
Abstract
Aging is the natural trace that time leaves behind on life during blossom and maturation, culminating in senescence and death. This process is accompanied by a decline in the healthy function of multiple organ systems, leading to increased incidence and mortality from diseases such as diabetes, cancer, cardiovascular disease, and neurodegeneration. Based on the fact that both sirtuin expression and activity appear to be upregulated in some types of cancer while they are being downregulated in others, there is quite some controversy stirring up as to the role of sirtuins, acting as cancer suppressors in some cases while under other circumstances they may promote cellular malignancy. It is therefore currently quite unclear as to what extent and under which particular circumstances sirtuin activators and/or inhibitors will find their place in the treatment of age-related disease and cancer. In this review, we take an effort to bring together the highlights of sirtuin research in order to shed some light on the mechanistic impact that sirtuins have on the pathogenesis of cellular malignancy.
Similar content being viewed by others


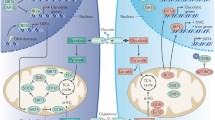
The sirtuin family of proteins
Seven human sirtuins (SIRT1–SIRT7) have been identified so far. Based on phylogenetic analyses, they are further grouped into four subclasses (Frye 1999, 2000; Voelter-Mahlknecht et al. 2006b). The main characteristic feature that distinguishes sirtuins from non-sirtuin HDACs is their unique enzymatic mechanism. The sirtuins are NAD+-dependent deacetylases and adenosine diphosphate (ADP) ribosyltransferases. Most sirtuins catalyze NAD+-dependent deacetylation (Imai et al. 2000; Landry et al. 2000; North et al. 2003; Tanner et al. 2000). While SIRT4 possesses NAD+-dependent mono-ADP-ribosyltransferase activity (Haigis et al. 2006; Liszt et al. 2005), SIRT1 and SIRT6 have been reported to exert both ADP ribosyltransferase and substrate-specific deacetylase activities (Michishita et al. 2008). For SIRT4 and SIRT7, a deacetylase activity has not been reported so far, but such activity may require a specific substrate, as for SIRT6 (Landry et al. 2000; Tanner et al. 2000). Sirtuin enzymatic activities have been linked to a variety of cellular processes such as heterochromatin silencing, differentiation, metabolism, neuronal protection, apoptosis, and cell survival due to the sirtuin ability to deacetylate both histone and numerous non-histone targets (Michan and Sinclair 2007). Three mammalian sirtuins are located within the mitochondria (SIRT3, SIRT4, and SIRT5), while the other sirtuins exert their functions within the cytosol (Sirt2), in the cell nucleus (Sirt1 and Sirt6), or within the nucleoli (Sirt7) (Table 1). A number of signal transduction pathways have been associated with the life-span-extending capacity of calorie restriction (CR). In this context, the sirtuins have been reported to play a key role, which is mostly based on their requirement of NAD+ as a co-factor for enzymatic activity, which in turn demonstrates the crucial link between sirtuins and the energy-dependent regulation of gene expression. In fact, studies in lower organisms such as yeast, Drosophila melanogaster, or Caenorhabditis elegans demonstrated that either the overexpression or hyperactivity of yeast SIR2 and its orthologs is coupled with prolonged life span (Table 1, Fig. 1; Longo and Kennedy 2006).

Transcriptional and post-transcriptional regulatory mechanisms of SIRT1. Four transcription factors (HIC1, p53, E2F1, and cMYC) have been identified to modulate SIRT1 expression under oxidative stress/DNA-damage conditions and/or nutrient deprivation. Post-transcriptional control of SIRT1: upon oxidative stress, SIRT1 mRNA is degraded due to a checkpoint-kinase-2-mediated dissociation of the RNA binding protein HuR. Via a feedback loop, SIRT1 regulates the activity of promoter-bound transcription factors through deacetylation. Green boxes: activating factors; red boxes: inactivating factors; green arrow: activation/increase; red line: inactivation/inhibition
Sirtuin 1
SIRT1, the closest human homolog to yeast SIR2, is the best characterized member within the family of sirtuins with regard to life span and age-related disease. It has been implicated to play a crucial role during the aging process for several reasons (Guarente and Picard 2005; Mahlknecht and Voelter-Mahlknecht 2009a; Saunders and Verdin 2007; Voelter-Mahlknecht et al. 2006b; Westphal et al. 2007). First, SIRT1 is downregulated in senescent cells (Sasaki et al. 2006) and during aging (Sommer et al. 2006). Secondly, calorie restriction induces SIRT1 expression in mammalian cells and humans, thereby promoting cell survival and proliferation, while it reduces cellular senescence (Cohen et al. 2004), whereas SIRT1-knockout mice fail to display a phenotype of CR (Chen et al. 2005). Consistent with this observation, phenotypes of sirt1-overexpressing mice partially display phenotypes of its calorie-restricted counterparts (Bordone et al. 2007). Also, an increase of SIRT1 activity and/or expression represses tumor suppressor and DNA-repair genes, including FOXO1/2/4, WRN, Rb, p73, MLH1, and NBS1 (Deng 2009; Vijg et al. 2008). In white adipose tissue, SIRT1 promotes fatty-acid mobilization through inhibition of peroxisome proliferation-activating receptor gamma (PPARγ) and upregulation of the production/secretion of adiponectin and FGF21 via FOXO1 and/or PPARγ (Imai and Guarente 2010; Liu et al. 2008). In addition, SIRT1 is involved in the upregulation of mitochondrial biogenesis due to its capability to deacetylate and thus activate the PPARγ co-activator-1α (PGC-1α; Rodgers et al. 2005; Zschoernig and Mahlknecht 2008), which stimulates mitochondrial activity and subsequently increases glucose metabolism, which in turn improves insulin sensitivity (Engel and Mahlknecht 2008; Lagouge et al. 2006). The maintenance of this delicate balance between sensitivity and secretion of insulin in major metabolic tissues (liver, skeletal muscle, white adipose tissue, and pancreatic β-cells) is essentially regulated by Sirt1, which regulates the production of glucose in the liver via PGC-1α, FOXO1, CRTC2, and STAT3, which seems to repress insulin sensitivity. On the other hand, SIRT1 increases insulin sensitivity in the skeletal muscle by increasing fatty-acid oxidation through PGC-1α and repression of PTB1B (Imai and Guarente 2010; Liu et al. 2008; Nie et al. 2009; Rodgers et al. 2005). The regulation of mitochondrial biogenesis and metabolism is widely accepted as a key component in the regulation of life span and aging (Lopez-Lluch et al. 2008). Furthermore, SIRT1 has not only been demonstrated to mimic calorie restriction but also to exert neuroprotective effects. The resveratrol-mediated activation promotes a SIRT1-induced resistance to axonal degeneration (Araki et al. 2004), and increasing evidence that SIRT1 protects neurons from apoptosis (Brunet et al. 2004) and is involved in the prevention of Alzheimer’s disease and amyotrophic lateral sclerosis disease models (Kim et al. 2007) has emerged. Interestingly, the pharmacological activation of SIRT1 recapitulates many of the observations that have been made in the context of a knockout or transgenic overexpression of SIRT1 in mice.
