agents (Kovalchuk et al. 2008; Liang et al. 2010). Additionally, activation of BRCA1 is disrupted by deficiencies in the DNA damage sensor, ATM (Wang et al. 2000). One possible mechanism mediating the decrease in ATM is via the direct targeting of the ATM gene transcript by miR-100 (Ng et al. 2010), which were found to be up-regulated in doxorubicin-resistant cell lines (Kovalchuk et al. 2008). This is likely to have broad repercussions for DNA repair because BRCA1 and ATM are intimately connected to the BRCA1-associated genome surveillance complex involved in the recognition and repair of aberrant DNA structures (Wang et al. 2000).
miRNAs in epithelial-to-mesenchymal transition
The conversion of cancer cells from an epithelial phenotype to a more aggressive mesenchymal phenotype through epithelial-to-mesenchymal transition (EMT) has been associated with multidrug resistance and poor clinical outcome (Hiscox et al. 2004; May et al. 2011; Nuyten et al. 2006). Indeed, one of the features of drug-resistant cells is enhanced invasiveness, arising from the altered expression of microtubule regulatory proteins, such as E-cadherin and actin, and the subsequent dysregulation of the extracellular matrix (Wang et al. 2010b)
The induction of EMT by transforming growth factor beta (TGF-β), in coordination with the Ras and Wnt pathways, is characterized by the dysregulation of several key mRNAs, which may contribute, in part, to the altered gene patterns observed during EMT (Kong et al. 2008; May et al. 2011; Wang et al. 2010b). Up-regulation of miR-21 and miR-155 by TGF-β, whose targets include RECK, MASPIN, TPM1, and the Ras homolog, RHOA, has been shown to promote tumor progression, metastasis, and multidrug resistance (Blower et al. 2007; Kong et al. 2008; Yu et al. 2010). In contrast, the enforced down-regulation of these miRNAs has suppressed TGF-β-induced EMT and subsequently sensitized breast cancer cells to taxol by reducing cell viability and invasiveness (Kong et al. 2008; Mei et al. 2010). Other miRNAs targeting the TGF-β signaling pathway include miR-15, miR-128, miR-141, and miR-211 (Burk et al. 2008; Martello et al. 2007; Masri et al. 2010; Wang et al. 2010a).
TGF-β-induced EMT is often accompanied by the loss of E-cadherin expression associated with epigenetic remodeling at the CDH1 promoter (Tryndyak et al. 2010), as well as post-translation repression by miRNAs (Ma et al. 2010). Indirect targeting of E-cadherin by the miR-200 family, through the action of the transcriptional co-repressors ZEB1 and ZEB2, has also been linked to more aggressive breast cancers (Burk et al. 2008). This TGF-β-driven mechanism (Korpal et al. 2008) may explain the low levels of E-cadherin and high expression of mesenchymal markers detected in breast cancer after chemotherapy (Creighton et al. 2009), an important observation since the loss of E-cadherin increases resistance to doxorubicin, actinomycin D, and paclitaxel (Gupta et al. 2009). Conversely, enforced expression of miR-200 inhibits EMT by restoring E-cadherin expression through the down-regulation of ZEB1 and ZEB2 and increases sensitivity to microtubule-targeting agents (Cochrane et al. 2009). Enhanced response to chemotherapeutic agents can also be induced by depleting other E-cadherin repressors, such as the transcription factor TWIST and the metastasis regulator miR-9 (Li and Zhou 2011; Ma et al. 2010). TWIST-induced activation of beta-catenin and AKT pathways has been shown to be necessary for the maintenance of EMT-associated cancers (Li and Zhou 2011), suggesting a complex interplay between mechanisms regulating cellular proliferation, apoptosis, and cell adhesion.
miRNAs in regulating epigenetics
It is widely known that DNA methylation and histone modifications play a prominent role in regulating the expression of genes involved in breast cancer progression and drug resistance; however, evidence for miRNA-mediated epigenetic changes has only been recently been brought to light.
