
Abstract
Chromatin remodeling and gene expression are regulated by histone deacetylases (HDACs) that condense the chromatin structure by deacetylating histones. HDACs comprise a group of enzymes that are responsible for the regulation of both cellular and viral genes at the transcriptional level. In mammals, a total of 18 HDACs have been identified and grouped into four classes, i.e., class I (HDACs 1, 2, 3, 8), class II (HDACs 4, 5, 6, 7, 9, 10), class III (Sirt1–Sirt7), and class IV (HDAC11). We review here the role of HDACs on viral replication and how HDAC inhibitors could potentially be used as new therapeutic tools in several viral infections.
Similar content being viewed by others


Introduction
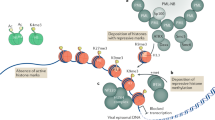
Chromatin is the complex combination of DNA and proteins that makes up chromosomes. It is divided between heterochromatin (condensed) and euchromatin (extended) forms. The major components of chromatin are DNA and histone proteins, although many other chromosomal proteins have prominent roles, too. The functions of chromatin are to package DNA into a smaller volume to fit in the cell, to strengthen the DNA to allow mitosis and meiosis, and to serve as a mechanism to control expression and DNA replication. Changes in chromatin structure are affected by chemical modifications of histone proteins and by non-histone DNA-binding proteins. The non-histone proteins that are found associated with isolated chromatin fall into several functional categories such as chromatin-bound enzymes, high mobility group proteins (HMG), transcription factors, and scaffold proteins. Thus, acetylation of histone and non-histone chromosomal proteins by histone acetyl transferases (HATs) leads to decondensation of the chromatin. Chromatin remodeling and gene expression is simultaneously regulated by histone deacetylases (HDACs) that condense the chromatin structure by deacetylating histones. HDACs comprise a group of enzymes that are responsible for the removal of acetyl groups from a e-N-acetyl lysine amino acids. As the organization and packaging of eukaryotic DNA is achieved through the addition of core histones H2A, H2B, H3, and H4, which form the chromatin, the modification of core histones is of fundamental importance to conformational changes in chromatin.
The level of acetylation influences transcription activity. The acetylation of histones results in the formation of shapes of DNA that will serve as specific recruitment platforms for other protein and enzyme complexes. These multiprotein complexes will result in enhanced transcriptional activity (euchromatin). By contrast, HDACs present in a repressor complex deacetylate histone proteins, leading to the recruitment of histone methyltransferase and resulting in subsequent methylation of histones. Ultimately, proteins such as heterochromatin protein 1 (HP1) are recruited to the repressor complex, leading to the formation of condensed heterochromatin. Thus, HDAC activities are a prerequisite step allowing further modification of histone proteins that promote the final heterochromatin formation.
HDAC family members
In mammals, a total of 18 HDACs have been identified and grouped into four classes, i.e., class I (HDACs 1, 2, 3, 8), class II (HDACs 4, 5, 6, 7, 9, 10), class III (Sirt1–Sirt7), and class IV (HDAC11) (Table 1). The members of class I HDACs contain a deacetylase domain and are the homologs of yeast RPD3. They exhibit 45% to 93% amino acid sequence identity and localize in the nucleus, except for HDAC3. Class II HDACs are homologs of yeast Hda1 and are present in both the nucleus and the cytoplasm (Martin et al. 2009; Martin et al. 2007). HDAC6 is an exception which contains two deacetylase domains, at both the N- and C-terminus. The molecular weights of class II are twofold higher than class I HDACs. The members of class III, sirtuins, are homologs of yeast Sir2 and form a structurally distinct class of NAD-dependent enzymes found in both the nucleus and cytoplasm. HDAC11 has properties of both class I and class II HDACs and represent class IV.
HDACs work in multi-subunit transcriptional co-repressor complexes that are recruited by sequence-specific transcription factors to promoter regions. There are several co-repressor complexes for distinct promoters, which recruit specific HDAC isoforms for silencing of target genes (Miska et al. 1999; Rayman et al. 2002). For example, HDACs 1, 2, and 3 are majorly responsible for catalytic core for different co-repressor complexes to achieve efficient transcriptional repression. HDAC1 and HDAC2 are present in the CoREST, Mi2/NuRD, and Sin3 complexes, whereas HDAC3 is responsible for the catalytic activity of the N-CoR and SMRT co-repressor complexes (Cress and Seto 2000; Mahlknecht et al. 2004; Ng and Bird 2000). HDACs cooperate with various other transcriptional regulators such as HDAC1 and HDAC2 associate with DNA methyltransferases (Burgers et al. 2002; Fuks et al. 2001) and histone methyltransferases (HMTs) (Czermin et al. 2001). Furthermore, HDAC1 interacts with topoisomerase II enzyme that is responsible for chromosome condensation (Tsai et al. 2000). However, little is known about the specificity of a particular histone deacetylase enzyme for a specific lysine residue. The preferential acetylation of H3K18 and H3K9 following knockdown of HDAC1 and HDAC3, respectively, has been reported (Zhang et al. 2004). Non-histone deacetylation-based gene expression involves the deacetylation of various transcription factors by HDACs. Deacetylation of sequence-specific transcription factors can decrease their DNA binding activity and subsequently may repress transcription. The covalent modifications of several transcriptional factors, including E2F, sp3, p53, GATA1, and TFIIF, have been reported (Ammanamanchi et al. 2003; Boyes et al. 1998; Braun et al. 2001; Gu and Roeder 1997; Imhof et al. 1997; Marzio et al. 2000). The specificity of HDAC1 for p53 deacetylation, resulting in the degradation of deacetylated p53 has been reported (Ito et al. 2002). HDAC2 deacetylates the glucocorticoid receptor and HDAC3 is needed for the deacetylation of myocyte enhancer factor-2 (MEF-2) (Gregoire et al. 2007; Ito et al. 2006). HDACs are also involved in the deacetylation of non-nuclear proteins like tubulin45 and HSP90 (Hubbert et al. 2002; Kovacs et al. 2005).
HDAC in viral diseases
HDACs are involved in the regulation of the replication of numerous viruses (Table 2). We will described here the role of HDACs in viral diseases including hepatitis B and C, HIV and HPV infections, and infections with Herpesviridae (HSV, HCMV, EBV) and Paramyxoviridae (RSV).