The most prominent activator of SIRT1 is resveratrol (3,4,5-trihydroxystilbene). Analysis in no-mammalian organisms revealed that treatment with resveratrol extends life span through direct activation of SIRT1 (Howitz et al. 2003; Wood et al. 2004) by increasing its substrate binding affinity (Borra et al. 2005). In addition, it retards cellular senescence in human diploid fibroblasts (Huang et al. 2008). In a study by Baur and co-workers, resveratrol treatment has been demonstrated to improve health and life span in mice in the presence of a high-calorie diet (Baur et al. 2006). Despite the fact that high-calorie-fed mice were obese, the group receiving resveratrol lived significantly longer and exhibited the characteristic molecular changes that have been observed in conjunction with increased life span including improved insulin sensitivity, reduced insulin-like growth factor 1 levels, increased PGC-1α activity, and an increased numbers of mitochondria. In addition to resveratrol and a number of agents including quercetin, fistein, butein, pyrroloquinoxaline, and oxazolopyridine that have been described a while ago (Haigis and Sinclair 2010), more recently, a number of highly specific SIRT1-activating compounds (SRT1460, SRT1720, and SRT2183) have been identified by a high-throughput fluorescence polarization analyses followed by high-throughput mass spectrometry (Milne et al. 2007). These activators are structurally unrelated to resveratrol and exhibit nanomolar to low micromolar potency towards SIRT1 in vitro. Notably, compound SRT1720 has not only been proven to be a useful activator of SIRT1 in vitro but also in three different in vivo models which displayed the characteristic changes of calorie restriction. In diet-induced obese as well as in genetically obese mice (Lepob/ob), treatment with SRT1720 significantly improved insulin sensitivity, decreased plasma glucose levels, and increased mitochondrial biogenesis. Consistent with these results, glucose homeostasis and insulin sensitivity in adipose tissue, skeletal muscle, and liver were markedly improved in Zucker fa/fa rats, a genetically obese rodent model. Taken together, SIRT1 activation by SRT1720 appears to mimic the effects of calorie restriction on metabolic and mitochondrial function and therefore constitutes a promising drug for the treatment of age-related diseases such as type 2 diabetes. Similarly, specific inhibition of SIRT1 activity by sirtinol, a cell-permeable 2-hydroxy-1-napthaldehyde derivate, has been reported to induce senescence-like growth arrest in human endothelial and cancer cells and to increase histone H3 lysine 14 (H3K14) and histone H4 lysine 16 (H4K16) as well as p53 acetylation levels, accompanied by an attenuated DNA synthesis, an increased SA-β-gal activity as well as senescence-like morphological changes ( Ota et al. 2006, 2007), which further supports the idea that SIRT1-activating compounds could be an effective strategy in the treatment of aging or age-related diseases (Figs. 1, 2, and 3).

Modulation of SIRT1 activity through direct interaction with activating/inactivating ligands. Green boxes: activating factors; red boxes: inactivating factors; green arrow: activation/increase; red line: inactivation/inhibition

In the classical model, SIRT1 inhibits tumor cell apoptosis and senescence while it increases cell proliferation through modulation of methylation at the histone level and promoter CpG islands as well as the deacetylation of histone and non-histone proteins
Sirtuin 2
SIRT2 is primarily found in the cytoplasm and co-localizes with microtubules, where it deacetylates tubulin (North et al. 2003; North and Verdin 2007). SIRT2 controls mitotic checkpoint functions during early metaphase in order to prevent chromosomal instability and increases considerably during mitosis. Both SIRT1 and SIRT2 deacetylate H3K56, which is an acetylation marker that is typically increased in several different types of cancer (Das et al. 2009; Inoue et al. 2007b, 2009). During the G2/M transition of the cell cycle, the SIRT2 protein is phosphorylated. p53 and histones H3 and H4 are SIRT2 substrates, and SIRT2 is involved in the regulation of the cell cycle (Heltweg et al. 2006; Inoue et al. 2007a), in adipocyte differentiation, and in the response to oxidative stress (Jing et al. 2007; North et al. 2003; Voelter-Mahlknecht et al. 2005; Wang et al. 2007). SIRT2 activation in the context of cancer could therefore be as undesirable and contradictory as the activation of SIRT1, since it catalyzes the deacetylation of p53 and consequently the inactivation of p53, which aggravates tumor growth, while on the other hand SIRT2 has been suggested to release mitotic arrest in critically damaged cells, allowing them to proceed to apoptosis (Inoue et al. 2009; North et al. 2003; North and Verdin 2007; Fig. 1). Also, SIRT2 is known to deacetylate FOXO3a, which is a regulator of transcription. The deacetylation of FOXO increases its DNA binding and consequently the expression of FOXO target genes such as p27Kip1, manganese superoxide dismutase, and BIM. SIRT2 therefore decreases the cellular levels of reactive oxygen species. Also, since BIM is a proapoptotic factor, SIRT2 does in fact promote cell death when cells are under severe stress (Wang et al. 2007). The expression of SIRT2 is highest in healthy brain tissue but appears to be severely reduced in a large number of brain tumor cell lines (Voelter-Mahlknecht et al. 2005). Interestingly, the yeast SIRT2 ortholog Hst2 has been reported to work concurrently with SIR2 in certain strains with regard to life span extension and rDNA silencing (Lamming et al. 2005). Most recently, one study on SIRT2 reported the therapeutic effectiveness of AGK2, which exhibits a >14-fold selective inhibition of SIRT2 relative to SIRT1 in the treatment of neurodegenerative diseases such as Parkinson’s (Outeiro et al. 2007) through formation of less toxic α-synclein aggregates (the exact mechanism remains however to be further elucidated). Another selective SIRT2 inhibitor is AC-93253, which is a small molecule that is cytotoxic in at least four different tumor cell lines (Zhang et al. 2009). Cambinol and salermide are compounds that inhibits both SIRT1 and SIRT2.