DNA methylation
Aberrant DNA methylation patterns are a prominent hallmark of cancer cells (Hanahan and Weinberg 2011). The global loss of genomic methylation, as well as regional hyper- and hypomethylation of genes involved in cell signaling, proliferation, and apoptosis, is thought to favor cell survival and tumor progression (Hanahan and Weinberg 2011; Pogribny and Beland 2009). There are three DNA methyltransferases (DNMTs) that have been shown to play a prominent role in cancer-specific DNA methylation, including maintenance DNMT1 and de novo DNA methyltransferases DNMT3A and DNMT3B. Although the mechanisms underlying elevated levels of DNMT remain unclear, recent reports suggest a role of miR-29, miR-148, miR-152, and miR-194 in mediating DNMT expression (Duursma et al. 2008; Fabbri et al. 2007; Meng et al. 2010; Pan et al. 2010) in drug-resistant breast cancer cell lines (Kutanzi et al., unpublished data; Pogribny et al. 2010). Importantly, the down-regulation of miR-148 has been shown to correlate with tumor stage in human breast cancer patients (Kutanzi, unpublished data), suggesting that the loss of this miRNA may contribute to methylation-specific patterns of tumor progression. Hypermethylation of miR-148 by DNMTs in the early stages of tumor development was shown to repress miR-148 gene expression (Lujambio et al. 2008), possibly indicating that a positive feedback loop exists to reinforce the overexpression of DNMTs in breast cancer. Furthermore, reactivation of miR-148 upon treatment with a DNA demethylating agent was associated with reduced tumor growth and inhibition of metastasis (Lujambio et al. 2008).
Targeting other components of the DNA methylation machinery has also been shown to influence methylation patterns and contribute to tumorigenesis (Pulukuri and Rao 2006; Wada et al. 2010; Yaqinuddin et al. 2008). Indeed, several miRNAs, such as miR-132 and miR-194, which target the methyl-binding protein MeCP2, were also found to be up-regulated in cell lines resistant to cisplatin and doxorubicin (Kovalchuk et al. 2008; Pogribny et al. 2010). Given that MeCP2 links DNA methylation with histone modifications (Fuks et al. 2003), these miRNAs have the potential to affect epigenetic-mediated drug resistance on a large scale.
Histones
Alterations in chromatin structure have been linked to the aberrant expression of oncogenes and tumor suppressor genes regulating tumor progression and chemoresistance (Baker and El-Osta 2003; Baylin 2011; Hanahan and Weinberg 2011). Aberrant expression of chromatin-modifying enzymes, which add functional groups to amino acid residues on core histone tails to alter the degree of DNA packaging, plays a key role in establishing the cancer epigenome (Baker and El-Osta 2003; Baylin 2011). Imbalances in histone acetyltransferase and deacetylase expression are mediated, in part, by a number of miRNAs, many of which are differentially expressed in parental and drug-resistant breast cancer cell lines (Kovalchuk et al. 2008; Liang et al. 2010; Pogribny et al. 2010), which suggests that the perturbation of chromatin remodeling complexes may contribute to the establishment of a drug-resistant phenotype. Furthermore, the up-regulation of miR-101, which targets EZH2, the enzyme responsible for trimethylating histone H3 lysine 27 to establish a repressive chromatin state, has been linked to tamoxifen and fulvestrant resistance (Rao et al. 2011; Sachdeva et al. 2011). Histone demethylation may also be affected by the down-regulation of miR-342, which is predicted to target JMJD1 demethylase, and has been linked to cisplatin resistance (Pogribny et al. 2010). It should also be pointed out that numerous other miRNAs aberrantly expressed in drug-resistant tumors (Chen et al. 2010; Kovalchuk et al. 2008; Pogribny et al. 2010; Xin et al. 2009; Zhao et al. 2008) have confirmed targets whose products bind to histone remodeling complexes to influence chromatin structure.
From a rather simplistic point of view, the observation that several different mechanisms of drug resistance exhibit a common theme of epigenetic regulation indicates that future studies analyzing the significance of cancer-specific miRNA-mediated chromatin remodeling may identify novel targets for therapeutic intervention.
miRNAs in deregulation of drug targets
The efficacy of chemotherapeutic agents is greatly influenced by cellular events that influence drug–target interactions. More specifically, alterations in drug target levels, including hormone and growth factor receptors, are often a determining factor of drug sensitivity. The identification of miRNAs that target these receptors and their cofactors has provided insight into the mechanisms of miRNA-mediated resistance to hormonal and targeted therapies.
Estrogen receptor-α
Estrogen receptor (ER) expression is an important determinant of endocrine responsiveness. Given that ∼70% of breast cancers are ER-positive, many endocrine therapies, including tamoxifen, target the steroid receptor to reduce estrogen-driven cellular proliferation (Ariazi et al. 2006). These drugs, however, are ineffective for treating breast tumors that lose expression of the ER, as it often happens in more advanced breast cancers (Ariazi et al. 2006). It has been suggested that increased expression of miR-101, miR-206, and miR-221/222, which translationally repress the ER, may be responsible for the decreased sensitivity of breast tumors to anti-estrogen drugs (Kondo et al. 2008; Rao et al. 2011; Sachdeva et al. 2011; Zhao et al. 2008). Profiling of ER-α-negative tumors has clearly identified miR-206 and miR-221/222 as being up-regulated (Kondo et al. 2008; Zhao et al. 2008), suggesting that a negative feedback mechanism reinforces the phenotype.