The hepatitis B virus x (HBx) protein plays a critical role in liver carcinogenesis. HBx has been reported to activate cyclin D1. HBx induces the expression of metastasis-associated protein 1 (MTA1), a protein closely related to tumor growth and metastasis in various cancers, especially in hepatocellular carcinoma (HCC). In addition, HBx has been reported to modulate hypoxia-inducible factor-1 alpha (HIF-1 alpha) expression and activity, and HIF-1 alpha increased expression has been correlated with tumor growth and poor patient prognosis (Semenza 2004). Under conditions of normoxia, post-translational modifications including the hydroxylation of proline residues and acetylation of a lysine residue within the oxygen-dependent degradation domain promote HIF-1α interaction with von Hipple–Lindau (pVHL) ubiquitin E3 ligase complex. This occurs concurrently with the hydroxylation of an asparagine residue by the aparaginyl hydroxylase FIH-1 and inhibits the binding of transcriptional co-activators p300 and CBP to HIF-1α. These events result in polyubiquitination and the proteosomal degradation of HIF-1α. In contrast, conditions of cellular hypoxia result in HIF-1α-stabilized expression by remaining unhydroxylated and deacetylated, especially via class II HDAC activation (Qian et al. 2006). Stabilized HIF-1α escapes pVHL-mediated degradation and is able to bind p300 and CBP where it translocates to the nucleus from the cytoplasm and heterodimerizes with HIF-1β to initiate the transcription of its target genes. During HBV infection, especially at the stage of HCC, hypoxia occurs, resulting in HIF-1alpha overexpression parallel to HBx upregulation. In the presence of HBx, HDAC1/2 and MTA1 physically associate with HIF-1 alpha in vivo. Thus, a positive cross-talk between HBx and the MTA1/HDAc complex in stabilizing HIF-1 alpha may play a critical role in angiogenesis and metastasis of HBV-associated HCC (Yoo et al. 2008).
HCV-induced oxidative stress suppresses the expression of hepcidin, a negative regulator of iron absorption. Hypoxia and chemical stabilizers of the HIF-1 alpha have also been shown to suppress hepcidin expression (Volke et al. 2009). Expression of hepcidin inhibitor, HIF, is increased by HDAC, suggesting that HCV-induced oxidative stress suppresses hepcidin expression through increased HDAC activity resulting in increased HIF-1 alpha activity (Miura et al. 2008).
After entry into the target cell and reverse transcription, HIV-1 genes are integrated into the host genome. It is now well established that the viral promoter activity is directly governed by its chromatin environment (Van Lint 2000). Epigenetic modifications and disruption of Nuc-1, a nucleosome located immediately downstream of the transcriptional initiation site that directly impedes LTR activity, are a prerequisite to the activation of LTR-driven transcription and viral expression (Van Lint 2000; Van Lint et al. 1996; Verdin et al. 1993). The compaction of chromatin and its permissiveness for transcription are directly dependent on the post-translational modifications of histones such as acetylation, methylation, phosphorylation, and ubiquitination (Fischle et al. 2003). In contrast to productively infected cells, latently infected cells frequently harbor HIV-1 genomes integrated in heterochromatic structures, which allows the viral persistence of silenced integrated proviruses (Jordan et al. 2003). These observations might at least partially explain how the virus can form viral reservoirs in some target cells and thereby fuel the progression of the disease (Finzi et al. 1997; Pierson et al. 2000). Some transcription factors, such as YY1 and LSF, repress the transcription from the HIV-1 LTR by recruiting HDAC1 to the repressor complex (Coull et al. 2000). In agreement with these data, HIV-1 gene transcription has been shown to be activated by trichostatin A (TSA) treatment, and several transcription factors bound to the viral LTR recruit class I or II HDAC (Coull et al. 2000; Van Lint et al. 1996; Williams et al.1 2006). HP1 is a transcriptional repressor that directly binds to the methylated lysine 9 residue of histone H3 (H3K9me), which is a hallmark histone modification for transcriptionally silenced heterochromatin (Zeng et al. 2010; Bannister et al. 2001). The levels of trimethylated histone H3 and HP1-alpha associated with HIV proviruses have been reported to fall rapidly after TNF alpha activation, indicating that epigenetic mechanisms targeting chromatin structures selectively restrict HIV transcription (Pearson et al. 2008).
COUP-TF interacting protein 2 (CTIP2) is a recently cloned transcriptional repressor that can associate with members of the COUP-TF family (Avram et al. 2000). CTIP2 has been reported to inhibit HIV-1 replication in human microglial cells (Marban et al. 2005; Marban et al. 2007; Rohr et al. 2003). Microglial cells constitute the central nervous system resident macrophages and have been described as latently HIV-1-infected cellular reservoirs (Barber et al. 2006). However, contrary to CD4+ T lymphocyte reservoirs (Marcello 2006), within macrophages and microglial cells CTIP2 inhibits HIV-1 gene transcription (reviewed in Redel et al. 2010). CTIP2 recruits HDAC1 and HDAC2 to promote local histone H3 deacetylation at the HIV-1 promoter region and also associates with the histone methyltransferase SUV39H1, which increases local histone H3 lysine 9 methylation. Thereafter, HP1 proteins are recruited to the viral promoter, leading to the formation of local heterochromatin and subsequently to HIV-1 silencing (Marban et al. 2007).
p21WAF1 is a major cell cycle regulator of the response to DNA damage, senescence, and tumor suppression and its induction upon HIV-1 infection favors HIV-1 replication in macrophages. CTIP2 is recruited to the p21 gene promoter, resulting in the silencing of p21 gene transcription through interactions with histone deacetylases and methyltransferases SUV39H1, and therefore could abolish Vpr-mediated stimulation of p21, thereby indirectly contributing to HIV-1 latency (Cherrier et al. 2009). Besides CTIP2, the AP-4 site within the HIV-1 LTR represses HIV-1 gene expression by recruiting HDAC1 (Imai and Okamoto 2006). Also, c-Myc and Sp1 transcription factors contribute to proviral latency by recruiting HDAC1 to the HIV-1 LTR (Jiang et al. 2007).