The regulation and the effects of SIRT2 activity are highly complex, and changes of SIRT2 activity may be beneficial to the cell under specific circumstances while they may be detrimental in others (Garske et al. 2007; Jing et al. 2007). Decreased levels of SIRT2 have been reported in gliomas and therefore an inhibition of SIRT2 may predispose cells to uncontrolled growth, while increasing activity of SIRT2 in certain cancers could be beneficial (Hiratsuka et al. 2003). Accordingly, overexpression of SIRT2 slows down or even arrests cell cycle progression and increases the number of multinucleated cells. This may happen in response to microtubule inhibitors such as nocodazole (Inoue et al. 2007b; Inoue et al. 2009). The activity of SIRT2 appears to depend on the phosphorylation of SIRT2Ser368 by CDK1 (Dransfeld et al. 2010; Jin et al. 2009; North and Verdin 2007). Well-balanced levels of SIRT2 are required for mitotic reliability, and therefore, depending on the circumstances, while the inhibition of SIRT2 may be useful in the treatment of some cancers, its activation may be useful in other types of cancer.
Calorie restriction and its effects on aging and age-related disease
Cellular senescence and organism aging are characterized by the progressive loss of physiological functions and metabolic processes which is often accompanied by age-associated disease, such as diabetes, cancer, cardiovascular disease, and neurodegeneration (Love 2005; Zschoernig and Mahlknecht 2008). Since the discovery of the first mammalian sirtuin, SIRT1, 10 years ago, major achievements in our understanding of the function and regulation of sirtuins and their effects on mammalian physiology and life span have been published. Based on the underlying biological complexities, we still lack a complete picture of the molecular mechanisms related to the aging process that takes place in humans. A number of factors are associated with the rate of aging such as variable genomic stability, metabolic control, changes in gene expression patterns and the production of reactive oxygen species (ROS; Gruber et al. 2008; Oberdoerffer and Sinclair 2007). As mitochondria constitute the major production site of ROS, this organelle most likely plays a key role in life span and aging. Interestingly, around 20% of mitochondrial proteins are suggested to be posttranslationally modified by reversible acetylation, especially those involved in life span and metabolism (Fig. 2; Kim et al. 2006).
Healthy aging is one of the ideals of modern society. Both the identification of the underlying molecular mechanisms and interventions regarding the aging process are of considerable interest. Up to now, CR, a phenomenon that was first described in the 1930s by McCay and co-workers, where an organism was provided with at least 20% less calories below ad libitum level, constitutes the most robust and reproducible way of extending health and longevity (McCay et al. 1989). CR has not only been shown to increase the median and maximum life span of a variety of organisms (Masoro 2000; Masoro and Austad 1996; Weindruch and Sohal 1997; Weindruch et al. 1986; Yu 1994), it is also associated with a decreased incidence or delayed rate of age-related disease as demonstrated in several rodent studies (Weindruch et al. 1986; Yu 1994). Also, the beneficial effects of CR are related to lower circulating insulin levels and increased insulin sensitivity, thereby reducing the predisposition to diabetes as well as other metabolic disorders which is also associated with life span extension based on experiments in animal models (Katic and Kahn 2005; Lane et al. 1995). A different key feature of CR is a lowered core body temperature that goes along with reduced and more efficient energy expenditure and thus increased life span (Lane et al. 1996; Roth et al. 1995). Regardless of the lack of long-term studies, there is emerging evidence that CR may contribute to life span extension in humans. Studies on dogs (Kealy et al. 2002), cows (Pinney et al. 1972), and non-human primates revealed that many of the physiological responses in these organisms look a lot like those observed for CR on rodents (Mattison et al. 2003; Roth et al. 2004). Most notably, the National Institute of Aging initiated short-term human CR studies (6–12 months) at the Washington University, Tufts University, and the Pennington Center at Louisiana State which already confirm, albeit on a preliminary basis, reduced plasma insulin levels and body temperature that are key features of the CR response repeatedly observed in animal studies (Hadley et al. 2005; Heilbronn and Ravussin 2003). In consideration of the already demonstrated positive effect on human health (Roth 2005; Roth et al. 2005), these data are the first evidence that humans might indeed benefit from CR not only due to disease protection but also in terms of increased survival.
The sirtuin connection to cancer
Even though calorie restriction appears to be the most effective way to prevent cancer in rodents and primates, which some view as an indication that sirtuins are tumor suppressors (Sinclair 2005), some sirtuins, such as SIRT1 and SIRT3, appear to improve cell survival, which could be a sign that they may in fact promote tumorigenesis (Brooks and Gu 2008, 2009). Cancer rates directly correlate with increasing age. Most cancers arise from genetic or epigenetic damage in renewable tissues, resulting in vigorous cell proliferation and survival on the basis of impaired apoptosis. While organ degeneration is accompanied by a loss of function, cancer goes along with a gain of aberrant function (Mahlknecht and Hoelzer 2000a; Mahlknecht et al. 2000b; Mei et al. 2004).
The level of SIRT1 is highly elevated in a number of cancer cell types. SIRT1 binds and deacetylates the androgen receptor and represses dihydrotestosterone-induced androgen receptor signaling in human prostate cancer (Fu et al. 2006). On the other hand, SIRT1 silencing induces growth arrest and apoptosis in human epithelial cancer cells (Ford et al. 2005). The ectopic induction of SIRT1 in beta-catenin-driven mouse model of colon cancer significantly reduces tumor formation, proliferation, and animal morbidity (Firestein et al. 2008). Conversely, SIRT1 may also stimulate TNF-α-induced apoptosis, which indicates that SIRT1 may not only suppress apoptosis but also promote apoptosis (Yeung et al. 2004).
