
Abstract
Zones of secondary contact provide a good opportunity to investigate the origin and dynamics of reproductive isolation between related populations. We analyzed genetic and phenotypic patterns and gene flow between two subspecies of the Eurasian magpie Pica pica s.l. which recently came into contact after presumably long periods of isolation. We describe the distribution of subspecies in a young contact zone at Argun’ river basin in southern Siberia where populations occur in parapatry and an older hybrid population in eastern Mongolia. Based on genome-wide SNP data, we analyzed patterns and strength of gene flow between the subspecies. Our results indicate occasional hybridization with backcrossing and asymmetric introgression along a wide range in Transbaikalia and locally in eastern Mongolia. Males of P. p. jankowskii apparently exhibit higher dispersal ability towards the west compared to P. p. leucoptera (towards the east). The former occasionally migrates to eastern Mongolia and Transbaikalia where introgression of nuclear, but not mitochondrial DNA was evident. Bioacoustic investigations showed differences between the subspecies in speed and structure of vocalization. We discovered intermediate calls of hybrid magpies and bilingual birds alternating calls that are typical for the two taxa. Furthermore, we found dramatically decreased reproductive success in hybridogeneous populations. By complementing our results with established phylogeographic patterns of P. pica s.l. based on a mitochondrial marker sequence, and considering indications of sterility of hybrids in the contact zone, we propose to elevate the two corresponding subspecies to species level: P. pica for the western form and P. serica for the eastern form.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Secondary contact and hybridization are important phenomena of evolutionary biology. Their study helps to understand many aspects of microevolution, such as the origins of reproductive isolation at early stages of speciation (Abbott et al., 2013, 2016; Harrison & Larson, 2014; Mallet, 2005; Taylor & Larson, 2019). When conspecific populations or closely related species have chance to meet after long periods of spatial isolation in glacial refugia (Hewitt, 2011)—or more recently due to often anthropogenic environmental changes (Seehausen et al., 2008; Crispo et al., 2011; Millette et al., 2020)—the outcome of such secondary contact is often unpredictable. In some cases, populations become reproductively isolated due to prezygotic and/or postzygotic isolation barriers from Bateson-Dobzhansky-Muller incompatibilities (Dobzhansky, 1940; Muller, 1942; Orr, 1996) or other types of incompatibilities (Abbott et al., 2013) that arose in isolation. In other cases, when backcrosses are successful, more or less pronounced introgression takes place (Seehausen, 2004). This may lead to admixture and fusion of populations and/or species in statu nascendi, which is documented by an increasing number of published examples of hybrid speciation (Lavrenchenko, 2014; Ottenburghs, 2018). While homoploid hybrid speciation is rare for higher animals (Mavarez & Linares, 2008; Schumer et al., 2014; Nieto et al., 2017), introgressive hybridization is a common phenomenon. It often occurs at the edges of distribution ranges of closely related species or populations, where population density is low and mate competition with parental form is weak (Abbott et al., 2013; Pfennig et al., 2016).
Isolating factors may be reinforced when populations experience continuous maladaptive hybridization in a secondary contact zone (Dobzhansky, 1940). For example, natural selection may favor traits that enhance assortative mating (Howard, 1993). Nonetheless, in most cases, the underlying evolutionary processes and genetic patterns remain poorly characterized and understood (Coyne & Orr, 2004). Overall, the shape and dynamics of a hybrid zone is determined by many factors such as dispersal rates, genetic incompatibilities, morphology, behavior, and ecology (Barton & Hewitt, 1985). Introgression can be adaptive (Taylor & Larson, 2019), but involve only a limited part of a genome and thus may be barely distinguishable from selection effects of local adaptation (Currat et al., 2008; Excoffier et al., 2009; Quilodrán et al., 2020). Hybridization often occurs during biological invasions or at the species borders and, under certain conditions, facilitates range expansions into new environments (Pfennig et al., 2016). The problem to distinguish between primary intergradation, i.e., clinal variation due to spatially varying selection along an environmental gradient and secondary contacts, can be solved in a hybrid zone study (Harrison & Larson, 2016). In general, the initial stages of such processes of secondary contact may provide important insights.
Whole-genome sequencing techniques, which now make it possible to study single nucleotide polymorphisms (SNPs) on a genome-wide scale, revolutionized phylogeography. Genomic data enable quantitatively characterizing hybridization and introgression (Edwards et al., 2015). They also shed light on the genomic architecture of traits under selection in order to find regions on increased differentiation, so-called “islands of speciation” (Ellegren et al., 2012; Nosil et al., 2009; Poelstra et al., 2014; Turner et al., 2005), and thus the genetic background of early divergence and on the origin of reproductive isolation. These new approaches have improved our understanding of evolution (Edwards et al., 2005; Ottenburghs et al., 2017). Nuclear SNPs became highly attractive as genetic markers. In particular, they were successfully used in studies of hybridization in numerous non-model animal groups, e.g., in woodpeckers (Gurgul et al., 2019), mountain and brown hares (Levanen et al., 2018), wild and domestic cats (Oliveira et al., 2015), Peromyscus mice (Garcia-Elfring et al., 2017), chiffchaffs (Shipilina et al., 2017), warblers (Brelsford et al., 2017), sparrows (Elgvin et al., 2017), and American crows (Slager et al., 2020).
Among the technical approaches to analyze SNPs and other genome-wide markers, restriction site-associated DNA sequencing (RAD-Seq) has proven to be cost-efficient and powerful for population genetic inference. It is based on a reduced representation of the genome, which is an advantage—particularly when working with non-model organisms (where reference genomes are often not available) or organisms with large genome sizes (Baird et al., 2008; Harvey et al., 2016). Accordingly, RAD-Seq has been successfully applied in hybrid zone studies (Dufresnes & Martínez-Solano, 2020; Razkin et al., 2016). In particular, double-digest restriction site-associated DNA (ddRAD) sequencing requires much less starting DNA, allows precise selection of fragments by size, and produces libraries at reduced costs (Peterson et al., 2012). It has therefore been successfully employed in many fields of population and evolutionary genetics. These include, for example, analyses of introgression across a contact zone in voles (Lin et al., 2018) and brown trout (Jacobs et al., 2018), hybrid zone of warblers (Toews et al., 2018), sparrows (Maxwell et al., 2021), and lizards (Grummer et al., 2021).
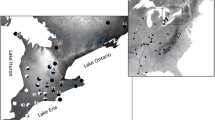
The magpie Pica pica s.l. (Linnaeus, 1758) serves as a good case of contact zones between subspecies or taxa with disputed rank. Among common bird species, Pica pica s.l. has one of the widest Holarctic distributions (Fig. 1) and exhibits clinal variation in size and coloration (Cramp & Perrins, 1994). Such phenotypic variability and the broad distribution throughout the Northern hemisphere may reflect its complicated evolutionary history fueled by known shifts of environmental conditions, especially in the Holocene. This is also reflected in its subdivision into numerous subspecies. Depending on the authors, the numbers range from a single species with 13 subspecies (Madge & Burn, 1999) to three species in the Palearctic and two species in the Nearctic (del Hoyo & Collar, 2016). In our previous paper (Kryukov et al., 2017), we accepted the following taxonomic scheme for Pica pica s.l. which formed the basis of the present paper: P. p. melanotos A.E. Brehm, 1857; P. p. mauritanica Malherbe, 1845; P. p. pica (Linnaeus, 1758); P. p. fennorum Lönnberg, 1927; P. p. hemileucoptera Stegmann, 1928; P. p. bactriana Bonaparte, 1850; P. p. leucoptera Gould, 1862; P. p. serica Gould, 1845; P. p. jankowskii Stegmann, 1928; P. p. anderssoni Lönnberg, 1923; P. p. bottanensis Delessert, 1840; P. p. camtschatica Stejneger, 1884; and P. p. asirensis Bates, 1936 in the Palearctic (the latter is a bit outside), in addition to P. p. hudsonia (Sabine, 1823) and P. p. nuttalli (Audubon, 1837) in the Nearctic, whereby the status of the latter two remains ambiguous.