HIV-1 integrase and INI1/hSNF5, a component of the SWI/SNF complex, bind SAP18 (Sin3a-associated protein, 18 kDa), a component of the Sin3a-HDAC1 complex, and selectively recruit components of Sin3a–HDAC1 complex into HIV-1 virions (Sorin et al. 2009). Thus, HIV-1 virion-associated HDAC1 is required for efficient early post-entry events.
HIV Tat is involved in binding to the CBP/p300 and cdk9/cyclin T1 complexes, resulting in Tat acetylation by the transcriptional coactivator p300, thereby facilitating transcription initiation. Acetylated Tat dissociates from the TAR RNA structure and recruits bromodomain-binding chromatin-modifying complexes such as p/CAF and SWI/SNF to facilitate transcription elongation (Agbottah et al. 2006). By contrast, Tat is deacetylated by human sirtuin 1 (SIRT1), a nicotinamide adenine dinucleotide-dependent class III protein deacetylase in vitro and in vivo (Pagans et al. 2005; Zhang et al. 2009), supporting a model in which cycles of Tat acetylation and deacetylation regulate HIV-1 transcription (Blazek and Peterlin 2008; Kwon et al. 2008).
Upon infection of permissive cells such as fibroblasts, epithelial cells, endothelial cells, and macrophages, human cytomegalovirus (HCMV) undergoes an ordered cascade of gene expression. The major immediate early (IE) genes are the first transcribed, resulting in two abundant proteins, IE1 p72 and IE2 p86 (Stenberg et al. 1989), which are able to autoregulate the major immediate early promoter (MIEP) (Cherrington and Mocarski 1989; Pizzorno et al. 1988). It is known that expression from the MIEP is at least partly determined by cellular factors such as NF-kB/rel, CREB/ATF, SP-1, AP-1, Bcl-3, serum response factor, and ELK-1, which all promote activity by binding one or more sites in the enhancer (Chan et al. 1996; Kowalik et al. 1993; Lang et al. 1992; Liu and Stinski 1992; Sambucetti et al. 1989; Wade et al. 1992; Khan et al. 2009). In contrast, the MIEP is also regulated by cellular repressors, such as MBFs, YY1, MRF and Gfi-1, which bind to the MIEP and all are preferentially expressed in undifferentiated cells (Huang et al. 1996; Liu et al. 1994; Shelbourn et al. 1989; Zweidler-Mckay et al. 1996). The chromatin structure around the MIEP changes with cellular differentiation and thus may play a role in controlling HCMV latency and reactivation. T2 cells are normally non-permissive for HCMV infection; however, following treatment with TSA, T2 cells can be rendered transiently permissive for HCMV (Meier 2001), suggesting that HDACs play a role in the repression of viral replication. HDACs have been reported as limiting HCMV infection as the relatively high levels of HDAC3 in T2 cells and monocytes correlate with non-permissiveness for HCMV (Murphy et al. 2002). Furthermore, fibroblasts, which are fully permissive for HCMV, contain little HDAC3, implying that the relative level of HDAC may be important for HCMV permissiveness in a range of cell types. HDAC3 acts as a repressor of the HCMV MIEP since superexpression of HDAC3 appeared most effective at inhibiting viral infection in permissive T2RA cells (Murphy et al. 2002).
Changes in chromatin structure have been implicated in the control of latency in herpesviruses. Latent herpes simplex virus type 1 (HSV-1) DNA is associated with nucleosomes in vivo (Deshmane and Fraser 1989). Following TSA treatment of neurons latently infected with a recombinant HSV-1, the activity of the viral immediate early promoter increased significantly (Arthur et al. 2001), suggesting that HDACs may play a role in controlling HSV-1 latency. The abundant and chromatin-associated protein HCF-1 is a critical player in HSV transcription. HCF-1 associates with the Sin3 HDAC and human trithorax-related Set1/Ash2 histone methyltransferase (Wysocka et al. 2003). HCF-1 tethers the Sin3 and Set1/Ash2 transcriptional regulatory complexes together even though they are generally associated with opposite transcriptional outcomes: repression and activation of transcription, respectively. Nevertheless, this tethering is context-dependent because the transcriptional activator VP16 selectively binds HCF-1 associated with the Set1/Ash2 HMT complex in the absence of the Sin3 HDAC complex (Wysocka et al. 2003). These results suggest that HCF-1 can broadly regulate transcription, both positively and negatively, through selective modulation of chromatin structure. HSV-1 immediate early regulatory protein ICP0 is important for stimulating the initiation of the lytic cycle and efficient reactivation of latent or quiescent infection. ICP0 inhibits cellular HDAC activity through an interaction with the HDAC-1 binding partner CoREST (Everett et al. 2009).
Like HSV-1, in latent Epstein–Barr virus (EBV), DNA is organized in nucleosomes (Dyson and Farrell 1985; Jenkins et al. 2000; Shaw et al. 1979) and the switch to productive EBV infection is believed to involve the activation of the IE gene BZLF1 (Countryman and Miller 1985). Although it was proposed that HDACs 4 and 5 are the HDACs which might mediate the repression of Zp (Gruffat et al. 2002), direct measurement of the expression of class II HDACs has shown that HDACs 4 and 5 are undetectable but HDAC7 is the most readily detected class II HDAC in Raji cells, suggesting that it may be involved in the repression of the Zp promoter of the EBV BZLF1 gene during latency (Bryant and Farrell 2002). Moreover, the promoter of BZLF1 is more associated with acetylated histones following activation of EBV-positive AK6 cells (Jenkins et al. 2000). During EBV latency, latent membrane protein (LMP)-1 expression induces several B lymphocyte activation markers, including intercellular adhesion molecule (ICAM)-1. HDAC inhibition induces the latent form of LMP-1 expression in EBV-positive cell lines, resulting in ICAM-1 expression upregulation and homotypic adhesion (Park and Faller 2002). The primary events in EBV reactivation involve dephosphorylation of the MEF-2D transcription factor. Dephosphorylation of MEF-2D is known to be associated with histone acetylase recruitment (Bryant and Farrell 2002). Histone deacetylation in response to phosphorylated MEF-2D can be mediated by class I or class II HDACs (Gruffat et al. 2002). Telomere repeat factor 2 (TRF2) binding at the EBV origin of plasmid replication (OriP) represses EBV replication. HDAC1 and HDAC2 form a stable complex with TRF2 at OriP and enhance EBV episome stability by providing a checkpoint that delays replication initiation at OriP (Zhou et al. 2009).