In addition to the deacetylation of histones, SIRT1 also deacetylates non-histone proteins such as various transcription factors that are involved in growth regulation, the response to stress, and apoptosis in the fundamental progression of cancer. The inhibition of SIRT1 goes along with an increase of H4K16, H3K9, and H1K26 acetylation at endogenous promoters and suffices to induce gene re-expression in cancer cells as shown for breast and colon cancer (Pruitt et al. 2006). SIRT1 also regulates the formation of heterochromatin via deacetylation of histone H1K26 and promotes the loss of H3K79me2, a marker associated with transcriptionally active chromatin. These are a few examples of how SIRT1 was speculated to be associated with the modulation of epigenetic hallmarks of cancer.
Despite of all its beneficial effects, SIRT1 activation may for some instances also be detrimental: the SIRT1-catalyzed deacetylation of the tumor suppressor protein p53 (Luo et al. 2001; Vaziri et al. 2001) goes along with the inactivation of p53, which allows cells to bypass p53-mediated apoptosis (Fig. 1). This is good for normal cells since it increases their survival and prolongs life span. In tumor cells, however, this effect is not at all desirable since it aggravates tumor growth. On the other hand, a number of tumor suppressor proteins including p53, HIC1, and DBC1 have been identified to negatively regulate SIRT1: HIC1 (hypermethylated in cancer 1) is a zinc-finger protein that is regulated by p53, which in turn binds to the SIRT1 promoter and thus represses the transcription of Sirt1 (102). Accordingly, the inactivation of HIC1 upregulates the transcription of SIRT1, thereby inactivating p53, which allows cells to bypass apoptosis after DNA damage. Interestingly, HIC1 appears to undergo hypermethylation at the promoter level as a consequence of aging, which in turn may at least in part explain the increasing susceptibility to cancer with increasing age. CtBP is a co-repressor that binds to HIC1 and the binding of CtBP to HIC1 is particularly strong during glycolysis. Therefore, if glycolysis is inhibited, the binding of the co-repressor CtBP to HIC1 is decreased, which in turn enhances SIRT1 expression (Fig. 1; Zhang et al. 2007). DBC1 (deleted in breast cancer) is another tumor suppressor that negatively regulates SIRT1 deacetylase activity (Anantharaman and Aravind 2008; Kim et al. 2008; Zhao et al. 2008). DBC1 knockdown by siRNA promotes the deacetylation of p53, which in turn allows cells to survive genotoxic stress, which is an effect that is mediated by SIRT1. DBC1 may therefore promote the development of breast cancer through activation of SIRT1, which then downregulates p53 and/or other tumor suppressor pathways. cMYC is a proto-oncogene that regulates cell proliferation, stem cell self-renewal, and apoptosis. cMYC binds to the SIRT1 promoter and induces SIRT1 expression, but through a feedback loop that could in fact avoid cellular transformation, SIRT1 deacetylates cMYC, which decreases cMYC stability and thus results in tumor suppression (Yuan et al. 2009; Zschoernig and Mahlknecht 2008, 2009; Fig. 1).
In fact, the expression of SIRT1 appears to be upregulated for most types of cancer such as in human prostate cancer (Huffman et al. 2007), acute myeloid leukemia (Bradbury et al. 2005), non-melanoma skin cancers (Hida et al. 2007; Lim 2006), primary colon cancer (Stunkel et al. 2007), and breast cancer (Haigis and Sinclair 2010). This and the capacity of SIRT1 to inactivate a number of proteins that are associated with tumor suppression and the repair of DNA damage explains why SIRT1 has been largely considered a tumor promoter (Ashraf et al. 2006; Fraga and Esteller 2007; Jung-Hynes et al. 2009; Lim 2006; Stunkel et al. 2007). The SIRT1-mediated silencing of E-cadherin through hypermethylation of a CpG island at the promoter level is such an example of how SIRT1 may contribute to carcinogenesis in epithelial cancers (O'Hagan et al. 2008; Pruitt et al. 2006). Also, the reactivation of p53 through inhibition of SIRT1, instead of stimulating it, could trigger tumor cell apoptosis. Whether elevated levels of SIRT1 are the cause or consequence of tumorigenesis is currently not clear. On the contrary, more recently, several studies have demonstrated SIRT1 levels to be reduced in some other types of cancers such as glioblastoma, cancers of the bladder, the prostate, the female breast, and ovary, and liver cancer when compared to the corresponding normal tissues (Wang et al. 2008a) and that SIRT1 deficiency may in fact result in genetic instability and tumorigenesis, while overexpression of SIRT1 attenuates cancer formation in mice that are heterozygous for tumor suppressor p53 or APC (Deng 2009), which on the other hand may be an indicator that SIRT1 could be a tumor suppressor rather than a promoter of cancer in these tissues. Also, recent publications have reported SIRT1 to be essential in the repair of DNA strand breaks and thus to prevent the development of cellular malignancy in mouse cancer models, and in fact, mice that carried additional copies of SIRT1 did not reveal any signs of premature death or increased cancers of any sort. In murine cancer models of leukemia and colon cancer, SIRT1 transgenic mice were reported to live longer (Deng 2009; Firestein et al. 2008; O'Hagan et al. 2008; Oberdoerffer et al. 2008). The question of whether SIRT1 acts primarily as an oncogene or as a tumor suppressor is still unanswered and remains still to be determined. It is however strongly evident that SIRT1 is a critical regulator in the pathogenesis of cancer.
Another example showing a direct beneficial effect of SIRT1 in cancer relates to its activity on the regulation of breast cancer cell apoptosis. In healthy cells, breast-cancer-associated gene 1 (BRCA1), which is a potent tumor suppressor gene, maintains the expression of SIRT1, which in turn, inhibits the expression of survivin, a protein that inhibits apoptosis. If BRCA1 is defective through a spontaneous or inherited mutation, this defective BRCA1 is no longer able to keep up sufficient levels of SIRT1, and consequently, the expression of survivin may no longer be sufficiently inhibited, which results in a resistance to apoptosis and thus continuous tumor cell growth (Wang et al. 2008b). At least in vitro and in animal models, the compound resveratrol, which is known to enhance SIRT1 activity, was able to strongly inhibit tumor growth in BRCA1-defective cells as a consequence of reduced survivin expression and subsequent apoptosis of BRCA1-deficient cancer cells (Wang et al. 2008b).