miRNA-mediated targeting of ER-α cofactors, which influence agonistic and antagonistic effects of ligand binding, has also been linked to impaired chemotherapeutic response. Indeed, one of the major co-activators for ER-α, amplified in breast cancer (AIB1), is a target for miR-17 and miR-106, which are found to be dysregulated in breast cancer cells exhibiting drug resistance (Kovalchuk et al. 2008; Liang et al. 2010). Similarly, the transcriptional co-repressor, receptor interacting protein 140 (RIP140), was found to be targeted by miR-346, which is down-regulated in endocrine-resistant cell lines (Xin et al. 2009), indicating a role for miRNAs in regulating estrogen-responsive genes.
Epidermal growth factor receptor
Overexpression of the human epidermal growth factor (HER) family members occurs in ∼20–25% of invasive breast cancer cases and correlates with poor patient prognosis (Murphy and Modi 2009). Over half of the breast cancer patients who undergo HER2-targeted therapy with trastuzumab fail to respond. It has been speculated that drug efficacy is reduced by the continuous overexpression of the drug target, a phenomenon that may be mediated by the down-regulation of miR-125 and miR-331 (Kovalchuk et al. 2008) whose normal function is to suppress HER2. Moreover, high expression of HER2 in cancer cells has been shown to induce ligand-independent constitutive activation of the receptor and its downstream MAPK and PI3K signaling pathways, thereby facilitating cross talk with the ER to induce multidrug resistance (Britton et al. 2006). miRNA-based suppression of EGFR-induced PI3K activation, including the repression of other EGFR members, such as HER3 by miR-205 (Iorio et al. 2009), may be an important avenue to explore for re-sensitizing breast cancer cells that have acquired cross-resistance to chemotherapeutic agents.
Limitations and future considerations
Clearly, aberrant expression of miRNAs is an important feature of cancer cells with an acquired drug-resistant phenotype and may be a critical factor contributing to its development. Indeed, miRNAs are implicated in several cellular responses to drug exposure, including, but not limited to, drug influx/efflux, cell cycle arrest, DNA repair, and apoptosis, all of which mediate cancer cell survival and tumor progression. The dysregulation of similar pathways in cancer cells exhibiting resistance to a wide range of drugs may explain the existence, and provide a mechanism, of cross-resistance to different types of chemotherapeutic agents.
Despite the growing evidence of cancer-specific miRNA patterns, the role of miRNAs in cancer is complex and difficult to unravel. The downstream effects of even a single miRNA are complicated by the number of targets it may have, with predictions ranging from one to hundreds of transcripts (Xin et al. 2009). Moreover, several miRNAs, which may be differentially regulated, can target the transcript of a single gene, making it difficult to predict the net result. It is believed, however, that mRNAs targeted by multiple down-regulated miRNAs are more likely to be overexpressed than mRNAs targeted by a single miRNA (Xin et al. 2009) and that mRNAs possessing multiple target sites for similarly expressed miRNAs have a greater probability of being affected than those with only one site (Xin et al. 2009). Furthermore, since a number of miRNAs can target DNA and histone-modifying enzymes, they are likely to affect gene expression on a much broader scope. For this reason, studies need to address the wide range of effects of even a single dysregulated miRNA in order to completely understand the complex network of feedback mechanisms and cross talk between pathways. To complicate matters further, recent papers demonstrate that miRNA function is tissue-specific and context-dependent, switching from repressors to activators in different circumstances (Liu and Kohane 2009; Vasudevan et al. 2007). From a clinical perspective, given that miRNAs can induce a diversity of effects, caution must be exercised when extrapolating findings from in vitro to in vivo.
Despite the difficulties to overcome, the value of miRNAs in clinical applications is projected to be monumental. Indeed, miRNA profiling has been convincingly demonstrated to classify tumor and non-tumor samples more effectively than gene expression profiling and, more importantly, identifying breast cancer subtypes (Blenkiron et al. 2007; Iorio et al. 2005; Lu et al. 2005). The diagnostic role of miRNAs is crucial since different breast cancer subtypes exhibit different responsiveness to chemotherapeutic agents (Carey et al. 2007; Rouzier et al. 2005). In the future, predicting responses to cytotoxic therapy will ideally involve a combination of gene expression data, miRNA profiling, and receptor status determination, with the goal of developing a personalized treatment strategy to improve patient outcome. Current studies are also investigating the value of miRNAs as early plasma biomarkers, with the expectation that they will exhibit a greater degree of specificity and sensitivity over the current biomarkers (Heneghan et al. 2010; Lodes et al. 2009; Roth et al. 2010; Zhao et al. 2010a). The detection of cancer-specific miRNAs in plasma provides a relatively noninvasive method for monitoring predictive markers (Heneghan et al. 2010; Lodes et al. 2009). Moreover, profiling of miRNAs that regulate genes known to be predictive of drug resistance can potentially be used in guiding drug treatment decisions.