Respiratory syncytial virus (RSV) is a paramyxovirus that produces airway inflammation, in part by inducing interleukin-8 (IL-8) expression, a CXC-type chemokine, via the NF-kB/RelA and STAT/IRF signaling pathways. In RSV-infected A549 cells, IL-8 transcription attenuates after 24 h in spite of ongoing viral replication and persistence of nuclear RelA, suggesting a mechanism for transcriptional attenuation (Jamaluddin et al. 2005). RSV infection induces Bcl-3 expression 6 to 12 h after viral infection, at times when IL-8 transcription is inhibited. Nuclear Bcl-3 associates with STAT1 and HDAC1, increasing HDAC1 recruitment to the IL-8 promoter. Treatment with TSA blocks the attenuation of IL-8 transcription (Jamaluddin et al. 2005). Thus, Bcl-3 is a RSV-inducible inhibitor of IL-8 transcription by interfering with the NF-kB and STAT/IRF signaling pathways by complexing with them and recruiting HDAC1 to attenuate target promoter activity.
The activities encoded by the early gene product E7 are essential to the oncogenic properties of human papillomavirus type 16 (HPV-16) (Longworth et al. 2005). HPV-16 E7 binds to cellular factors involved in cell cycle regulation and differentiation. The cdc25A tyrosine phosphatase is required for G(1)/S transition, and its deregulation is associated with carcinogenesis. HPV-16 E7 can directly target cdc25A transcription and maintains cdc25A gene expression by disrupting Rb/E2F/HDAC1 repressor complexes (Nguyen et al. 2002). The interaction of E7 with HDACs is essential for extending the life span of keratinocytes and for stable maintenance of viral genomes (Longworth and Laimins 2004). E7 activates the E2F2 transcription in suprabasal keratinocytes through its ability to bind HDACs (Longworth et al. 2005). Other studies have found that growth arrest of HPV-positive cells after HDAC inhibition is independent of E6/E7 oncogene expression (Finzer et al. 2002). HPV E7 protein is functionally associated with the interferon regulatory factor (IRF)-1 tumor suppressor in cervical carcinogenesis. Under TSA treatment, the repressing activity of E7 is released in a dose-dependent manner (Park et al. 2000), suggesting that HPV E7 interferes with the transactivation function of IRF-1 by recruiting HDAC to the promoter.
HDACs: new therapeutic targets in viral diseases?
There are several HDAC inhibitors. The “classical” HDAC inhibitors act exclusively on class I and class II HDACs by binding to the zinc-containing catalytic domain of the HDACs. These classical HDAC inhibitors fall into several groupings:
-
1.
hydroxamic acids, such as trichostatin A
-
2.
cyclic tetrapeptides (such as trapoxin B) and the depsipeptides
-
3.
benzamides
-
4.
electrophilic ketones
-
5.
the aliphatic acid compounds such as phenylbutyrate and valproic acid
The “second-generation” HDAC inhibitors include the hydroxamic acids vorinostat (SAHA), belinostat (PXD101), LAQ824, and panobinostat(LBH589) and the benzamides entinostat (MS275), CI994, and mocetinostat (MGCD0103). The sirtuin class III HDACs are NAD+ dependent and are therefore inhibited by nicotinamide, as well derivatives of NAD, dihydrocoumarin, naphthopyranone, and 2-hydroxynaphaldehydes.
HDAC inhibitors (HDACi) could potentially be used as new therapeutic tools in several viral infections. First, HDACi could reactivate the latent virus present in infected cells and thereby favor the clearance of the virus from cellular reservoirs such as in HIV and HCMV infections. Second, HDACi could favor the apoptosis of infected cells and also could thereby lead to the elimination of infected cells. This mechanism could be used to kill transformed cells infected by viruses such as HPV and EBV.
HDACi as therapeutic tools to reactivate latent virus from cellular reservoirs
The ability of HIV to establish a latent infection causes life-long virus persistence, even after long-term highly active antiretroviral therapy (HAART). The role that latency is playing in preventing the clearance of the virus infection has become evident in recent years. Patients who have been successfully treated with HAART, having undetectable levels of viral RNA (below 40 copies/ml) in the plasma for years, experience rapid virus rebound on withdrawal of therapy. Activation of latent proviruses from the infected cells in combination with HAART is a therapeutic strategy that may lead to the complete elimination of HIV infection (Colin and Van Lint 2009). Combination therapy with an HDAC inhibitor such as TSA and intensified HAART has been shown to safely accelerate the clearance of HIV from resting CD4+ T cells in vivo, suggesting a new and practical approach to eliminate HIV infection in this persistent reservoir (Lehrman et al. 2005). By contrast, more recent data indicate that the stability of the latent reservoir despite intensive therapy requires new strategies to eradicate HIV-1 from this reservoir (Gandhi et al. 2010; Sagot-Lerolle et al. 2008; Archin et al. 2010).
HDACs act on histones within the nucleosome-bound promoter of HIV-1 to maintain proviral latency. HDAC inhibition leads to promoter expression and the escape of HIV from latency. After exposure to pyrrole–imidazole polyamides that specifically block HDAC1 recruitment by the transcription factor LSF to the HIV promoter, replication-competent HIV is recovered from the cultures of resting CD4+ T cells in HIV-infected patients whose viremia had been suppressed by therapy (Ylisastigui et al. 2004a). Class I HDAC inhibitors such as suberoylanilide hydroxamic acid (SAHA, Vorinostat) have been tested ex vivo on HIV-1-infected cells isolated from the peripheral blood of patients (Quivy et al. 2007). In the resting CD4+ T cells of antiretroviral-treated aviremic HIV-infected patients, clinically achievable exposures to SAHA induced virus outgrowth ex vivo (Archin et al. 2009a; Edelstein et al. 2009). Valproic acid (VPA) induces acetylation at the integrated HIV proviral promoter, but CD4 cells exposed to VPA do not become activated or more permissive for de novo HIV infection. VPA induces outgrowth of HIV from the resting CD4+ T cells of aviremic patients at concentrations achievable in vivo (Ylisastigui et al. 2004b). In addition, combination therapy with oral VPA (500–750 mg, twice daily) added to intensified HAART for 3 months safely accelerates the clearance of HIV from resting CD4+ T cells in vivo, suggesting a way to eliminate HIV infection in the persistent reservoir (Lehrman et al. 2005). Class I HDACi are a strikingly efficient inducer of virus outgrowth from resting CD4+ T cells of aviremic patients, whereas HIV is rarely recovered from patient’s cells exposed to class II HDACi (Archin et al. 2009b). Since global HDAC inhibition can induce host cytotoxicity, the RNA expression and protein expression and compartmentalization of HDAC1 to HDAC11 in the resting CD4+ T cells of HIV-1-positive aviremic patients were recently assessed. Although all HDACs were detected in resting CD4+ T cells, targeted inhibition of HDACs by small-interfering RNA demonstrated that mostly HDAC2 and HDAC3 contribute to the repression of HIV-1 LTR expression (Keedy et al. 2009). Therefore, HDACi specific for a limited number of class I HDACs may offer a targeted approach to the disruption of persistent HIV-1 infection. Thus, potent selective HDACi may allow improved targeting of persistent proviral infection in HIV-infected patients.