In addition to its activity as a deacetylase, SIRT1 is known to localize to the promoters of quite a few aberrantly silenced tumor suppressor genes whose DNA is hypermethylated (Jones and Baylin 2002). This is particularly essential if a DNA break is initiated within a CpG island because SIRT1 then appears to be required for the transient recruitment of DNA methyltransferase 3B and the subsequent silencing of this DNA region by methylation (O'Hagan et al. 2008; Oberdoerffer et al. 2008; Wang et al. 2008a; Yuan and Seto 2007). The localization of SIRT1 to DNA breaks and is essential for efficient DNA-break repair (Oberdoerffer et al. 2008; Wang et al. 2008a): cells and mice that lack SIRT1 are more prone to DNA-damage-induced aneuploidy, and the efficiency of DNA-break repair and the maintenance of genome stability is impaired by 50% (Oberdoerffer et al. 2008). Cells that lack SIRT1 are not able to effectively recruit DNA-repair factors subsequent to DNA damage (Fig. 3; Deng 2009).
The other sirtuins, SIRT3–SIRT7, and their relation to cancer
Sirtuin 3
For SIRT3–SIRT7, only a few hints indicate that these enzymes may be important in the pathogenesis of cancer. SIRT3 may be proapoptotic in HCT116 cells through JNK2. This pathway is independent from SIRT1 (Allison and Milner 2007). In other situations, however, such as in response to DNA damage when NAD+ levels in mitochondria are low, SIRT3 and SIRT4 can in fact be antiapoptotic (Yang et al. 2007). SIRT3 is the first sirtuin that was reported to be localized to the mitochondrial matrix of mammalian cells and represents the major mitochondrial deacetylase (Lombard et al. 2007; Michishita et al. 2005; Schwer et al. 2006; Voelter-Mahlknecht and Mahlknecht 2003). Despite the controversy that has arisen on the possibility that SIRT3 might also be present in the nucleus, it should be emphasized that, whatsoever, the bulk of SIRT3, both mouse and human, is localized in the mitochondrion (Hallows et al. 2008). SIRT3 appears to be the only sirtuin that is linked genetically to life span in humans (Rose et al. 2003): in human population studies, analyses of SIRT3-related polymorphisms suggested a SIRT3 G477T transversion, which does in fact not affect the amino acid sequence, to be associated with increased survival in elderly males (Bellizzi et al. 2005; Rose et al. 2003). Similarly, a loss of enhancer activity due to a variable number of tandem repeats within sirt3 intron 5 was correlated with increased survival rates in males beyond the age of 90 years (Bellizzi et al. 2005). Even though these findings need to be further validated in larger population cohorts, they further strengthen the rationale that the expression of SIRT3 may promote longevity in humans.
Surprisingly, when deleted in the mouse, Sirt3 does not exhibit any signs of disorder as long as the animals are without stress (Lombard et al. 2007). Several lines of evidence suggest that SIRT3 is important in regulating the response to stress, and thus, there is a close connection between SIRT3 and the regulation of cellular energy metabolism, the regulation of fatty-acid oxidation during fasting, and cellular aging: SIRT3 is abundantly expressed in brown adipose tissue and its expression is further increased during fasting (Hirschey et al. 2010; Shi et al. 2005). In addition, two independent studies showed that the mitochondrial form of acetyl CoA synthetase 2 (ACS2) is a direct SIRT3 target, which is activated upon deacetylation (Hallows et al. 2006; Schwer et al. 2006). ACS2 is the nuclear source of acetyl CoA for histone acetylation, an intermediate of the citric acid cycle, and is required for cholesterol and fatty-acid synthesis. SIRT3 is therefore a key player that regulates the entry of carbons from acetate into central metabolic pathways.
SIRT3 plays an important role in the pathogenesis of cancer: SIRT3 activates the expression of MnSOD and catalase by promoting the translocation of cytosolic FOXO3a to the nucleus (Saunders and Verdin 2007). FOXO3a affects the expression of the antioxidant MnSOD, which degrades mitochondrial superoxide into H2O2, which appears to regulate cellular transformation. Therefore, a loss or aging-associated decrease in the expression of SIRT3 leads to increased FOXO3a phosphorylation, triggering its nuclear export and thus promoting oncogenic transformation by enhancing mitochondrial ROS, which may induce genetic instability and the stabilization of hypoxia-inducible factor (Kim et al. 2010; Lanza et al. 2008). Thus, SIRT3 functions as a tumor suppressor, and the nuclear/cytosolic functions of SIRT3, rather than the mitochondrial, mediate its role in the regulation of antioxidant activity and cellular transformation, and it may therefore be worth to consider whether NAD administration could drive the cells in a reverse direction along the transformation pathway in tumors where SIRT3 is either being lost or decreased. Since increased levels of SIRT3 mRNA have been associated with breast cancer and thyroid cancer, it is currently not clear as to what extent Sirt3 acts as a tumor suppressor rather than a tumor promoter (Ashraf et al. 2006; Frye 2002).
Sirtuin 4
SIRT4 is another mitochondrial protein that exerts strong ADP ribosyltransferase activity but lacks almost any deacetylase activity (Haigis et al. 2006; Mahlknecht and Voelter-Mahlknecht 2009b; Michishita et al. 2005). A number of key regulators of the cellular metabolism have been identified to be regulated by SIRT4: the activity of glutamate dehydrogenase (GDH), a mitochondrial enzyme which is involved in the conversion of glutamate to α-ketoglutarate, is repressed by SIRT4-mediated ADP ribosylation which represents an important mechanism in the regulation of amino-acid-stimulated insulin secretion (Haigis et al. 2006). Also, SIRT4-knockout mice have been reported to be viable; pancreatic islets isolated from these mice did however secrete higher levels of insulin, which is consistent with the role of SIRT4 as a downregulator of insulin secretion through the repression of GDH activity (Haigis et al. 2006). A loss of SIRT4 function could therefore contribute to the development of diabetes due to higher levels of insulin that are known to increase the predisposition to both diabetes and other metabolic disorders (Katic and Kahn 2005). The identification of both insulin-degrading enzymes and adenine nucleotide transporters to be substrates of SIRT4 further supports the thought that SIRT4 plays a direct role in the maintenance of physiological levels of insulin in response to glucose (Ahuja et al. 2007).