Map of Pica pica s.l. subspecies. The study area in the contact zone is indicated by a rectangle. Question marks indicate regions of unclear subspecies distribution. The white star indicates the terra typica for Pica pica anderssoni in eastern China
In the past, intraspecies genetic variation of Pica pica s.l. has been studied with several molecular approaches. Microsatellite analysis of 15 European magpie populations revealed large-scale population structure but extensive gene flow between nearby populations (Martinez et al., 1999). In early studies, high levels of differentiation between P. p. camtschatica and the Nearctic P. p. hudsonia were found by restriction fragment analysis of mtDNA (Zink et al., 1995). Later, analyses of the mitochondrial (mt) genes for 16S rRNA, tRNA-Leu, and NADH dehydrogenase subunit 1 (ND1) revealed a close relationship between P. p. camtschatica and the European nominate form, both being distant from the clade of P. p. hudsonia and P. p. nuttalli. All the above form the sister group of East Asian P. p. serica (Lee et al., 2003). More recently, in the study of genetic structure and population demography of P. p. serica in eastern China, using three mt genes and two nuclear introns, Zhang et al. (2012) found no genetic structure. Newly developed nuclear microsatellite loci (Martín-Gálvez et al., 2009) helped to assess genetic diversity and the continental origin of two Japanese island populations (Mori et al., 2014).
In our previous papers based on dense and broad geographic sampling which included most subspecies inhabiting the Palearctic, we explored the phylogeographic and intraspecific variation in P. pica s.l. employing the mt cytochrome b gene and the mt control region (CR) (Haring et al., 2007; Kryukov et al., 2004, 2017). We identified a group of haplotypes from the western subspecies (Clade West) highly diverged from the East Asian group (Clade East) with a mean p-distance of 0.049. Despite the limited sample size of P. p. hudsonia and P. p. mauritanica, these studies showed that P. p. camtschatica appeared to be the sister group of Clade West, and P. p. hudsonia was the sister lineage of these two clades. Finally, P. p. mauritanica was found to be very distant from the abovementioned groups splitting from the basal node in the tree. Moreover, Kryukov et al. (2017) reported a good correlation between the general phylogeographic pattern and bioacoustic characteristics, namely the rate of chatter calls, which clearly differed between the main mt lineages. More recently, Song et al. (2018) analyzed two mt genes and two nuclear introns and essentially confirmed the previously established Holarctic phylogeographic picture, where the composition and distinctness of Clades West and East are undisputed. Based on Song et al.’s results, several subspecies were elevated to full species in the current editions of Birds of the World (Madge et al., 2020) and IOC World Bird List (Gill et al., 2021). This includes P. mauritanica, P. asirensis, P. bottanensis, and P. serica. Nonetheless, given the unclear reasoning for this reclassification and for the sake of consistency with other subspecies, we initially refer to the Eurasian forms as subspecies when presenting our results in this manuscript.
Throughout the wide species’ range (Fig. 1), many subspecies intergrade, but some forms such as camtschatica, asirensis, and mauritanica are allopatric and separated by wide gaps. In contrast, other groups of subspecies are presumably parapatric or the extent of the geographic gaps has not been investigated or is questionable. Such a situation was found in southern Siberia and adjacent Mongolia, where, ~ 90 years ago, the distribution ranges of P. p. leucoptera and P. p. jankowskii were separated by a gap that stretched over 600 km from west to east (Rustamov, 1954; Stegmann, 1932; Stepanyan, 2003). In eastern Mongolia (Tugarinov, 1932) and northern China (Cheng, 1987), however, the two subspecies might have already been in contact in the early twentieth century. In the last decades, with increasing numbers of settlements, villages, and roads, magpies have been expanding their populations (Goroshko et al., 2018)—leucoptera from the west and jankowskii from the east. Nonetheless, the degree of contact between leucoptera and jankowskii as well as the magnitude of the potential hybrid zone, its dynamics, and structure remain unclear. Breeding success and hybridization (or its frequency) among different populations was unexplored. Since these two subspecies represent terminal geographic positions of the major mt lineages (Clade West, Clade East; Haring et al., 2007; Kryukov et al., 2004, 2017), analyzing this zone is important to assess evolutionary consequences of their secondary contact.
The focus of this study is the genetic and phenotypic variation and its evolutionary significance in the contact zone of P. p. leucoptera and P. p. jankowskii in southern Siberia and adjacent Mongolia and China. To determine the parameters of their secondary contact, we evaluated the extent of their ranges through field observations, counted hybrids and mixed pairs in several populations, and estimated their breeding success. To genetically detect hybrids and estimate the level of introgression, we carried out comprehensive population genetic analyses based on genome-wide nuclear SNPs. The results of these analyses from the contact zone and adjacent regions were embedded in an extended mt-based phylogeography of Pica pica s.l. along its total range in the Palearctic. Using the mtDNA data set, we also traced the population history by Bayesian modeling. Because acoustic signals may be an important part of species-specific mating behavior, we further analyzed bioacoustic characteristics in admixed and pure populations. Based on these findings which contribute to our understanding of the complex evolutionary history of Pica pica, we discuss the evolutionary significance of limited hybridization, as well as a possible fate of the populations in the contact zone. We then suggest changes to the current taxonomy. Specifically, we discuss whether certain subspecies or groups of subspecies deserve species status, considering their genetic and phenotypic divergence and level of reproductive isolation.
Material and methods
Field work and sampling
We conducted several field expeditions in the years 2014 to 2021 in the presumed contact zone between P. p. leucoptera and P. p. jankowskii (hereafter, except for the “Taxonomic considerations,” we provide only subspecific epithets) and adjacent territories in Transbaikalia (southern Siberia), eastern Mongolia, and northern China. We collected tissue samples for genetic analyses, made field observations, and took more than 1250 photos of magpies, examined 89 nests, and recorded the voices of 68 birds.
The subspecies were identified in the field mainly made on coloration and vocalization. Adult leucoptera have white tops of the first primaries, greenish secondaries, and a wide white rump, whereas jankowskii has black tops of the first primaries, blueish secondaries, a narrow dark gray rump, and smaller body (Madge & Burn, 1999). We used binoculars with eightfold magnification, telescope with 25–75-fold magnification, and cameras with 150–500-mm lenses. Alarm calls of leucoptera sound like fast chattering calls, whereas jankowskii performs slow chattering with separated elements. Intermediates by appearance and/or calls were considered as putative hybrids. Populations were defined as “hybridogeneous” if they contained a pronounced number of hybrids which correspond to “rare hybridization” in Short’s (1969) classification of hybridization and as “admixed” if both parental phenotypes co-occurred with single hybrids.