Recently, a strong synergistic activation of HIV-1 production by clinically used histone deacetylase inhibitors (HDACis) combined with prostratin, a non-tumor-promoting nuclear factor (NF)- κB inducer, was shown using the latently infected U1 monocytic cell line and latently infected J-Lat T cell clones (Reuse et al. 2009). Mechanistically, HDACis increased prostratin-induced DNA-binding activity of nuclear NF-κB and degradation of cytoplasmic NF-κB inhibitor, IκBα. Moreover, the combined treatment prostratin+HDACi caused a more pronounced nucleosomal remodeling in the U1 viral promoter region than the treatments with the compounds alone. This more pronounced remodeling correlated with a synergistic reactivation of HIV-1 transcription following the combined treatment prostratin+HDACi. The physiological relevance of the prostratin+HDACi synergism has been shown in CD8+-depleted peripheral blood mononuclear cells from HAART-treated patients with undetectable viral load (Reuse et al. 2009). Moreover, this combined treatment reactivated viral replication in resting CD4+ T cells isolated from similar patients. Viral reactivation assays have shown in cell cultures prepared from HAART-treated HIV-1-infected individuals with undetectable viral load that a combination prostratin+VPA or prostratin+SAHA can induce HIV-1 recovery in CD8+-depleted PBMC cultures tested. Moreover, the combination prostratin+SAHA can reactivate viral replication in HLA DR− CD4+ T cell cultures prepared from additional patients (Reuse et al 2009). Altogether, these results suggest that combinations of different kinds of proviral activators may have important implications for reducing the size of latent HIV-1 reservoirs in HAART-treated patients, not only in CD4+ T cells but also in monocytes/macrophages. Interestingly, resveratrol, a SIRT1 activator, has been recently reported to inhibit in vitro Tat-induced HIV-1 LTR transactivation via NAD(+)-dependent SIRT1 activity (Zhang et al. 2009). Besides HDACi, resveratrol and related compounds may represent as potential candidates for novel anti-HIV therapeutics.
Beside HIV, other viruses could potentially be targeted by HDACi to clear cellular reservoirs of virions. HCMV has been reported to replicate at low levels in infected cells and to be reactivated upon immune suppression, especially in transplant recipients (Coaquette et al. 2004). Recently, we reported that the activation of p52/Bcl-3 NF-kB complexes in primary macrophages and p50/p65 NF-kB complexes in fibroblasts in response to HCMV infection could explain the low-level growth of the virus in macrophages versus efficient growth in fibroblasts (Khan et al. 2009). The low level sustained replication of HCMV in macrophages could favor the formation of reservoirs of virions, and the use of HDACi could reactivate the virus which could be targeted with antiviral compounds such as ganciclovir. Thus, the use of HDACi in addition to antiviral treatment could be a new promising approach in several viral infections including HIV-1, but also herpesviruses such as HCMV and others.
HDACi as therapeutic tools to kill virus-induced transformed cells
Cervical cancer cells are addicted to the expression of the HPV oncoproteins E6 and E7. The oncogenicity of E6 is mediated in part by targeting p53 and PDZ-family tumor suppressor proteins for rapid proteosomal degradation, whereas the E7 oncoprotein acts in part by coopting HDAC1/2. Immunohistochemical analysis reveals elevated HDAC1/HDAC2 expression in cervical dysplasia and cervical carcinoma versus normal cervical epithelium.
HDACi sodium butyrate and TSA arrest HPV-positive carcinoma cells in G1 to S transition of the cell cycle, which is paralleled by an upregulation of the cyclin-dependent kinase inhibitors (CKIs) p21CIP1 and p27KIP1 as well as the complete loss of cdk2 activity. HDAC inhibition triggered an E7-dependent degradation of pRb, while the levels of E2F remained unaffected. The presence of free intracellular E2F and the concomitant upregulation of CKIs during G1 arrest render the cells to undergo apoptosis (Finzer et al. 2001). In addition, HDACi induce an intrinsic type of apoptosis in HPV-positive cells by disrupting the mitochondrial transmembrane potential (Finzer et al. 2004). The HPV oncogenic proteins E6 and E7 are able to inactivate p53 and pRb proteins, which results in malignant transformation. Apicidin HDACi significantly reduces HPV16-E6 and -E7 protein levels in cervical cancer cells (Luczak and Jagodzinski 2008).
The combination of bortezomib, a proteosome inhibitor, and HDACi TSA or SAHA shows the synergistic killing of HPV-positive, but not HPV-negative, cervical cancer cell lines (Lin et al. 2009). Thus, a combination of HDAC and proteosome inhibitors, including SAHA and bortezomib, respectively, warrants exploration for the treatment of cervical cancer.