Evidence of miRNA-mediated reversal of multidrug resistance in human cancer (Zhang et al. 2010; Zhao et al. 2010c) warrants further studies of miRNA-based approaches for treating drug-resistant tumors. One of the most promising therapeutic targets is miR-21, commonly found to be up-regulated in breast cancer, which is associated with enhanced metastatic potential and drug resistance. In a systematic search for miRNAs mediating drug efficacy, miR-21 was identified as having the capacity to alter the potency of a wide range of chemotherapeutic agents by up to fourfold (Blower et al. 2007), which suggests that targeting miR-21 may enhance sensitivity to multiple chemotherapeutic drugs. Indeed, enforced down-regulation of miR-21 has been shown to increase levels of PTEN and caspase-3, which are associated with an increased number of apoptotic cells and reduced invasiveness leading to drug sensitivity (Bourguignon et al. 2009; Mei et al. 2010; Ren et al. 2010). Preliminary evidence has also demonstrated the viability of delivering both miRNA and drug treatments at the same time to improve the response to chemotherapeutic agents. A study by Mei et al. (2010) showed that a G5-PAMAM dendrimer could be utilized to deliver a miR-21 inhibitor and taxol simultaneously to effectively reduce tumor growth and invasiveness. In the future, miRNA-based treatments, in combination with traditional chemotherapy, may be a new strategy for the clinical management of drug-resistant breast cancers.
References
Aebi S, Kurdi-Haidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, Howell SB (1996) Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res 56:3087–3090
Aigner A (2011) MicroRNAs (miRNAs) in cancer invasion and metastasis: therapeutic approaches based on metastasis-related miRNAs. J Mol Med 89:445–457
Allen JD, van Dort SC, Buitelaar M, van Tellingen O, Schinkel AH (2003) Mouse breast cancer resistance protein (Bcrp1/Abcg2) mediates etoposide resistance and transport, but etoposide oral availability is limited primarily by P-glycoprotein. Cancer Res 63:1339–1344
Andorfer CA, Necela BM, Thompson EA, Perez EA (2011) MicroRNA signatures: clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol Med 17(6):313–319.
Ariazi EA, Ariazi JL, Cordera F, Jordan VC (2006) Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 6:181–202
Baguley BC (2010) Multiple drug resistance mechanisms in cancer. Mol Biotechnol 46:308–316
Baker EK, El-Osta A (2003) The rise of DNA methylation and the importance of chromatin on multidrug resistance in cancer. Exp Cell Res 290:177–194
Baker EK, Johnstone RW, Zalcberg JR, El-Osta A (2005) Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene 24:8061–8075
Bandrés E, Cubedo E, Agirre X, Malumbres R, Zárate R, Ramirez N, Abajo A, Navarro A, Moreno I, Monzó M, García-Foncillas J (2006) Identification by real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer 5:29–38
Bar N, Dikstein R (2010) miR-22 forms a regulatory loop in PTEN/AKT pathway and modulates signaling kinetics. PLoS One 5(5):e10859
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–97
Baylin SB (2011) Resistance, epigenetics and the cancer ecosystem. Nat Med 17:288–289
Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, Barbosa-Morais NL, Teschendorff AE, Green AR, Ellis IO, Tavaré S, Caldas C, Miska EA (2007) MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol 8:R214
Blower PE, Verducci JS, Lin S, Zhou J, Chung JH, Dai Z, Liu CG, Reinhold W, Lorenzi PL, Kaldjian EP, Croce CM, Weinstein JN, Sadee W (2007) MicroRNA expression profiles for the NCI-60 cancer cell panel. Mol Cancer Ther 6:1483–1491
Borchert GM, LanierW DBL (2006) RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 13:1097–1101
Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E (2009) Hyaluronan–CD44 interaction with protein kinase C (epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J Biol Chem 284:26533–26546
Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, Gee JM, Nicholson RI (2006) Bidirectional cross talk between ERa and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat 96:131–146
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9:582–589
Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM (2007) The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 13:2329–2334
Chekhun VF, Kulik GI, Yurchenko OV, Tryndyak VP, Todor IN, Luniv LS, Tregubova NA, Pryzimirska TV, Montgomery B, Rusetskaya NV, Pogribny IP (2006) Role of DNA hypomethylation in the development of the resistance to doxorubicin in human MCF-7 breast adenocarcinoma cells. Cancer Lett 231:87–93