Nest searches were carried out to study the impact of a pair content and hybridization on breeding success. During the breeding periods of 2015–2021, we observed 119 nesting pairs, but were only able to check the nest content of 89 from these pairs: 52 in Transbaikalia, 33 in eastern Mongolia, and 4 in northern China. We identified subspecies of 168 nesting magpies by field observations, taking photos, and recording call. For nests that contained clutches, we estimated brooding success. First, the eggs were candled to check the development and then put into a cup of water to examine viability. When an egg showed no signs of embryo development, it was checked again after 7–14 days. If a translucent egg floated up, this indicated that it was unfertilized or the embryo had died at a very early stage of development and thus the egg had dried out. Such eggs were opened to confirm this conclusion and none of them contained any remains of even small embryos. We considered the progeny of a given pair as viable, if their nest contained any chicks or fertilized eggs at a late stage, and treated the pair as infertile, if all eggs in a clutch were non-viable. In addition, for subspecies identification based on morphology and vocalizations, 164 non-breeding 1-year-old birds were observed in Transbaikalia, 43 in eastern Mongolia, and 276 in northern China.
Blood and feather samples from living fledglings were collected, fixed in 96% ethanol, and stored until laboratory procedures. For analysis of mtDNA, we used 58 freshly collected samples from Transbaikalia, 26 from Mongolia, 16 from northern China, and 16 from the Amur region. We also obtained 15 tissue samples from Korea and 14 from Spain. We used one sample per clutch because all chicks in the same nest usually share the same mitochondrial genome. In total, we isolated and analyzed mtDNA from 256 tissue samples (Table S1, Fig. 2) by including 111 sequences of CR generated in a previous paper (Kryukov et al., 2017). For the ddRAD analysis, we selected samples of 89 individuals (feather or muscle samples preserved in ethanol or dried blood samples) collected from the contact zone between leucoptera and jankowskii in South Siberia and Mongolia as well as from adjacent regions presumably without introgression (Table S1, Fig. 3).

Map of sampling localities for mitochondrial DNA analysis in the zone of contact between Pica pica leucoptera and Pica pica jankowskii. Distribution of haplotypes is indicated by colours: Pica pica leucoptera is shown with blue dots, and Pica pica jankowskii with red dots. Numbers of dots correspond to Table S1. Some dots correspond to several specimens due to closeness of their coordinates

Map of sampling localities for nuclear SNP analysis. Distribution of genotypes of nuclear SNPs revealed by ddRAD analysis are shown with blue circles for Pica pica leucoptera and red circles for Pica pica jankowskii. Bi-colored circles mean mixed (hybrid) genotype: portion of blue/red corresponds to probability of it belonging to one of the groups, as revealed in the Structure analysis. Numbers of circles correspond to Table S1. Some circles correspond to several specimens due to closeness of their coordinates. Conditional populations are indicated by Roman numerals I–VI. I—central Mongolia; II—eastern Mongolia; III—central Transbaikalia; IV—Argun’ river basin; V—northern China; VI—Amur river basin
Analysis of morphological traits
Morphometric differences between subspecies were analyzed in museum samples from the collections at the Zoological Museum of Moscow State University and the Zoological Institute of the Russian Academy of Sciences, St. Petersburg. Seventy-one skins of leucoptera were measured in the current study and compared to 130 skins of the serica group, published in Red’kin et al. (2021). For each sample, eight measurements of wing, tail, tarsus, and bull were taken (see Table S2). Measurements were carried out using a metal ruler and Vernier caliper. Statistica 7.0 was used for t-test and Microsoft Office Excel to process and analyze the data.
Analysis of mtDNA
Laboratory procedures
The primer set for amplification of the mt CR and all laboratory procedures were previously described in Kryukov et al. (2017). In brief, the complete mt CR was amplified with primers binding in the adjacent genes (tRNA-Glu, tRNA-Phe; amplicon length 1455 bp). In some cases, when the complete CR fragment could not be obtained, it was amplified in two overlapping sections (primers published first in Kryukov et al., 2004). The CR was sequenced in the Federal Scientific Center of the East Asia Terrestrial Biodiversity, Far Eastern Branch of the Russian Academy of Sciences (FSC EATB), on an ABI PRISM 3130 automatic laser sequencer (Applied Biosystems, USA/Hitachi, Japan). Sequences determined in the present study are deposited in GenBank under the accession numbers OK317735–OK317879 and OL539421–OL539424 (Table S1).
Analysis of sequences
The sequences were assembled using the Staden v.1.53 software package (Bonfield et al., 1995) and aligned using ClustalW implemented in MEGA X, v.10.1.7 (Kumar et al., 2018). All alignments were edited manually in BioEdit v.6.0.7 (Hall, 1999). Due to various lengths of readable sequences, the alignment was trimmed at both ends yielding a total sequence length of 1326 bp. Polymorphism parameters such as number of polymorphic sites, haplotype, and nucleotide diversity were evaluated in DnaSP v.5.10.01 (Librado & Rozas, 2009). MEGA X was used to estimate mean p-distances between haplogroups and subspecies and to calculate a phylogenetic maximum likelihood (ML) tree, based on the HKY + G substitution model, which was determined as the optimal model under the BI criterion. Bootstrap support was assessed with 1000 replicates. The time-calibrated Bayesian tree was constructed with Beast v.2.6.2 (Bouckaert et al., 2014), using the HKY substitution model and constant population model as a prior, with Markov chain Monte Carlo (MCMC) length as 108, stored every 10,000 generations, and discard the first 107 trees of each run as burn-in. The molecular clock model was set as strict and the substitution rate was estimated as 0.025 substitutions/site/million years, as it was suggested and applied previously for bird mitochondrial CR (Fok et al., 2002; Freeland & Boag, 1999; Kryukov et al., 2004; Omland et al., 2006). To check if each parameter reached the effective sample size of 200, we used Tracer v.1.7.1 (Rambaut et al., 2018). The maximum clade credibility tree was obtained with TreeAnnotator v.2.6.2 (Drummond et al., 2012) and a target tree was visualized with FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree). A median-joining (MJ) network (Bandelt et al., 1999) was constructed with Network v.4.612 (available at www.fluxus-engineering.com). To reduce the number of cycles in the network, we down-weighted several rapidly mutating positions as recommended in the software manual. We treated haplogroups as distinct if they differed by 7 substitutions or more.
Tests of neutrality, population demography, and Bayesian skyline plots
The following neutrality tests were conducted using DnaSP: Tajima’s D (Tajima, 1989), Fu’s Fs (Fu, 1997), and Ramos-Onsins and Rozas’ R2 (Ramos-Onsins & Rozas, 2002). The program used 1000 coalescence simulations of random mutations to determine significance of these tests. Arlequin v.3.5.2.2 (Excoffier & Lischer, 2010) was used to re-estimate neutrality tests D and Fs and to conduct mismatch analysis (Rogers & Harpending, 1992) with 1000 bootstrap replicates. Raggedness (r-) index (Harpending, 1994) was calculated to test if the observed process fits to the expected mismatch distribution (MMD) under the population expansion model. Furthermore, the sum of squared deviation (SSD) method was employed with models of sudden and spatial expansions as implemented in Arlequin. Significance of r was estimated by means of coalescent simulations as described above, and in both r and SSD methods, insignificant deviation from zero means not rejecting the hypothesis of population expansion. The τ parameter of MMD (expansion parameter) was used to evaluate the time after expansion measured in mutations. We used the formula T = τ/2µk, where T is the time since expansion in years, µ is the substitution rate per site per year, and k is the sequence length (Rogers & Harpending, 1992). Mismatch distribution graphs were constructed to analyze past demographic history under the model of population growth-decline in DnaSP.