Latent infection of the EBV is strongly associated with the pathogenesis of several tumor types. The restricted expression of the latent EBV antigens is critical for EBV-associated tumors to escape from immune surveillance. TSA causes cell cycle arrest at low concentrations and induces apoptosis at higher (>300 nM) concentrations in the EBV-transformed lymphoblastoid cell lines (LCLs) (Seo et al. 2008). Following TSA treatment, a reduced expression of cyclin D2 and an induction of p21 may play an essential role for G1 arrest in LCLs. In addition, TSA induces EBV lytic replication cycle in LCL cells (Seo et al. 2008; Ye et al. 2007). Thus, TSA may exert an enhanced anti-tumor effect for EBV-associated tumors not only by inducing a cell cycle arrest and apoptosis but also by triggering an EBV lytic cycle. Finally, combining the use of a chemotherapeutic agent with oncolytic virotherapy is a useful way to increase the efficiency of the treatment of cancer. Replication-conditional (oncolytic) mutants of HSV are considering promising therapeutic alternatives for human malignancies. VPA pretreatment improves the propagation and therapeutic efficacy of oncolytic HSV in a human glioma xenograft model in vivo (Otsuki et al. 2008). TSA can be used as an enhancing agent for oncolytic virotherapy for oral squamous cell carcinoma with gamma(1)34.5 gene-deficient HSV-1 (Katsura et al. 2009). Altogether, HDACi can improve the efficacy of tumor virotherapies.
Hepatitis B oncoprotein, HBx, estrogen receptor (ER) alpha, and HDAC1 form a ternary complex (Han et al. 2006). Tricostatin A restores the transcriptional activity of ERalpha inhibited by HBx. Since estrogen, which exerts its biological function through ER, can inhibit HBV replication, ERalpha agonists may be developed for HCC therapy.
Although the killing of infected cells via apoptosis using HDACi has shown a “proof-of-concept” in oncology, additional experiments have to be performed to determine if this approach is translatable to patients without malignancy and/or if it will succeed in the absence of a second drug and/or an alternative treatment. Future studies will be needed to further test this hypothesis.
Conclusion
HDACs are critical factors involved in the control of viral replication. The molecular mechanisms underlying the role of HDACs in viral latency, viral reactivation, and carcinogenesis are progressively unveiled. The use of HDAC inhibitors as therapeutic tools to reactivate latent virus from cellular reservoirs may have important implications for reducing the size of latent viral reservoirs in patients infected with HIV and herpesviruses (HCMV, HSV, EBV). In addition, HDACi could be promising drugs which will kill virus-induced transformed cells especially in HPV and EBV infections. A better understanding of the role of HDACs in viral diseases will lead to the development of new drugs, especially HDAC inhibitors that will target cellular and viral proteins actively involved in numerous viral diseases.
References
Agbottah E, Deng L, Dannenberg LO, Pumfery A, Kashanchi F (2006) Effect of SWI/SNF chromatin remodelling complex on HIV-1 Tat activated transcription. Retrovirology 3:48
Ammanamanchi S, Freeman JW, Brattain MG (2003) Acetylated sp3 is a transcriptional activator. J Biol Chem 278:35775–35780
Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM (2009a) Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses 25:207–212
Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM (2009b) Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 23:1799–1806
Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, Eron J, Cohen M, Margolis DM (2010) Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS ONE 5:e9390
Arthur JL, Scarpini CG, Connor V, Lachmann RH, Tolkovsky AM, Efstathiou S (2001) Herpes simplex virus type 1 promoter activity during latency establishment, maintenance, and reactivation in primary dorsal root neurons in vitro. J Virol 75:3885–3895
Avram D, Fields A, Pretty On Top K, Nevrivy DJ, Ishmael JE, Leid M (2000) Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J Biol Chem 275:10315–10322
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124
Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE (2006) Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus–macaque model. J Infect Dis 193:963–970
Blazek D, Peterlin BM (2008) Tat-SIRT1 tango. Mol Cell 29:539–540
Boyes J, Byfield P, Nakatani Y, Ogryzko V (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396:594–598
Braun H, Koop R, Ertmer A, Nacht S, Suske G (2001) Transcription factor Sp3 is regulated by acetylation. Nucleic Acids Res 29:4994–5000
Bryant H, Farrell PJ (2002) Signal transduction and transcription factor modification during reactivation of Epstein–Barr virus from latency. J Virol 76:10290–10298
Burgers WA, Fuks F, Kouzarides T (2002) DNA methyltransferases get connected to chromatin. Trends Genet 18:275–277
Chan YJ, Chiou CJ, Huang Q, Hayward GS (1996) Synergistic interactions between overlapping binding sites for the serum response factor and ELK-1 proteins mediate both basal enhancement and phorbol ester responsiveness of primate cytomegalovirus major immediate-early promoters in monocyte and T-lymphocyte cell types. J Virol 70:8590–8605
Cherrier T, Suzanne S, Redel L, Calao M, Marban C, Samah B, Mukerjee R, Schwartz C, Gras G, Sawaya BE, Zeichner SL, Aunis D, Van Lint C, Rohr O (2009) p21(WAF1) gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 28:3380–3389
Cherrington JM, Mocarski ES (1989) Human cytomegalovirus ie1 transactivates the alpha promoter–enhancer via an 18-base-pair repeat element. J Virol 63:1435–1440
Coaquette A, Bourgeois A, Dirand C, Varin A, Chen W, Herbein G (2004) Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin Infect Dis 39:155–161
Colin L, Van Lint C (2009) Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6:111
Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM (2000) The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74:6790–6799
Countryman J, Miller G (1985) Activation of expression of latent Epstein–Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc Natl Acad Sci USA 82:4085–4089
Cress WD, Seto E (2000) Histone deacetylases, transcriptional control, and cancer. J Cell Physiol 184:1–16
Czermin B, Schotta G, Hulsmann BB, Brehm A, Becker PB, Reuter G, Imhof A (2001) Physical and functional association of SU(VAR)3-9 and HDAC1 in Drosophila. EMBO Rep 2:915–919
Deshmane SL, Fraser NW (1989) During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 63:943–947
Dyson PJ, Farrell PJ (1985) Chromatin structure of Epstein–Barr virus. J Gen Virol 66(Pt 9):1931–1940
Edelstein LC, Micheva-Viteva S, Phelan BD, Dougherty JP (2009) Short communication: activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res Hum Retroviruses 25:883–887
Everett RD, Parsy ML, Orr A (2009) Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J Virol 83:4963–4977