Chekhun VF, Lukyanova NY, Kovalchuk O, Tryndyak VP, Pogribny IP (2007) Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethylated targets. Mol Cancer Ther 6:1089–1098
Chen GQ, Zhao ZW, Zhou HY, Liu YJ, Yang HJ (2010) Systematic analysis of microRNA involved in resistance of the MCF-7 human breast cancer cell to doxorubicin. Med Oncol 27:406–415
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102(39):13944–13949
Climent J, Dimitrow P, Fridlyand J, Palacios J, Siebert R, Albertson DG, Gray JW, Pinkel D, Lluch A, Martinez-Climent JA (2007) Deletion of chromosome 11q predicts response to anthracycline-based chemotherapy in early breast cancer. Cancer Res 67:818–826
Cochrane DR, Spoelstra NS, Howe EN, Nordeen SK, Richer JK (2009) MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther 8:1055–1066
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC (2009) Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA 106:13820–13825
Dar AA, Majid S, de Semir D, Nosrati M, Bezrookove V, Kashani-Sabet M (2011) miRNA-205 suppresses melanoma cell proliferation and induces senescence via regulation of E2F1 protein. J Bio Chem 286(19):16606–16614
Del Bufalo D, Biroccio A, Leonetti C, Zupi G (1997) Bcl-2 overexpression enhances the metastatic potential of a human breast cancer line. FASEB J 11:947–953
Di Leva G, Gasparini P, Piovan C, Ngankeu A, Garofalo M, Taccioli C, Iorio MV, Li M, Volinia S, Alder H, Nakamura T, Nuovo G, Liu Y, Nephew KP, Croce CM (2010) MicroRNA cluster 221–222 and estrogen receptor α interactions in breast cancer. J Natl Cancer Inst 102:706–721
Duursma AM, Kedde M, Schrier M, le Sage C, Agami R (2008) miR-148 targets human DNMT3b protein coding region. RNA 14:872–877
Egger G, Liang G, Aparicio A, Jones PA (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429:457–463
Ellis LM, Hicklin D (2009) Resistance to targeted therapies: refining anticancer therapy in the era of molecular oncology. Clin Cancer Res 15:7471–7478
Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 104:15805–15810
Feng L, Xie Y, Zhang H, Wu Y (2011) miR-107 targets cyclin-dependent kinase 6 expression, induces cell cycle G1 arrest and inhibits invasion in gastric cancer cells. Med Oncol. doi:10.1007/s12032-011-9823-1
Filippov V, Solovyev V, Filippova M, Gill SS (2000) A novel type of RNase III family proteins in eukaryotes. Gene 245:213–221
Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T (2003) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278:4035–4040
Gatti L, Zunino F (2005) Overview of tumor cell chemoresistance mechanisms. Methods Mol Med 111:127–148
Garofalo M, Croce CM (2011) microRNAs: master regulators as potential therapeutics in cancer. Ann Rev Pharmacol Toxicol 51:25–43
Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Tacioli C, Pichiorri F, Alder H, Secchiero P, Gasparini P, Gonelli A, Costinean S, Acunzo M, Condorelli G, Croce CM (2009) miR-221 & 222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 16(6):498–509
Glasspool RM, Teodoridis JM, Brown R (2006) Epigenetics as a mechanism driving polygenic clinical drug resistance. Br J Cancer 94:1087–1092
Gottesman MM, Ling V (2006) The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 580:998–1009
Greene SB, Gunaratne PH, Hammond SM, Rosen JM (2010) A putative role for microRNA-205 in mammary epithelial cell progenitors. J Cell Sci 123(Pt 4):606–618
Guo HA, Ingolia NT, Weissman JS, Bartel DP (2010a) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466(7308):835–840
Guo XB, Guo L, Ji J, Zhang J, Zhang J, Chen X, Cai Q, Li J, Gu Q, Liu B, Zhu Z, Yu Y (2010b) miRNA-331-3p directly targets E2F1 and induces growth arrest in human gastric cancer. Biochem Biophys Res Commun 398(1):1–6
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138:645–659
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Heneghan HM, Miller N, Lowery AJ, Sweeney KJ, Newell J, Kerin MJ (2010) Circulating microRNAs as novel minimally invasive biomarkers for breast cancer. Ann Surg 251:499–505
Hiscox S, Morgan L, Barrow D, Dutkowskil C, Wakeling A, Nicholson RI (2004) Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib (‘Iressa’, ZD1839). Clin Exp Metastasis 21:201–212
Ingelman-Sundberg M, Rodriguez-Antona C (2005) Pharmacogenetics of drug-metabolizing enzymes: implications for a safer and more effective drug therapy. Philos Trans R Soc Lond B Biol Sci 360:1563–1570
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM (2005) MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65:7065–7070
Iorio MV, Casalini P, Piovan C, Di Leva G, Merlo A, Triulzi T, Ménard S, Croce CM, Tagliabue E (2009) microRNA-205 regulates HER3 in human breast cancer. Cancer Res 69:2195–2200