To infer past population dynamics through time and estimate the time of the most recent common ancestor (tMRCA), we constructed Bayesian skyline plots (BSPs) (Drummond et al., 2005) based on mt CR data employing the BEAST v.2.6.3 package which includes BEAUti2 (Bouckaert et al., 2014). The best-fitting substitution model, selected in MEGA X based on BIC, was the HKY model for all populations and haplogroups. The clock model was set as strict and the substitution rate was estimated as 0.025 substitutions/site/million years, as above. We used BEAST to run MCMC with a chain length of 10,000,000 generations and logs sampled every 1000 generations. The first 1,000,000 generations were discarded as a burn-in period. The dimension of population and group sizes was set to 4 as recommended in the online tutorial for BEAST v.2.5 by Muller and du Plessis (2020). For reconstructing the Bayesian skyline plots, we used Tracer v.1.7.1 (Rambaut et al., 2018). After checking the values of effective sample sizes (ESS), where all statistic parameters had ESS > 200, we obtained plots of population dynamics (i.e., effective female population size per time) which were visualized in Excel.
ddRAD analysis
Genomic DNA was extracted from blood, feather, or muscle samples with DNeasy Blood & Tissue Kit (Qiagen). After preliminary quality control on a fluorometer (Qubit 2.0 Fluorometer, Invitrogen, Carlsbad, CA), we sent 1–10 µg per DNA samples at concentrations ranging from 30 to 330 ng/µl to IGATech (Italy). ddRAD libraries were produced by IGATech following Peterson’s double-digest restriction site-associated DNA preparation protocol (Peterson et al., 2012) with modifications. Enzyme combinations were preselected by in silico analyses of restriction sites based on three closely related species: Corvus cornix (GCA_000738735.2_ASM73873v2_genomic.fna and GCA_002023255.2_ASM202325v2_genomic.fna), C. brachyrhynchos (GCA_000691975.1_ASM69197v1_genomic.fna), and C. hawaiiensis (GCA_003402825.1_Coha_1.0_genomic.fna). Concentrations of genomic DNA were fluorometrically quantified and then normalized. After that, 300 ng was double digested with 2.4 U of both PstI and EcoRI endonucleases (New England BioLabs) in 30 µl reaction supplemented with CutSmart Buffer and incubated first at 37 °C for 90 min and then at 65 °C for 20 min. Fragmented DNA was purified with 1.5 volumes of AMPureXP beads (Agencourt) and subsequently ligated with 200 U of T4 DNA ligase (New England BioLabs) to 2.5 pmol of overhang barcoded adapter for rare cut sites and to 5 pmol of overhang barcoded adapter for frequent cut sites in 50 µl reaction. These samples were incubated at 23 °C for 60 min and at 20 °C for 60 min followed by 20 min at 65 °C and subsequently pooled in multiplexed batches and bead purified as described before. For each pool, targeted fragment length distribution was evaluated on a BluePippin instrument (Sage Science Inc.) and only fragments within the range of 530–650 bp were used for sequencing. Gel-eluted DNA fragments were amplified with indexed primers to increase the DNA volume for sequencing using Phusion High-Fidelity PCR Master Mix (New England BioLabs) in a final volume of 50 µl and subjected to the following thermal protocol: [95 °C, 3 min] − [95 °C, 30 s −60 °C, 30 s −72 °C, 45 s] × 10 cycles − [72 °C, 2 min]. Products were purified with 1 volume of AMPureXP beads. The resulting libraries were checked with both Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and Bioanalyzer DNA assay (Agilent technologies, Santa Clara, CA) prior to paired-end sequencing with 125 cycles on a HiSeq 2500 sequencer following the manufacturer’s instructions (Illumina, San Diego, CA). This yielded a total of 241,818,224 raw sequences. Pools were demultiplexed to individual FASTQ files by removing barcode sequences and by trimming to a final length of 110 bp using Stacks v.2.0 (Catchen et al., 2013). Paired-end reads (110 bp) were then clustered to define reference locus sequences for each fragment flanked by two restriction sites using the de novo pipeline from the Stacks package with default parameters. Each locus thus represents a 220-bp-long sequence, which was obtained by clustering reads in pairs. The size selection prevents overlap between each pair and eventually reduces the number of sampled bases per locus. Using the “populations” tool of Stacks, we retained loci that were successfully genotyped in at least 75% of the samples. For 28,292 genotyped loci, we assembled paired-end contigs and merged them with the single-end locus, aligned reads to the locus, and called SNPs. Overall, 135,872 polymorphic sites were obtained and stored as VCF and other file types for downstream population genetics analyses. These files can be downloaded from https://github.com/nhmvienna/PicaPica_Introgression.
We first carried out Bayesian model-based cluster analyses with the software Structure v.2.3.4 (Pritchard et al., 2000) using the following parameters: burn-in period = 10,000 and MCMC = 20,000, allele frequencies = independent, admixture model, iterations = 5, K = 1–5 (two of the initial 91 samples were excluded due to unstable results across several runs of Structure). To evaluate the parameter K with highest likelihood, which indicates the prospective number of independent genetic groups in our data set, we run Harvester Web v.0.6.94 (http://taylor0.biology.ucla.edu/structureHarvester/; Earl & von Holdt, 2012) on the results of the Structure analyses.
We then used the PLINK software (https://www.cog-genomics.org/plink/1.9/) to prune linked loci from the dataset because these could potentially bias results of subsequent clustering analyses. From 135,872 SNPs, 102,876 correlated pairs of loci were identified and deleted, yielding 32,996 unlinked loci. To visualize and estimate patterns of genetic differentiation along with sample clustering as well as to identify potential outlier samples, we applied a principal component analysis (PCA) on the unlinked SNP dataset (Patterson et al., 2006) and visualized scores of PC axes 1–4 as scatterplots with PLINK in LibreOffice Calc. This analysis permits identifying sample clustering, reveals outliers, and shows axes of maximal variation.
Vocalization
Sounds were recorded during field work in 2014–2018. Portable recorders (ZOOM H5 and Sound Devices 702) were used with microphones of the types SGH-6 (ZOOM) and Telinga Stereo MK2. The sampling frequency was set to 44.1 kHz with 16-bit. We recorded the chatter call of 9 birds in central Transbaikalia, 11 in the Argun’ river basin, 16 in eastern Mongolia, 28 in Vladivostok (Russian Far East), and 4 in the Altai Mountains. Spectrograms were obtained and developed with Raven v.1.3 (Charif et al., 2008) and Syrinx v.2.6 h (Burt, 2006).
We focused on the speed of a chatter call which is used for communication and as a general alarm signal (Birkhead, 1991) as our main parameter to assess individual variation and population differentiation. The number of elements in a series varies from 2–3 to dozens and the performance speed usually slows down towards the end of a series. Accordingly, we estimated the duration of the first five elements in a series, i.e., intervals between the beginning of the first and the sixth elements in chattering were measured, including intervals between elements. Furthermore, the total length of a series was measured. The number of elements per second was calculated for both these methods. Samples greatly varied in their size, and the assumption of homogeneity of variances was violated. For the cases of unequal variances, the Games-Howell test with ANOVA was used (Games & Howell, 1976; Sokal & Rohlf, 2012). For visualization of results, the graphical package “ggstatplot” (Patil, 2021) was used.