Finzer P, Kuntzen C, Soto U, zur Hausen H, Rosl F (2001) Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene 20:4768–4776
Finzer P, Ventz R, Kuntzen C, Seibert N, Soto U, Rosl F (2002) Growth arrest of HPV-positive cells after histone deacetylase inhibition is independent of E6/E7 oncogene expression. Virology 304:265–273
Finzer P, Krueger A, Stohr M, Brenner D, Soto U, Kuntzen C, Krammer PH, Rosl F (2004) HDAC inhibitors trigger apoptosis in HPV-positive cells by inducing the E2F-p73 pathway. Oncogene 23:4807–4817
Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF (1997) Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300
Fischle W, Wang Y, Allis CD (2003) Binary switches and modification cassettes in histone biology and beyond. Nature 425:475–479
Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J 20:2536–2544
Gandhi RT, Bosch RJ, Aga E, Albrecht M, Demeter LM, Dykes C, Bastow B, Para M, Lai J, Siliciano RF, Siliciano JD, Eron JJ, AIDS Clinical Trials Group A5173 Team (2010) No evidence for decay of the latent reservoir in HIV-1-infected patients receiving intensive enfuvirtide-containing antiretroviral therapy. J Infect Dis 201:293–296
Gregoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ (2007) Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol 27:1280–1295
Gruffat H, Manet E, Sergeant A (2002) MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep 3:141–146
Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90:595–606
Han J, Ding L, Yuan B, Yang X, Wang X, Li J, Lu Q, Huang C, Ye Q (2006) Hepatitis B virus X protein and the estrogen receptor variant lacking exon 5 inhibit estrogen receptor signaling in hepatoma cells. Nucleic Acids Res 34:3095–3106
Huang TH, Oka T, Asai T, Okada T, Merrills BW, Gertson PN, Whitson RH, Itakura K (1996) Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res 24:1695–1701
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417:455–458
Imai K, Okamoto T (2006) Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J Biol Chem 281:12495–12505
Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, Wolffe AP, Ge H (1997) Acetylation of general transcription factors by histone acetyltransferases. Curr Biol 7:689–692
Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP (2002) MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J 21:6236–6245
Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM (2006) Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med 203:7–13
Jamaluddin M, Choudhary S, Wang S, Casola A, Huda R, Garofalo RP, Ray S, Brasier AR (2005) Respiratory syncytial virus-inducible BCL-3 expression antagonizes the STAT/IRF and NF-kappaB signaling pathways by inducing histone deacetylase 1 recruitment to the interleukin-8 promoter. J Virol 79:15302–15313
Jenkins PJ, Binne UK, Farrell PJ (2000) Histone acetylation and reactivation of Epstein–Barr virus from latency. J Virol 74:710–720
Jiang G, Espeseth A, Hazuda DJ, Margolis DM (2007) c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol 81:10914–10923
Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877
Katsura T, Iwai S, Ota Y, Shimizu H, Ikuta K, Yura Y (2009) The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther 16:237–245
Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM (2009) A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J Virol 83:4749–4756
Khan KA, Coaquette A, Davrinche C, Herbein G (2009) Bcl-3 regulated transcription of major immediate early promoter of human cytomegalovirus (HCMV) in monocyte-derived macrophages. J Immunol 182:7784–7794
Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18:601–607
Kowalik TF, Wing B, Haskill JS, Azizkhan JC, Baldwin AS Jr, Huang ES (1993) Multiple mechanisms are implicated in the regulation of NF-kappa B activity during human cytomegalovirus infection. Proc Natl Acad Sci USA 90:1107–1111
Kwon HS, Brent MM, Getachew R, Jayakumar P, Schnolzer M, McBurney MW, Marmorstein R, Green WC, Ott M (2008) Human immunodeficency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 3:158–167
Lang D, Fickenscher H, Stamminger T (1992) Analysis of proteins binding to the proximal promoter region of the human cytomegalovirus IE-1/2 enhancer/promoter reveals both consensus and aberrant recognition sequences for transcription factors Sp1 and CREB. Nucleic Acids Res 20:3287–3295
Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, Coffin JM, Bosch RJ, Margolis DM (2005) Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549–555
Lin Z, Bazzaro M, Wang MC, Chan KC, Peng S, Roden RB (2009) Combination of proteasome and HDAC inhibitors for uterine cervical cancer treatment. Clin Cancer Res 15:570–577
Liu B, Stinski MF (1992) Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J Virol 66:4434–4444
Liu R, Baillie J, Sissons JG, Sinclair JH (1994) The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res 22:2453–2459
Longworth MS, Laimins LA (2004) The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 78:3533–3541
Longworth MS, Wilson R, Laimins LA (2005) HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J 24:1821–1830
Luczak MW, Jagodzinski PP (2008) Apicidin down-regulates human papillomavirus type 16 E6 and E7 transcripts and proteins in SiHa cervical cancer cells. Cancer Lett 272:53–60
Mahlknecht U, Will J, Varin A, Hoelzer D, Herbein G (2004) Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression. J Immunol 173:3979–3990
Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, Leid M, Aunis D, Schaeffer E, Rohr O (2005) COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res 33:2318–2331
Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O (2007) Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J 26:412–423
Marcello A (2006) Latency: the hidden HIV-1 challenge. Retrovirology 3:7
Martin M, Kettmann R, Dequiedt F (2007) Class IIa histone deacetylases: regulating the regulators. Oncogene 26:5450–5467
Martin M, Kettmann R, Dequiedt F (2009) Class IIa histone deacetylases: conducting development and differentiation. Int J Dev Biol 53:291–301
Marzio G, Wagener C, Gutierrez MI, Cartwright P, Helin K, Giacca M (2000) E2F family members are differentially regulated by reversible acetylation. J Biol Chem 275:10887–10892
Meier JL (2001) Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J Virol 75:1581–1593
Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18:5099–5107
Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA (2008) Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 48:1420–1429
Murphy JC, Fischle W, Verdin E, Sinclair JH (2002) Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 21:1112–1120
Ng HH, Bird A (2000) Histone deacetylases: silencers for hire. Trends Biochem Sci 25:121–126
Nguyen DX, Westbrook TF, McCance DJ (2002) Human papillomavirus type 16 E7 maintains elevated levels of the cdc25A tyrosine phosphatase during deregulation of cell cycle arrest. J Virol 76:619–632