Iwasa Y, Nowak MA, Michor F (2006) Evolution of resistance during clonal expansion. Genetics 172:2557–2566
Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D (1996) Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res 56:988–994
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D, Xu L (2008) Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 8:266
Kastl L, Brown I, Schofield AC (2011) miRNA-34a is associated with docetaxel resistance in human breast cancer cells. Breast Cancer Res Treat. doi:10.1007/s10549-011-1424-3
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139
Kondo N, Toyama T, Sugiura H, Fujii Y, Yamashita H (2008) miR-206 expression is down-regulated in estrogen receptor alpha-positive human breast cancer. Cancer Res 68:5004–5008
Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ (2008) MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 28:6773–6784
Konig J, Nies AT, Cui Y, Leier I, Keppler D (1999) Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta 1461:377–394
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914
Kovalchuk O, Filkowski J, Meservy J, Ilnytskyy Y, Tryndyak VP, Chekhun VF, Pogribny IP (2008) Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther 7:2152–2159
Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, Lodish HF, Lim B (2009) MicroRNA-125b is a novel negative regulator of p53. Genes Dev 23:862–876
Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, Kschischo M, Swanton C (2011) Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 71:1858–1870
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23(20):4051–4060
Li J, Zhou BP (2011) Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 11:49
Li LM, Hou DX, Guo YL, Yang JW, Liu Y, Zhang CY, Zen K (2011) Role of microRNA-214-targeting phosphatase and tensin homolog in advanced glycation end product-induced apoptosis delay in monocytes. J Immunol 186(4):2552–25560
Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang K, Wagar N, Yoon Y, Cho HT, Scala S, Shim H (2010) Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem Pharmacol 79:817–824
Liu H, Kohane IS (2009) Tissue and process specific microRNA-mRNA co-expression in mammalian development and malignancy. PLoS One 4(5):e5436
Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2008) miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res 36(16):5391–5404
Lodes MJ, Caraballo M, Suciu D, Munro S, Kumar A, Anderson B (2009) Detection of cancer with serum miRNAs on an oligonucleotide microarray. PLoS One 4:e6229
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838
Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM, Esteller M (2008) A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 105:13556–13561
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA (2010) miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 12:247–256
Martello G, Zacchigna L, Inui M, Montagner M, Adorno M, Mamidi A, Morsut L, Soligo S, Tran U, Dupont S, Cordenonsi M, Wessely O, Piccolo S (2007) MicroRNA control of nodal signalling. Nature 449:183–188
Masri S, Liu Z, Phung S, Wang E, Yuan YC, Chen S (2010) The role of microRNA-128a in regulating TGFbeta signaling in letrozole-resistant breast cancer cells. Breast Cancer Res Treat 124:89–99
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA (2011) Epithelial–mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 13:202
Mei M, Ren Y, Zhou X, Yuan XB, Han L, Wang GX, Jia Z, Pu PY, Kang CS, Yao Z (2010) Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat 9:77–86
Meng Z, Fu X, Chen X, Zeng S, Tian Y, Jove R, Xu R, Huang W (2010) miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology 52:2148–2157
Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S (2008) MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem 283:29897–29903
Murphy CG, Modi S (2009) HER2 breast cancer therapies: a review. Biologics 3:289–301
Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, Danes CG, Guo H, Lan KH, Ensor J, Hittelman W, Hung MC, Yu D (2009) 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res 69:3425–3432
Niemantsverdriet M, Wagner K, Visser M, Backendorf C (2008) Cellular functions of 14-3-3 zeta in apoptosis and cell adhesion emphasize its oncogenic character. Oncogene 27:1315–1319
Ng WL, Yan D, Zhang X, Mo YY, Wang Y (2010) Over-expression of miR-100 is responsible for the low-expression of ATM in the human glioma cell line:M059J. DNA Repair (Amst) 9(11):1170–1175
Nuyten DS, Kreike B, Hart AA, Chi JT, Sneddon JB, Wessels LF, Peterse HJ, Bartelink H, Brown PO, Chang HY, van de Vijver MJ (2006) Predicting a local recurrence after breast-conserving therapy by gene expression profiling. Breast Cancer Res 8:R62