Results
Field observations—nesting pairs and reproductive success
In central Transbaikalia and the Argun’ river basin, Russia, magpies occupy forest-steppe and steppe habitats with more or less scarce wooded vegetation. In general, we found no difference in habitat preferences between the subspecies. They nested usually at 3–7 m above ground on medium-sized trees or bushes. In the steppes of eastern Mongolia and adjacent northern China, the conditions are more severe, and magpies were observed only in valleys of lakes or rivers such as Kerulen, Uldz Gol, and Orshun Gol. Their nests were found on low willow bushes and elm trees commonly at a height of 2–4 m near rivers and on planted poplar trees in sparsely distributed human settlements. Magpie population density was about 3.5 pairs per km2 on average at the Onon river valley in central Transbaikalia, but much lower in Mongolia, Argun’, and adjacent China. In eastern Mongolia, magpies mainly accompanied shepherd’s camps and yurts located along the Kerulen River. The number of magpies declined sharply towards and became complete absent in a 15-km-broad belt near the state border with China, where people and livestock were also absent.
We found several magpies with intermediate appearance in plumage coloration and/or mixed alarm call in eastern Mongolia, one at the west side of the Argun’ river basin, and none in China. In eastern Mongolia, we recorded 33 breeding pairs, at least 11 of which were mixed: one partner was presumably a hybrid or jankowskii by appearance and/or alarm calls and the other one was leucoptera. The remaining pairs were either both leucoptera or their phenotypes were uncertain. Three of the mixed pairs were recorded at the Uldza River and eight within app. 30 km along the eastern part of narrow Kerulen River valley close to the Mongolian-Chinese border. Thus, hybrids seem to be concentrated in this region, while the remaining part of Kerulen population was inhabited by pure leucoptera and northern China by pure jankowskii. In addition, we observed non-breeding birds: 20 leucoptera, 8 presumed hybrids, and one jankowskii in eastern Mongolia; 158 leucoptera and 6 hybrids or jankowskii in Transbaikalia (the latter solely at the Argun’ river basin); and 276 jankowskii in northern China.
In the Argun’ river basin, we determined 59 nesting birds from 34 pairs at the subspecies level, 11 among them were jankowskii, two mixed pairs, and the remaining 21 were most probably leucoptera. Those nesting pairs of leucoptera were encountered 70–100 km north of the breeding range of jankowskii in the Argun’ floodplain. According to information from locals and based on our own data, this form appeared here only 20–30 years ago. One of the two presumably mixed pairs in Argun’ river basin was found on June 25, 2017, at the nest and was comprised of a putative hybrid female (identified by coloration and intermediate alarm call) and a male that was most probably leucoptera. This female had 8 unfertilized dried eggs in the nest and we were therefore unable to obtain any usable genetic samples. The location was near point #54 in Figs. 2 and 3. The next nearest nest of pure leucoptera was 4 km away, and the closest nest of pure jankowskii was found 78 km to the south-east, at point #104 near Priargunsk settlement (Goroshko et al., 2018). In 2018, we found pairs #50 and #108, both with five fledglings, nesting app. 5 km away from each other. Their chicks were later genotyped based on mtDNA markers as leucoptera and jankowskii, respectively, but the male of pair #108 had typical leucoptera alarm calls and plumage, so that we considered this pair as intersubspecific. The other nearest nesting points of pure leucoptera (#49) and of jankowskii (#104) were found in 25 and 73 km away, respectively (Fig. 2). Thus, the nests of both pure subspecies were always separated and no direct contact during the nesting period was recorded.
We analyzed clutch sizes and indirectly evaluated offspring survival rates of all nests observed in order to compare breeding success in different populations. In every population, a certain portion of nests contained some unfertilized or died eggs. For example, in our data set, 22% of the nests contained non-viable eggs in central Transbaikalia (n = 23 analyzed pairs at inhabited nests), 14% in Argun’ (n = 29), and 70% in eastern Mongolia (n = 33) (Table S3). More importantly, some nests contained only non-viable eggs, implying infertility of the nesting pair. Among 33 nests in eastern Mongolia, 19 had viable offspring (at least one living chick or big embryo), but the remaining 14 (42%) contained non-viable eggs only. In the Russian part, we found only 2% of such pairs (one pair in Argun’; n = 52). The content of all dead eggs was investigated, but they contained no embryo in the middle or late stage. In many cases, the females still long time brooded such non-viable eggs that had dried out. Considering fertile pairs only, all dead eggs, or a portion of dead eggs, were recorded in 47% such nests in eastern Mongolia, 11% in Argun’, 22% in central Transbaikalia, and none in China (Table S3).
Remarkably, 12 of those 14 nests in Mongolia which consisted of dead clutches were found at the eastern part of the contact zone, at a belt along the Kerulen River, approx. 30 km away from the Mongolian–Chinese border. Among 14 infertile pairs, we were able to check plumage and morphology of 7 pairs from a short distance. At least 6 contained a putative hybrid, while the other partner was leucoptera. In Kerulen, all 9 pairs of pure leucoptera had chicks. In contrast, among nests of 11 mixed pairs in Kerulen, 5 were found with chicks or eggs with large embryos and 6 with entirely non-viable clutches without embryos. In the 5 successful mixed pairs observed, one partner was intermediate in appearance and the other one a typical leucoptera. In Argun’ region, among 29 inspected nests, only one of the two mixed pairs described above had all dead eggs, while the other one had 5 chicks; all other 27 pure pairs of leucoptera and jankowskii had chicks or embryos in eggs. In neighboring northern China, all four nesting pairs of pure jankowskii were successful. In central Transbaikalia, all 47 nesting pairs of pure leucoptera were fertile and had chicks or embryos in eggs.
Altogether, our findings clearly show that breeding success in the hybridogeneous east Mongolian (Kerulen) population was drastically decreased due to failed fertilization or early embryonic mortality. Within the contact zone, the portion of non-viable clutches positively correlates with number of mixed pairs, and hybrids tend to occur close to each other. This part of the population may thus be considered as a “hybrid swarm” (Short, 1969).
Morphological differences between subspecies
We compared the newly obtained morphometric data on P. p. leucoptera with recently published measurements from three subspecies of the serica group (Red’kin et al., 2021). We found that leucoptera has remarkably bigger body size than jankowskii: wing length in adult leucoptera males was 224.3 ± 0.3 mm (n = 24) versus 209.5 ± 0.4 mm (n = 12) in jankowskii and tail length was 303.7 ± 0.7 mm versus 256.3 ± 0.9 mm, correspondingly. The same kind of difference was also evident for both sexes and age classes (Table S2). All differences were statistically significant (t-test; P < 0.001).