Otsuki A, Patel A, Kasai K, Suzuki M, Kurozumi K, Chiocca EA, Saeki Y (2008) Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol Ther 16:1546–1555
Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, Hruby H, Jung M, Verdin E, Ott M (2005) SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol 3:e41
Park JH, Faller DV (2002) Epstein–Barr virus latent membrane protein-1 induction by histone deacetylase inhibitors mediates induction of intercellular adhesion molecule-1 expression and homotypic aggregation. Virology 303:345–363
Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ (2000) Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem 275:6764–6769
Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J (2008) Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol 82:12291–12303
Pierson T, McArthur J, Siliciano RF (2000) Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol 18:665–708
Pizzorno MC, O'Hare P, Sha L, LaFemina RL, Hayward GS (1988) Trans-activation and autoregulation of gene expression by the immediate-early region 2 gene products of human cytomegalovirus. J Virol 62:1167–1179
Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, Pili R (2006) Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1α. Cancer Res 66:8814–8821
Quivy V, De Walque S, Van Lint C (2007) Chromatin-associated regulation of HIV-1 transcription: implications for the development of therapeutic strategies. Subcell Biochem 41:371–396
Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, te Riele H, Dynlacht BD (2002) E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev 16:933–947
Redel L, Le Douce V, Cherrier T, Marban C, Janossy A, Aunis D, Van Lint C, Rohr O, Schwartz C (2010) HIV-1 regulation of latency in the monocyte–macrophage lineage and in CD4+ T lymphocytes. J Leuk Biol 87:575–588
Reuse S, Calao M, Kabeya K, Guiguen A, Gatot JS, Quivy V, Vanhulle C, Lamine A, Vaira D, Demonte D, Martinelli V, Veithen E, Cherrier T, Avettand V, Poutrel S, Piette J, de Launoit Y, Moutschen M, Burny A, Rouzioux C, De Wit S, Herbein G, Rohr O, Collette Y, Lambotte O, Clumeck N, Van Lint C (2009) Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS ONE 4:e6093
Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, Leid M, Schaeffer E (2003) Recruitment of Tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol 77:5415–5427
Sagot-Lerolle N, Lamine A, Chaix ML, Boufassa F, Aboulker JP, Costagliola D, Goujard C, Pallier C, Delfraissy JF, Lambotte O, ANRS EP39 Study (2008) Prolonged valproic acid treatment does not reduce the size of latent HIV reservoir. AIDS 22:1125–1129
Sambucetti LC, Cherrington JM, Wilkinson GW, Mocarski ES (1989) NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J 8:4251–4258
Semenza GL (2004) Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 19:176–182
Seo JS, Cho NY, Kim HR, Tsurumi T, Jang YS, Lee WK, Lee SK (2008) Cell cycle arrest and lytic induction of EBV-transformed B lymphoblastoid cells by a histone deacetylase inhibitor, trichostatin A. Oncol Rep 19:93–98
Shaw JE, Levinger LF, Carter CW Jr (1979) Nucleosomal structure of Epstein–Barr virus DNA in transformed cell lines. J Virol 29:657–665
Shelbourn SL, Kothari SK, Sissons JG, Sinclair JH (1989) Repression of human cytomegalovirus gene expression associated with a novel immediate early regulatory region binding factor. Nucleic Acids Res 17:9165–9171
Sorin M, Cano J, Das S, Mathew S, Wu X, Davies KP, Shi X, Cheng SW, Ott D, Kalpana GV (2009) Recruitment of a SAP18–HDAC1 complex into HIV-1 virions and its requirement for viral replication. PLoS Pathog 5:e1000463
Stenberg RM, Depto AS, Fortney J, Nelson JA (1989) Regulated expression of early and late RNAs and proteins from the human cytomegalovirus immediate-early gene region. J Virol 63:2699–2708
Tsai SC, Valkov N, Yang WM, Gump J, Sullivan D, Seto E (2000) Histone deacetylase interacts directly with DNA topoisomerase II. Nat Genet 26:349–353
Van Lint C (2000) Role of chromatin in HIV-1 transcriptional regulation. Adv Pharmacol 48:121–160
Van Lint C, Emiliani S, Ott M, Verdin E (1996) Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15:1112–1120
Verdin E, Paras P Jr, Van Lint C (1993) Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J 12:3249–3259
Volke M, Gale DP, Maegdefrau U, Schley G, Klanke B, Bosserhoff AK, Maxwell PH, Eckardt KU, Warnecke C (2009) Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS ONE 4:e7875
Wade EJ, Klucher KM, Spector DH (1992) An AP-1 binding site is the predominant cis-acting regulatory element in the 1.2-kilobase early RNA promoter of human cytomegalovirus. J Virol 66:2407–2417
Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC (2006) NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149
Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W (2003) Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev 17:896–911
Ye J, Gradoville L, Daigle D, Miller G (2007) De novo protein synthesis is required for lytic cycle reactivation of Epstein–Barr virus, but not Kaposi's sarcoma-associated herpes virus, in response to histone deacetylase inhibitors and protein kinase C agonists. J Virol 81:9279–9291
Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM (2004a) Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS 18:1101–1108
Ylisastigui L, Coull JJ, Rucker VC, Melander C, Bosch RJ, Brodie SJ, Corey L, Sodora DL, Dervan PB, Margolis DM (2004b) Polyamides reveal a role for repression in latency within resting T cells of HIV-infected donors. J Infect Dis 190:1429–1437
Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK, Lee MO (2008) Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene 27:3405–3413
Zeng W et al. (2010) HP1: heterochromatin binding proteins working the genome. Epigenetics 5:287–292
Zhang X, Wharton W, Yuan Z, Tsai SC, Olashaw N, Seto E (2004) Activation of the growth-differentiation factor 11 gene by the histone deacetylase (HDAC) inhibitor trichostatin A and repression by HDAC3. Mol Cell Biol 24:5106–5118
Zhang HS, Zhou Y, Wu MR, Zhou HS, Xu F (2009) Resveratrol inhibited Tat-induced HIV-1 LTR transactivation via NAD(+)-dependent SIRT1 activity. Life Sci 85:484–489
Zhou J, Snyder AR, Lieberman PM (2009) Epstein–Barr virus episome stability is coupled to a delay in replication timing. J Virol 83:2154–2162
Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN (1996) Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol 16:4024–4034
Acknowledgements
The work of the authors is supported by institutional funds from the Franche-Comte University (to GH) and from the Association for Macrophage and Infection Research (AMIR) (to GH).
Conflict of interest
The authors have no conflict of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Herbein, G., Wendling, D. Histone deacetylases in viral infections. Clin Epigenet 1, 13–24 (2010). https://doi.org/10.1007/s13148-010-0003-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-010-0003-5