O’Driscoll L, Clynes M (2006) Biomarkers and multiple drug resistance in breast cancer. Curr Cancer Drug Tar 6:365–384
Ovcharenko D, Kelnar K, Johnson C, Leng N, Brown D (2007) Genome-scale microRNA and small interfering RNA screens identify small RNA modulators of TRAIL-induced apoptosis pathway. Cancer Res 67:10782–10788
Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, Li J, Zhou H, Tang Y, Shen N (2010) MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol 184:6773–6781
Pan YZ, Gao W, Yu AM (2009) MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab Dispos 37:2112–2117
Park SY, Lee JH, Ha M, Nam JW, Kim VN (2009) miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol 16(1):23–29
Pogribny IP, Beland FA (2009) DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci 66:2249–2261
Pogribny IP, Filkowski JN, Tryndyak VP, Golubov A, Shpyleva SI, Kovalchuk O (2010) Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int J Cancer 127:1785–1794
Pulukuri SM, Rao JS (2006) CpG island promoter methylation and silencing of 14-3-3sigma gene expression in LNCaP and Tramp-C1 prostate cancer cell lines is associated with methyl-CpG-binding protein MBD2. Oncogene 25:4559–4572
Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, Burow ME, Ivan M, Croce CM, Nephew KP (2011) MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 30:1082–1097
Reardon JT, Vaisman A, Chaney SG, Sancar A (1999) Efficient nucleotide excision repair of cisplatin, oxaliplatin, and bis-aceto-ammine-dichlorocyclohexylamine–platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res 59:3968–3971
Ren Y, Zhou X, Mei M, Yuan XB, Han L, Wang GX, Jia ZF, Xu P, Pu PY, Kang CS (2010) MicroRNA-21 inhibitor sensitizes human glioblastoma cells U251 (PTEN-mutant) and LN229 (PTEN-wild type) to taxol. BMC Cancer 10:27
Roberti A, La Sala D, Cinti C (2006) Multiple genetic and epigenetic interacting mechanisms contribute to clonally selection of drug-resistant tumors: current views and new therapeutic prospective. J Cell Physiol 207:571–581
Roth C, Rack B, Müller V, Janni W, Pantel K, Schwarzenbach H (2010) Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res 12:R90
Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M, Anderson K, Hess KR, Stec J, Ayers M, Wagner P, Morandi P, Fan C, Rabiul I, Ross JS, Hortobagyi GN, Pusztai L (2005) Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res 11:5678–5685
Ruan K, Fang X, Ouyang G (2009) MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett 285:116–126
Sachdeva M, Wu H, Ru P, Hwang L, Trieu V, Mo YY (2011) MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 30:822–831
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA (2006) Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9(6):435–443
Salter KH, Acharya CR, Walters KS, Redman R, Anguiano A, Garman KS, Anders CK, Mukherjee S, Dressman HK, Barry WT, Marcom KP, Olson J, Nevins JR, Potti A (2008) An integrated approach to the prediction of chemotherapeutic response in patients with breast cancer. PLoS One 3:e1908
Schultz J, Lorenz P, Gross G, Ibrahim S, Kunz M (2008) microRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res 18(5):549–557
Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455:58–63
Shen J, DiCioccio R, Odunsi K, Lele SB, Zhao H (2010) Novel genetic variants in miR-191 gene and familial ovarian cancer. BMC Cancer 10:47
Siddik ZH (2002) Biochemical and molecular mechanisms of cisplatin resistance. Cancer Treat Res 112:263–284
Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22:7265–7279
Snyder LL, Ahmed I, Steel LF (2009) RNA polymerase III can drive polycistronic expression of functional interfering RNAs designed to resemble microRNAs. Nucleic Acids Res 37:E127
Sorrentino A, Liu CG, Addario A, Peschle C, Scambi G, Ferlini C (2008) Role of microRNAs in drug resistant ovarian cancer cells. Gynecol Oncol 111:478–486
Starlard-Davenport A, Tryndyak VP, James SR, Karpf AR, Latendresse JR, Beland FA, Pogribny IP (2010) Mechanisms of epigenetic silencing of the Rassf1a gene during estrogen-induced breast carcinogenesis in ACI rats. Carcinogenesis 31:376–381
Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2008) Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett 582(10):1564–1568
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H (2007) Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 104:15472–15477
Thangaraju M, Kaufmann SH, Couch FJ (2000) BRCA1 facilitates stress-induced apoptosis in breast and ovarian cancer cell lines. J Biol Chem 275:33487–33496