Phylogeography based on mt control region sequences
A phylogenetic network based on the 1326-bp alignment of 256 individuals (Fig. 4) clearly separates two highly diverged main clades (corresponding to the previously described Clades East and West; Haring et al., 2007; Kryukov et al., 2017)—each subdivided into further groups. These main clades are diverged by at least 66 substitutions. The predominantly western Clade West includes populations from most parts of Europe, as well as from Siberia, Mongolia, and Kamchatka, and contains four groups. Group #1 (from Europe to Siberia) comprises leucoptera, and four additional subspecies: pica, fennorum, hemileucoptera, and bactriana. Group #2 (separated by 7 substitutions from group #1) includes leucoptera only (populations of Transbaikalia, Mongolia, and the Argun’ river). Thus, leucoptera is highly variable and distributed among the two haplogroups, whereas the other four subspecies are found in only one of them. The melanotos population from the Iberian Peninsula (Iberia herein) forms a distinct and rather variable lineage (group #3) separated by at least 8 substitutions from the European–Siberian groups. The even more distinct group #4 is formed by camtschatica and separated by at least 19 substitutions from the European–Siberian group. It is rather homogeneous and comprised of only three haplotypes. Clade East consists of two haplogroups, each containing both jankowskii and serica individuals. Moreover, even individuals from the same population appear in both groups. Thus, we failed to find a clear geographic or subspecific pattern. For example, the Korean samples are distributed across both groups (Fig. S1). One of these groups (#6) has a star-like structure with one central, very frequent haplotype, representing 4 populations, while the other one (#5) has several less pronounced main haplotypes. Both groups include samples from all populations involved, with one exception: all insular Kyushu population samples share the same haplotype which is found also in one sample from Korea. In summary, among the six haplogroups of the network three are heterogeneous regarding the subspecies content (#1, 5, and 6) and three homogeneous, including only one subspecies each (#2: leucoptera, #3: melanotos, and #4: camtschatica). This is also reflected by the distances between subspecies (Table S4) compared to distances between haplogroups 1–6 (Table S5). The designation of groups #1–6 was used in all subsequent analyses.

Phylogenetic median-joining network based on 256 mitochondrial control region sequences. Sizes of circles correspond to the number of birds sharing this haplotype; branch lengths are proportional to the number of substitutions and those over 2 are shown at the branches. Haplogroups 1–6 are indicated by numbers
The time-calibrated Bayesian inference tree (Fig. 5) shows a very similar topology to the network. All six haplogroups are clearly separated from each other and highly supported. Relationships among groups #1, 2, 3, and 4 are not resolved and thus considered as polytomy. Our Bayesian time-calibrated tree indicated that the Clades East and West diverged as early as ~ 1.68 Ma ago (95% highest posterior density (HPD) interval 1.26–2.16) in the early Pleistocene. Subclades #3 (melanotos) and #4 (camtschatica) emerge in the admixed Clade West and represent the only two reciprocally monophyletic subspecies. Subclade #1 includes five subspecies: pica, fennorum, bactriana, hemileucoptera, and some of leucoptera (see Fig. 4); and also the clearly distinct subclades #5 and 6 within Clade East are admixed with respect to subspecies (jankowskii and serica).

Time-calibrated Bayesian tree based on mitochondrial control region sequences of Pica pica. Numbers at the branches indicate Bayesian posterior probability values (left) and bootstrap values of the ML analysis (right, in percent). Triangle widths are proportional to specimen numbers. Blue bars next to nodes indicate 95% credibility intervals for their age estimates. The figures in bold and the time scale below are in million years (Ma) before present
mtDNA diversity, neutrality tests, and population demography
Based on the data set of 256 CR sequences, nucleotide and haplotype diversities were calculated. The haplotype and nucleotide diversity values in all six haplogroups proved to be rather similar, ca. 0.9 and 0.3%, respectively, with the exception of a very low diversity in camtschatica (Table 1) and zero diversity in serica from Kyushu (Table S6).
Tajima’s D estimated by DnaSP was negative and significantly different from zero (which usually indicates departure from the null hypothesis of neutrality) for groups #1, 5, and 6. This was also supported by the coalescent simulation method in DnaSP and the infinite site model in Arlequin. Similarly, Fu’s Fs was significant between the observed and simulation distribution for all haplogroups except for melanotos and camtschatica, as well as for fennorum, leucoptera, and jankowskii (the latter two taken in total population sets) when calculated in DnaSP. Using Arlequin, significance of Fs was confirmed for all haplogroups and subspecies. The R2 statistic, which is preferred for small sample sizes (Ramos-Onsins & Rozas, 2002), was significant and complemented the above findings from all haplogroups except for group #2 and most subspecies (Table 1, Table S4). In total, all haplogroups have significant estimates of at least part of the neutrality tests used, and thus they may be considered in the context of population dynamics.
Mismatch distribution (MMD) analysis was conducted for all haplogroups (Fig. 6). In all these cases, this yielded similar unimodal left-shifted curves, implying recent population size expansions. This is in accordance with the data on either D, Fs, or R2 statistics for all groups. At the same time, the SSD method showed insignificant deviation from the expected MMD for all groups, while the raggedness r-index was insignificant for four groups: #1, 2, 4, and 5 in the coalescence analysis (Table 1). No significant difference implies a good fit of observed data to simulations of expansion. For the haplogroups #2–6, the spatial model of expansion was supported by r-index, and in addition, the sudden expansion model was adopted for melanotos only. For camtschatica, we obtained an extreme right-skewed unimodal curve in mismatch analysis, reflecting very low nucleotide diversity (Fig. 6, Table 1). In contrast, all populations of leucoptera, jankowskii, and serica showed two peaks, corresponding to the pattern in the network (Fig. S2). The population of serica from Kyushu was not appropriate for this analysis because it appeared to be highly inbred (only a single mt haplotype).

Mismatch distribution of nucleotide differences in populations representing different haplogroups as at Figs. 4 and 5. X-axis—number of nucleotide differences; Y-axis—proportion (frequency). Solid lines—expected distributions (under expectation of population growth); dashed lines—observed distributions. a Haplogroup 1: Europe, Siberia, and Mongolia, n = 49. b Haplogroup 2: Transbaikalia, Mongolia, and Argun’, n = 61. c Haplogroup 3: P. p. melanotos, Iberia, n = 14. d Haplogroup 4: P. p. camtschatica, Kamchatka, n = 17. e Haplogroup 5: Russian Far East, Argun’, China, Japan, Korea, Taiwan, n = 69. f Haplogroup 6: the same, n = 46
We calculated Bayesian skyline plots (BSP) for the six haplogroups (Fig. 7a) that we defined in the phylogeographic analysis of the mt sequences (Figs. 4 and 5). The other set of BSPs represents the six subspecies under study, with one plot for each subspecies (Fig. 7b). The sets where the subspecies jankowskii and leucoptera were represented by all distinct populations analyzed are shown in Fig. 7c, d. Most haplogroups, subspecies, and populations proved to be different regarding coalescent times. The shortest time to the most recent common ancestor (tMRCA) was found for camtschatica as ~ 1300 years before present. The related European–Siberian groups #1 and #2 had tMRCA of ~ 35 and ~ 67 kyr, respectively. Two others, the eastern groups #5 and #6, showed ~ 38 and ~ 31 kyr, respectively. The group #3 (melanotos) from Iberia had a longer history compared to the European–Siberian group #1 (Fig. 7a) and also longer than the subspecies fennorum (Fig. 7b). Within subspecies, populations also differed by tMRCA (Fig. 7c, d). We were unable to provide any reasonable estimations of split times and Ne for the recently introduced population of Hokkaido because equilibrium in mutation rates, migration, and genetic drift was not yet established, as pointed out for such cases by Rius and Turon (2020).