Sirtuin 5
SIRT5 is a mitochondrial sirtuin that that exerts only weak deacetylase activity and which has only been barely studied so far (Michishita et al. 2005; Schuetz et al. 2007). Lately, two mitochondrial substrates have been identified: cytochrome c, a key regulator of oxidative metabolism and apoptosis initiation (Schlicker et al. 2008) and the carbamoyl phosphatase synthetase 1 (CPS1), which catalyzes the first step of the urea cycle and which is being activated by SIRT5 mediation (Nakagawa et al. 2009). During CR, increased CPS1 activity has been correlated with hypo-acetylation, and a 50% increase in mitochondrial NAD+ points out the role of SIRT5 in the upregulation of the urea cycle for ammonia disposal. In a recent study that assessed the mRNA expression of sirtuins in pancreatic cancer, only Sirt5 was consistently upregulated (Mahlknecht et al. 2006a; Ouaissi et al. 2008).
Sirtuin 6
SIRT6 is a nuclear protein that carries weak deacetylase and strong ADP ribosyltransferase activity (Liszt et al. 2005; Mahlknecht et al. 2006b). Studies on SIRT6-deficient knockout mice suggested SIRT6 to fundamentally influence the aging process since these mice displayed genomic instability and premature aging symptoms, dying several weeks after birth (Mostoslavsky et al. 2006). Such observations are ascribed to deficiency in a specific DNA-repair mechanism base excision repair (BER). Accordingly, SIRT6−/− MEFs show impaired proliferation and enhanced sensitivity to DNA-damaging agents, resulting in multiple chromosomal alterations (fragmentation, detached chromosomes, gaps, and translocations) on the basis of a BER defect (Mostoslavsky et al. 2006). Consistent with these results, the proper regulation of genomic stability is widely accepted to protect against tumor formation and premature aging (Lombard et al. 2005). SIRT6, which is an important key player in the maintenance of genomic stability, specifically interacts with GCIP, which is a potential tumor suppressor that is downregulated in colon, breast, and prostate cancers (Ma et al. 2007; Michishita et al. 2008; Mostoslavsky et al. 2006).
Sirtuin 7
The protein SIRT7 localizes to the nucleolus of human cells (Ford et al. 2006; Michishita et al. 2005; Voelter-Mahlknecht et al. 2006a). So far, neither a deacetylase nor an ADP ribosyltransferase activity has been detected for SIRT7. Nevertheless, it appears that SIRT7 is also involved in life span extension. SIRT7 knockdown induces apoptosis in human cells, indicating that SIRT7 is required for cell survival (Ford et al. 2006). This observation is based on the observation that SIRT7 is a positive regulator of RNA polymerase I transcription and therefore ribosome biogenesis. In addition, life span was reduced in SIRT7-deficient mice, which had also enhanced inflammatory cardiomyopathy compared to wild-type mice. In such mice, a reduction of SIRT7 levels was accompanied by increased p53 activity, subsequently resulting in enhanced cardiomyocyte apoptosis (Vakhrusheva et al. 2008; Voelter-Mahlknecht et al. 2006a).
Conclusions and perspectives
Quite some confusion has been stirring up as to the role of the sirtuins acting primarily as suppressors for some types of cancers, while they seem to promote cancer under other circumstances. It is therefore currently unclear as to what extent and under which particular circumstances sirtuin activators and/or inhibitors will find their place in the treatment of age-related disease and cancer. While SIRT1 expression and activity are repressed under non-malignant conditions through tumor suppressors, SIRT1 expression and activity may be increased once oncogenes are overexpressed or if the activity of tumor suppressor proteins is reduced, which may finally block senescence and apoptosis, while on the other hand it may induce angiogenesis and stimulate cell growth and go along with resistance to chemotherapy. Future investigations regarding the concerted interplay of the different sirtuins will therefore not only contribute to a more detailed understanding of the aging process but might also lead to the development of novel strategies in the treatment of cancer and other age-related diseases.
References
Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E (2007) Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem 282:33583–33592
Allison SJ, Milner J (2007) SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 6:2669–2677
Anantharaman V, Aravind L (2008) Analysis of DBC1 and its homologs suggests a potential mechanism for regulation of sirtuin domain deacetylases by NAD metabolites. Cell Cycle 7:1467–1472
Araki T, Sasaki Y, Milbrandt J (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305:1010–1013
Ashraf N, Zino S, Macintyre A, Kingsmore D, Payne AP, George WD, Shiels PG (2006) Altered sirtuin expression is associated with node-positive breast cancer. Br J Cancer 95:1056–1061
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342
Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, Passarino G, De Benedictis G (2005) A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 85:258–263
Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L (2007) SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 6:759–767
Borra MT, Smith BC, Denu JM (2005) Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 280:17187–17195
Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM (2005) Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19:1751–1759
Brooks CL, Gu W (2008) p53 activation: a case against Sir. Cancer Cell 13:377–378
Brooks CL, Gu W (2009) How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 9:123–128
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Chen D, Steele AD, Lindquist S, Guarente L (2005) Increase in activity during calorie restriction requires Sirt1. Science 310:1641
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305:390–392
Das C, Lucia MS, Hansen KC, Tyler JK (2009) CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459:113–117
Deng CX (2009) SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci 5:147–152
Dransfeld CL, Alborzinia H, Wolfl S, Mahlknecht U (2010) SIRT3 SNPs validation in 640 individuals, functional analyses and new insights into SIRT3 stability. Int J Oncol 36:955–960
Engel N, Mahlknecht U (2008) Aging and anti-aging: unexpected side effects of everyday medication through sirtuin1 modulation. Int J Mol Med 21:223–232
Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA (2008) The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3:e2020
Ford J, Jiang M, Milner J (2005) Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res 65:10457–10463
Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L (2006) Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev 20:1075–1080
Fraga MF, Esteller M (2007) Epigenetics and aging: the targets and the marks. Trends Genet 23:413–418
Frye RA (1999) Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun 260:273–279
Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273:793–798
Frye R (2002) “SIRT8” expressed in thyroid cancer is actually SIRT7. Br J Cancer 87:1479
Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, Powell MJ, Yang T, Gu W, Avantaggiati ML, Pattabiraman N, Pestell TG, Wang F, Quong AA, Wang C, Pestell RG (2006) Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol 26:8122–8135