Trompeter HI, Abbad H, Iwaniuk KM, Hafner M, Renwick N, Tuschl T, Schira J, Muller HW, Wernet P (2011) microRNAs miR-17, miR-20a, and miR-106b act in concert to modulate E2F activity on cell cycle arrest during neuronal lineage differentiation of USSC. PLoS One 6(1):e16138
Tryndyak V, Beland FA, Pogribny IP (2010) E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int J Cancer 126:2575–2583
Tsang WP, Kwok TT (2008) Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis 13:1215–1222
Tsukamoto Y, Nakada C, Noguchi T, Tanigawa M, Nguyen LT, Uchida T, Hijiya N, Matsuura K, Fugjioka T, Seto M, Moriyama M (2010) microRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res 70(6):2339–2349
Valeri N, Gasparini P, Braconi C, Paone A, Lovat F, Fabbri M, Sumani KM, Alder H, Amadori D, Patel T, Nuovo GJ, Fishel R, Croce CM (2010) MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc Natl Acad Sci USA 107:21098–21103
Vasudevan S, Tong Y, Steitz JA (2007) Switching from repression to activation: microRNAs can up-regulate translation. Science 318:1931–1934
Wada R, Akiyama Y, Hashimoto Y, Fukamachi H, Yuasa Y (2010) miR-212 is downregulated and suppresses methyl-CpG-binding protein MeCP2 in human gastric cancer. Int J Cancer 127:1106–1114
Wang FE, Zhang C, Maminishkis A, Dong L, Zhi C, Li R, Zhao J, Majerciak V, Gaur AB, Chen S, Miller SS (2010a) MicroRNA-204/211 alters epithelial physiology. FASEB J 24:1552–1571
Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14:927–939
Wang Z, Li Y, Ahmad A, Asmi AS, Kong D, Banerjee S, Sarkar FH (2010b) Targeting miRNAs involved in cancer stem cell and EMT regulation: an emerging concept in overcoming drug resistance. Drug Resist Updat 13:109–118
Winograd-Katz SE, Levitzki A (2006) Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 25:7381–7390
Xin F, Li M, Balch C, Thomson M, Fan M, Liu Y, Hammond SM, Kim S, Nephew KP (2009) Computational analysis of microRNA profiles and their target genes suggests significant involvement in breast cancer antiestrogen resistance. Bioinformatics 25:430–434
Yaqinuddin A, Abbas F, Naqvi SZ, Bashir MU, Qazi R, Qureshi SA (2008) Silencing of MBD1 and MeCP2 in prostate-cancer-derived PC3 cells produces differential gene expression profiles and cellular phenotypes. Biosci Rep 28:319–326
Yao E, Ventura A (2011) A new role for miR-182 in DNA repair. Mol Cell 41:135–137
Yu Y, Wang Y, Ren X, Tsuyada A, Li A, Liu LJ, Wang SE (2010) Context-dependent bidirectional regulation of the MutS homolog 2 by transforming growth factor β contributes to chemoresistance in breast cancer cells. Mol Cancer Res 8:1633–1642
Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG (2008) A cyclin D1/microRNA 17/12 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Boil 182:509–517
Zhang B, Steelwag EJ, Pan X (2009) Large-scale analysis reveals unique features of microRNAs. Gene 443:100–109
Zhang H, Li M, Han Y, Hong L, Gong T, Sun L, Zheng X (2010) Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma. Dig Dis Sci 55:2545–2551
Zhao JJ, Lin J, Yang H, Kong W, He L, Ma X, Coppola D, Cheng JQ (2008) MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J Biol Chem 283:31079–31086
Zhao H, Shen J, Medico L, Wang D, Ambrosone CB, Liu S (2010a) A pilot study of circulating miRNAs as potential biomarkers of early stage breast cancer. PLoS One 5:e13735
Zhao JJ, Lin J, Lwin T, Yang H, Guo J, Kong W, Dessureault S, Moscinski LC, Rezania D, Dalton WS, Sotomayor E, Tao J, Chang JQ (2010b) microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lympohoma. Blood 115(13):2630–2639
Zhao X, Yang L, Hu J, Ruan J (2010c) miR-138 might reverse multidrug resistance of leukemia cells. Leuk Res 34:1078–1082
Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, Xiong W, Li G, Lu J, Fodstad O, Riker AI, Tan M (2010) MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem 285:21496–21507
Zhu S, Sachdeva M, Wu F, Lu Z, Mo YY (2010a) Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene 29(12):1763–1772
Zhu W, Shan X, Wang T, Shu Y, Liu P (2010b) miR-181b modulates multidrg resistance by targeting BLC2 in human cancer cell lines. Int J Cancer 128(11):2520–2529
Zunino F, Cassinelli G, Polizzi D, Perego P (1999) Molecular mechanisms of resistance to taxanes and therapeutic implications. Drug Resist Updat 2:351–357
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
The views expressed in this paper do not necessarily represent those of the U.S. Food and Drug Administration.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kutanzi, K.R., Yurchenko, O.V., Beland, F.A. et al. MicroRNA-mediated drug resistance in breast cancer. Clin Epigenet 2, 171–185 (2011). https://doi.org/10.1007/s13148-011-0040-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-011-0040-8