Bayesian skyline plots (BSPs) for effective female population sizes for haplogroups, subspecies, and populations of Pica pica. a Comparison of 6 haplogroups, depicted in the network Fig. 4. b Comparison of 6 subspecies. c Comparison of 4 populations of P. p. jankowskii. d Comparison of 3 populations of P. p. leucoptera. X-axis—time in thousand years; Y-axis—effective female population size (Ne), log scale for clarity. Lines represent median estimates. Light blue columns depict LGM period. Short lines corresponding to camtschatica at a and b are poorly visible near the Y-axis
The curves also differ in shape. Almost all groups and populations, with exception of Mongolian leucoptera, experienced more or less intensive growth even before and during the Last Glacial Maximum (LGM), but the dynamics of effective population sizes differ among haplogroups, subspecies, and populations (Fig. 7a–d). Among the West European subspecies, the population of the marginal subspecies melanotos grew much faster than fennorum (Fig. 7b). When comparing total jankowskii and leucoptera data sets, jankowskii demonstrated intensive long-lasting growth from ~ 50 kyr BP to ~ 25 kyr BP, while leucoptera growth started to accelerate much later, ~ 10 kyr BP (Fig. 7b). When investigating the populations separately, the marginal northernmost Amur river population of jankowskii appeared to have expanded much faster than Argun’, Primorsky, and Chinese populations, in this order (Fig. 7c). In leucoptera, the Transbaikalia population showed the most pronounced increase, while the population from the Argun’ river basin was moderately growing, and that of Mongolia appeared stable and drastically declining during the last 20 kyr (Fig. 7d).
Differentiation with nuclear SNPs: results of ddRAD analysis
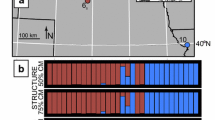
SNP analyses were performed based on population samples from the contact zone of leucoptera and jankowskii as well as from adjacent populations. We preliminarily divided all samples into six provisional populations according to geography: I—central Mongolia; II—eastern Mongolia; III—central Transbaikalia; IV—Argun’ river basin; V—northern China; and VI—Amur river basin (Fig. 3). In all runs of Structure and Harvester, delta K was maximal with K = 2, confirming two major genetic clusters—Western and Eastern (Fig. 8). In central Mongolia (Ulaanbaatar), all genotypes were of pure western type. In eastern Mongolia, along the Kerulen river, three admixed genotypes were found while all others were of the western type (Fig. 8). All admixed genotypes were obtained at sites closely located to each other from birds nesting along the low Kerulen river (#58, 61, and 62 at Fig. 3). Two individuals were very similar with respect to the proportion of eastern alleles (17.0–17.2% probability assignment to the eastern genetic group), while the third bird was predominantly of the eastern type (74.6%). In Transbaikalia, where the samples were collected across a broad geographic range, six admixed genotypes were identified. While one bird showed only low level of interbreeding (4% of eastern alleles, point #16, Fig. 3), the five other individuals had a higher portion of eastern alleles (35.6–40.6%). Three of these were nestlings from the same clutch, which was found in the northern-most and geographically quite remote rural site in Transbaikalia (#9–11, Fig. 3). Two more admixed individuals with 35.5–39.8% of jankowskii alleles were sampled in the south of Transbaikalia (#14–15).

Genetic structure of the contact zone and its vicinities by mtDNA and SNPs. Above—distribution of haplotypes of mitochondrial CR along conditional population numbers I–VI depicted in Fig. 3. Green—western haplotype; red—eastern haplotype. Below—graphical representation of individual contributions from nuclear SNPs obtained with ddRAD and Structure analysis. The number of clusters was determined as K = 2. Each individual is represented by a vertical bar and length of each colored segment assigns at the Y-axis the probability of belonging to one of the two genetic clusters (colored as above)
The population at the Argun’ river was subdivided by allelic state into two groups each consisting of individuals which possessed one of the two pure SNP genotypes (Figs. 3 and 8). Birds with eastern type prevailed in the southern part of the Argun’ river basin. Almost no admixed individuals were found, except for one sample with a trace (1%) of western genotype (#105, Fig. 3). Thus, in this region, both genotypes are sharply separated by geography. In northern China, represented in our data set by only one collecting point near Lake Wulan, pure eastern genotypes were found with the exception of one sample (#83, Fig. 3) with a trace of the western genotype (2.4%). Similarly, the Amur river population contained one admixed sample only (#117, Fig. 3), in which the indication of introgression was slightly more pronounced (7.9% of the western genotype).
A PCA analysis of unlinked SNP loci showed the distribution of individual genotypes from the same six populations studied. The first principal component, explaining most variation (23.3%), clearly discriminates the three leucoptera populations from three jankowskii populations (Fig. 9a). The second component (7.4% explained) divides the geographically remote Amur population, from the other two jankowskii populations. The single outlier from eastern Mongolia may represent a hybrid. The 3rd and 4th components, explaining 6.6 and 4.3% of the variation, respectively, showed a more distinct pattern (Fig. 9b). Individuals are mainly distributed along the third component and cluster in five groups, three of which are pure and two are admixed. In addition, several individuals are positioned far distant from their main clusters. While most samples from the Argun’ population cluster with individuals from northern China, four belong to the cluster of Transbaikalia. This perfectly matches our results from the Structure analysis (Fig. 8) and confirms that the Argun’ population is composed of two subspecies. Samples of the Amur population of jankowskii are distinct, but close to the other samples of jankowskii from China and Argun’. Rather unexpectedly, all three groups of leucoptera (i.e., I, II, and III in Fig. 9b) proved to be very distinct from each another. Especially, the central Mongolian group I, geographically remote, is distinct across both axes. Three samples from the hybridogeneous population of eastern Mongolia (Kerulen river) appear as outliers, two of these are positioned between the clusters of leucoptera of eastern Mongolia and the group of three populations of jankowskii IV–VI. They may represent hybrid samples from eastern Mongolia, detected with Structure analyses. Also, a hybrid origin may be assumed for the three outliers from the Transbaikalia population.

Population genetic structure based on unlinked SNP markers. Scatter plots of principal component analysis (PCA) show individual variation in components one and two (a) and three and four (b). The amount of variance explained by each PC is shown in parentheses. I—leucoptera, central Mongolia; II—leucoptera × jankowskii, eastern Mongolia; III—leucoptera, central Transbaikalia; IV—leucoptera + jankowskii, Argun’ river basin; V—jankowskii, northern China; VI—jankowskii, Amur river basin
Bioacoustics
The fundamental frequency of all alarm calls is about 2 kHz for both leucoptera and jankowskii, but the calls differ considerably in chattering speed (Figs. 10, 11, and 12; Table 2). We compared the speed of elements of chattering calls in different populations and individuals. The scatter plot in Fig. 11a shows averaged data for each bird in order to compare variation within and between the studied populations (Table 2). The clouds, corresponding to the two most distant populations (jankowskii from Vladivostok and leucoptera from Altai), are widely separated (slow chattering ~ 3–5 elements per second (eps) vs. fast chattering ~ 11–12 eps along the Y-axis, Fig. 11a). The latter forms a common cluster together with individuals from the leucoptera of Transbaikalia (9–12 eps). The hybridogeneous population from eastern Mongolia (Kerulen river) proved to be very variable in terms of chattering speed (ranging from ~ 7–12 eps). Half of the individuals belonged to the leucoptera cloud and the remaining individuals were located between the jankowskii and leucoptera clusters. The most remote points #3 and #5 correspond to birds with intermediate appearance, and #4 to the pair (#24) consisting of a hybrid male and a female leucoptera, from the nest with unfertilized eggs. Furthermore, most points corresponding to the admixed population from the Argun’ river were located within the leucoptera cloud, except two points (#1 and 2 in Fig. 11a), which were close to each other within the jankowskii cloud. The two corresponding birds were phenotypically jankowskii. Other records obtained from birds of this population phenotypically resemble leucoptera individuals. In total, both hybrid and admixed populations (Kerulen and Argun’) exhibit very high variability in chattering speed (Table 2), and, in particular, data points of the birds from the Kerulen population considerably extend to the jankowskii cloud. This pattern is summarized in Fig. 12 which depicts high variability of both admixed populations, the hybridogeneous population of Kerulen and the admixed population of Argun’. In both groups, the averaged estimates of chattering rates are similar (Fig. 12, Table 2), but for different reasons: due to hybrid birds in the former and due to the presence of pure birds from both subspecies in the latter.