Garske AL, Smith BC, Denu JM (2007) Linking SIRT2 to Parkinson's disease. ACS Chem Biol 2:529–532
Gruber J, Schaffer S, Halliwell B (2008) The mitochondrial free radical theory of ageing—where do we stand? Front Biosci 13:6554–6579
Guarente L, Picard F (2005) Calorie restriction—the SIR2 connection. Cell 120:473–482
Hadley EC, Lakatta EG, Morrison-Bogorad M, Warner HR, Hodes RJ (2005) The future of aging therapies. Cell 120:557–567
Haigis MC, Sinclair DA (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295
Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126:941–954
Hallows WC, Lee S, Denu JM (2006) Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A 103:10230–10235
Hallows WC, Albaugh BN, Denu JM (2008) Where in the cell is SIRT3? Functional localization of an NAD+-dependent protein deacetylase. Biochem J 411:e11–e13
Heilbronn LK, Ravussin E (2003) Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr 78:361–369
Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA, Bedalov A (2006) Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res 66:4368–4377
Hida Y, Kubo Y, Murao K, Arase S (2007) Strong expression of a longevity-related protein, SIRT1, in Bowen's disease. Arch Dermatol Res 299:103–106
Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, Watanabe T, Ohama E, Tahimic CG, Kurimasa A, Oshimura M (2003) Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun 309:558–566
Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425:191–196
Huang J, Gan Q, Han L, Li J, Zhang H, Sun Y, Zhang Z, Tong T (2008) SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE 3:e1710
Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR (2007) SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res 67:6612–6618
Imai S, Guarente L (2010) Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci 31:212–220
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800
Inoue T, Hiratsuka M, Osaki M, Oshimura M (2007a) The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle 6:1011–1018
Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, Yamaguchi S, Nakano S, Katoh M, Ito H, Oshimura M (2007b) SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 26:945–957
Inoue T, Nakayama Y, Yamada H, Li YC, Yamaguchi S, Osaki M, Kurimasa A, Hiratsuka M, Katoh M, Oshimura M (2009) SIRT2 downregulation confers resistance to microtubule inhibitors by prolonging chronic mitotic arrest. Cell Cycle 8:1279–1291
Jin L, Wei W, Jiang Y, Peng H, Cai J, Mao C, Dai H, Choy W, Bemis JE, Jirousek MR, Milne JC, Westphal CH, Perni RB (2009) Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem 284:24394–24405
Jing E, Gesta S, Kahn CR (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 6:105–114
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Jung-Hynes B, Nihal M, Zhong W, Ahmad N (2009) Role of sirtuin histone deacetylase SIRT1 in prostate cancer. a target for prostate cancer management via its inhibition? J Biol Chem 284:3823–3832
Katic M, Kahn CR (2005) The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci 62:320–343
Kealy RD, Lawler DF, Ballam JM, Mantz SL, Biery DN, Greeley EH, Lust G, Segre M, Smith GK, Stowe HD (2002) Effects of diet restriction on life span and age-related changes in dogs. J Am Vet Med Assoc 220:1315–1320
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23:607–618
Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J 26:3169–3179
Kim JE, Chen J, Lou Z (2008) DBC1 is a negative regulator of SIRT1. Nature 451:583–586
Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17:41–52
Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122
Lamming DW, Latorre-Esteves M, Medvedik O, Wong SN, Tsang FA, Wang C, Lin SJ, Sinclair DA (2005) HST2 mediates SIR2-independent life-span extension by calorie restriction. Science 309:1861–1864
Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A 97:5807–5811
Lane MA, Ball SS, Ingram DK, Cutler RG, Engel J, Read V, Roth GS (1995) Diet restriction in rhesus monkeys lowers fasting and glucose-stimulated glucoregulatory end points. Am J Physiol 268:E941–E948
Lane MA, Baer DJ, Rumpler WV, Weindruch R, Ingram DK, Tilmont EM, Cutler RG, Roth GS (1996) Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc Natl Acad Sci U S A 93:4159–4164
Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS (2008) Endurance exercise as a countermeasure for aging. Diabetes 57:2933–2942
Lim CS (2006) SIRT1: tumor promoter or tumor suppressor? Med Hypotheses 67:341–344
Liszt G, Ford E, Kurtev M, Guarente L (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 280:21313–21320
Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J 3rd, Olefsky J, Guarente L, Montminy M (2008) A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456:269–273
Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW (2005) DNA repair, genome stability, and aging. Cell 120:497–512
Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27:8807–8814
Longo VD, Kennedy BK (2006) Sirtuins in aging and age-related disease. Cell 126:257–268
Lopez-Lluch G, Irusta PM, Navas P, de Cabo R (2008) Mitochondrial biogenesis and healthy aging. Exp Gerontol 43:813–819
Love R (2005) Calorie restriction may be neuroprotective in AD and PD. Lancet Neurol 4:84
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W (2001) Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107:137–148
Ma W, Stafford LJ, Li D, Luo J, Li X, Ning G, Liu M (2007) GCIP/CCNDBP1, a helix-loop-helix protein, suppresses tumorigenesis. J Cell Biochem 100:1376–1386
Mahlknecht U, Hoelzer D (2000a) Histone acetylation modifiers in the pathogenesis of malignant disease. Mol Med 6:623–644
Mahlknecht U, Voelter-Mahlknecht S (2009a) Chromosomal characterization and localization of the NAD+-dependent histone deacetylase gene sirtuin 1 in the mouse. Int J Mol Med 23:245–252
Mahlknecht U, Voelter-Mahlknecht S (2009b) Fluorescence in situ hybridization and chromosomal organization of the sirtuin 4 gene (Sirt4) in the mouse. Biochem Biophys Res Commun 382:685–690
Mahlknecht U, Ottmann OG, Hoelzer D (2000b) When the band begins to play: histone acetylation caught in the crossfire of gene control. Mol Carcinog 27:268–271
Mahlknecht U, Ho AD, Letzel S, Voelter-Mahlknecht S (2006a) Assignment of the NAD-dependent deacetylase sirtuin 5 gene (SIRT5) to human chromosome band 6p23 by in situ hybridization. Cytogenet Genome Res 112:208–212
Mahlknecht U, Ho AD, Voelter-Mahlknecht S (2006b) Chromosomal organization and fluorescence in situ hybridization of the human sirtuin 6 gene. Int J Oncol 28:447–456
Masoro EJ (2000) Caloric restriction and aging: an update. Exp Gerontol 35:299–305
Masoro EJ, Austad SN (1996) The evolution of the antiaging action of dietary restriction: a hypothesis. J Gerontol A Biol Sci Med Sci 51:B387–B391
Mattison JA, Lane MA, Roth GS, Ingram DK (2003) Calorie restriction in rhesus monkeys. Exp Gerontol 38:35–46
McCay CM, Crowell MF, Maynard LA (1989) The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 5:155–171, discussion 172
Mei S, Ho AD, Mahlknecht U (2004) Role of histone deacetylase inhibitors in the treatment of cancer (review). Int J Oncol 25:1509–1519
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13
Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16:4623–4635
Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, Gozani O, Chua KF (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452:492–496
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450:712–716
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124:315–329
Nakagawa T, Lomb DJ, Haigis MC, Guarente L (2009) SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137:560–570
Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q (2009) STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol 11:492–500
North BJ, Verdin E (2007) Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J Biol Chem 282:19546–19555
North BJ, Marshall BL, Borra MT, Denu JM, Verdin E (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11:437–444
Oberdoerffer P, Sinclair DA (2007) The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol 8:692–702
Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, Wright SM, Mills KD, Bonni A, Yankner BA, Scully R, Prolla TA, Alt FW, Sinclair DA (2008) SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135:907–918
O'Hagan HM, Mohammad HP, Baylin SB (2008) Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 4:e1000155
Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y, Kaneki M (2006) Sirt1 inhibitor, sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25:176–185
Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y (2007) Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol 43:571–579
Ouaissi M, Sielezneff I, Silvestre R, Sastre B, Bernard JP, Lafontaine JS, Payan MJ, Dahan L, Pirro N, Seitz JF, Mas E, Lombardo D, Ouaissi A (2008) High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol 15:2318–2328
Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG (2007) Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science 317:516–519
Pinney DO, Stephens DF, Pope LS (1972) Lifetime effects of winter supplemental feed level and age at first parturition on range beef cows. J Anim Sci 34:1067–1074
Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB (2006) Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet 2:e40
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, BonaFe M, Franceschi C, Tan Q, Boiko S, Yashin AI, De Benedictis G (2003) Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol 38:1065–1070
Roth GS (2005) Caloric restriction and caloric restriction mimetics: current status and promise for the future. J Am Geriatr Soc 53:S280–S283
Roth GS, Ingram DK, Lane MA (1995) Slowing ageing by caloric restriction. Nat Med 1:414–415
Roth GS, Mattison JA, Ottinger MA, Chachich ME, Lane MA, Ingram DK (2004) Aging in rhesus monkeys: relevance to human health interventions. Science 305:1423–1426
Roth GS, Lane MA, Ingram DK (2005) Caloric restriction mimetics: the next phase. Ann NY Acad Sci 1057:365–371
Sasaki T, Maier B, Bartke A, Scrable H (2006) Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell 5:413–422
Saunders LR, Verdin E (2007) Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 26:5489–5504
Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C (2008) Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 382:790–801
Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A, Loppnau P, Vedadi M, Bochkarev A, Sternglanz R, Plotnikov AN (2007) Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 15:377–389
Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E (2006) Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A 103:10224–10229
Shi T, Wang F, Stieren E, Tong Q (2005) SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 280:13560–13567
Sinclair DA (2005) Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev 126:987–1002
Sommer M, Poliak N, Upadhyay S, Ratovitski E, Nelkin BD, Donehower LA, Sidransky D (2006) DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle 5:2005–2011
Stunkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto-Tellez M, Ni B, Entzeroth M, Wood J (2007) Function of the SIRT1 protein deacetylase in cancer. Biotechnol J 2:1360–1368
Tanner KG, Landry J, Sternglanz R, Denu JM (2000) Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci U S A 97:14178–14182
Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E (2008) Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res 102:703–710
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107:149–159
Vijg J, Maslov AY, Suh Y (2008) Aging: a sirtuin shake-up? Cell 135:797–798
Voelter-Mahlknecht S, Mahlknecht U (2003) Cloning and structural characterization of the human histone deacetylase 6 gene. Int J Mol Med 12:87–93
Voelter-Mahlknecht S, Mahlknecht U (2006) Cloning, chromosomal characterization and mapping of the NAD-dependent histone deacetylases gene sirtuin 1. Int J Mol Med 17:59–67
Voelter-Mahlknecht S, Ho AD, Mahlknecht U (2005) FISH-mapping and genomic organization of the NAD-dependent histone deacetylase gene, sirtuin 2 (Sirt2). Int J Oncol 27:1187–1196
Voelter-Mahlknecht S, Letzel S, Mahlknecht U (2006) Fluorescence in situ hybridization and chromosomal organization of the human sirtuin 7 gene. Int J Oncol 28:899–908
Wang F, Nguyen M, Qin FX, Tong Q (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6:505–514
Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng ZM, Appella E, Wang XW, Ried T, Deng CX (2008a) Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14:312–323
Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX (2008b) Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell 32:11–20
Weindruch R, Sohal RS (1997) Seminars in medicine of the Beth Israel Deaconess medical center. Caloric intake and aging. N Engl J Med 337:986–994
Weindruch R, Walford RL, Fligiel S, Guthrie D (1986) The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr 116:641–654
Westphal CH, Dipp MA, Guarente L (2007) A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci 32:555–560
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430:686–689
Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130:1095–1107
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23:2369–2380
Yu BP (1994) How diet influences the aging process of the rat. Proc Soc Exp Biol Med 205:97–105
Yuan Z, Seto E (2007) A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle 6:2869–2871
Yuan J, Minter-Dykhouse K, Lou Z (2009) A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 185:203–211
Zhang Q, Wang SY, Fleuriel C, Leprince D, Rocheleau JV, Piston DW, Goodman RH (2007) Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc Natl Acad Sci U S A 104:829–833
Zhang Y, Au Q, Zhang M, Barber JR, Ng SC, Zhang B (2009) Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem Biophys Res Commun 386:729–733
Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W (2008) Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451:587–590
Zschoernig B, Mahlknecht U (2008) SIRTUIN 1: regulating the regulator. Biochem Biophys Res Commun 376:251–255
Zschoernig B, Mahlknecht U (2009) Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem Biophys Res Commun 381:372–377
Acknowledgments
We apologize to all authors whose work was not discussed or cited in this manuscript.
Competing interests statement
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Voelter-Mahlknecht, S., Mahlknecht, U. The sirtuins in the pathogenesis of cancer. Clin Epigenet 1, 71–83 (2010). https://doi.org/10.1007/s13148-010-0008-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-010-0008-0