Spectrograms of chatter calls of pure P. p. leucoptera, pure P. p. jankowskii, and hybrids. a leucoptera, pair #4, Choibalsan, eastern Mongolia. b jankowskii, pair #6, Vladivostok, Russian Far East. c, d Bilingual hybrid, the same bird from the pair #24, Kerulen, eastern Mongolia. e Hybrid with mixed call, pair #39, Kerulen. f Presumably hybrid with mixed call, pair #5, Kerulen. X-axis—time in s; Y-axis—frequency in kHz

Rates of chatter calls in different magpie populations and individuals. a Each mark represents average chattering rate for a single bird from five populations indicated by colours. Figures are numbers for the outliers: 1, 2—jankowskii from the mixed population of Argun’; 3, 4, 5—hybrid birds from the hybridogeneous population of Kerulen. b Each mark represents average chattering rate for a series of chatterings of one selected individual representing jankowskii, leucoptera, and hybrid birds, respectively. Green mark—pair #6 jankowskii from Vladivostok; gray—pair #43 leucoptera from Tsasuchei, Transbaikalia; blue—pair #24 hybrids from Kerulen, eastern Mongolia. X-axis—number of elements per second in a total series of chattering; Y-axis—number of elements per second in a series of 5 elements of chattering

Violin plot diagram of the chatter call speed (elements per second) of Eurasian magpie populations across regions. X-axis presents a set of populations; Y-axis—elements per second. Box outlines the interquantile range (25%, 75%), whiskers represent range without outliers, central bar is the median, red dot is the mean, and figure shape is the probability density. The brackets on the top denote statistically significant pairwise differences (Games-Howell test, p < 0.05)
In addition to the above data, which were averaged across birds, we also analyzed call variation within individuals. Figure 11b shows values of chattering rates in each series performed by three selected individuals from the leucoptera population from Transbaikalia, jankowskii from Vladivostok, and a hybrid male from the pair #24 from Mongolia. The latter bird alternated both fast and slow chattering calls representing the typical vocal patterns of either the western or the eastern form, and this individual may thus be considered bilingual.
Besides differences in chattering speed, the studied subspecies as well as the hybrid individuals differ in call structure. A spectrographic analysis revealed that the separate sound of leucoptera appears as a single-syllable note (Fig. 10a). In contrast, the signal of jankowskii is clearly divided into two parts separated by a very short pause (about 30 ms; Fig. 10b). This difference is easily perceived by ear and thus allows distinguishing the different patterns in the field without additional gear. Both variants were recorded in the hybrid male from the mixed pair #24 mentioned above (Fig. 10c, d). It alternated both kinds of calls and may thus be defined as bilingual in this respect as well. The other two examples were recorded from the other two hybrids (from the pairs #39 and 5), also from Kerulen, Mongolia (Fig. 10e, f). In these individuals, the spectrograms are intermediate or similar to the acoustic shape of the western form, but are clearly intermediate in terms of chattering speed.
Discussion
Impact of Pleistocene refugia on magpie phylogeographic patterns
Despite the growing number of studies focusing on genetic variation in the Holarctic magpie species complex, the starting point of the present study was the lack of profound understanding of genetic differentiation and levels of gene flow among subspecies. Our phylogeographic study based on broad geographic sampling represents a combination of population genetic analyses of a mitochondrial marker (256 densely sampled individuals from the whole species’ distribution) and genome-wide nuclear ddRAD-Seq analysis (89 samples from the contact zone and adjacent areas). This considerably extends previous efforts. The results confirm the general pattern of a major East/West division, which resembles that of other vertebrate species with a similar subdivision (in extreme with disjunct ranges) (e.g., Latinne et al., 2020; Matyushkin, 1976). Nonetheless, previous assumptions concerning glacial refugia in P. pica (Haring et al., 2007; Kryukov et al., 2004, 2017) should be adapted and the subdivision of the mt network into six haplogroups may be interpreted by a history shaped in several refugia.
In Europe, three prominent southern refugia (Iberia, Apennines, and Balkans; Hewitt, 1999, 2001) are often considered as general source areas for northwards colonization after the Pleistocene Ice Ages. For example, the relic Iberian population of rook Corvus frugilegus was presumably the source of other European and West Siberian populations. As shown by microsatellite data, however, current gene flow with other regions is rather restricted (Salinas et al., 2021). In contrast, for magpies, the assumption of an Iberian refuge, from which a postglacial expansion started, appears unreasonable, based on the genetic variability and distinctness of melanotos which shares no haplotype with the main European–Siberian group #1. Only in the Pyrenees does P. p. melanotos meet with P. p. pica and intermediates were found in this area (Martinez, 2016). This region is also a contact zone for populations of Chorthippus parallelus, Alnus glutinosa, Fagus sylvatica (Hewitt, 1999), Picus viridis (Pons et al., 2019), and Emys orbicularis (Poschel et al., 2018). Thus, the Pyrenees are considered to be a so-called suture hybrid zone (Remington, 1968).
Apart from Iberia, independent expansions and colonization routes may be assumed for magpies in Central Europe. Our results suggest at least two additional independent refugia in Central Europe. One of these might have been located in the Carpathian region, which was shown for several vertebrate species (e.g., Hewitt, 2011; Sommer & Nadachowski, 2006) and might have served as the refuge area for group #1. In addition, the southern Urals and Caucasus region, where certain other species survived the LGM (Schmitt & Varga, 2012), appear to have been appropriate refugia. Group #1 has a star-like pattern (Fig. 4) and is composed of five subspecies; compared to group #2, it is also less variable regarding haplotypes (Table 1). Group #2 consists of solely leucoptera and has no clearly star-like pattern. In examining the still small sample of group #1 and its geographic distribution, note that, while we hypothesize on the Pleistocene distribution of lineages (haplogroups), the origin of subspecies remains unclear.
The eastern groups (#5 and 6) could be the result of admixture after a phase of isolation in two putative refugia in the Russian Far East/Primorye/north-east China (Mandshuria) vs. Korean Peninsula and/or south-east China, which have previously been described as refugia for various other organisms (e.g., Nazarenko, 1982; Zhang et al., 2008; Lee et al., 2008; Chung et al., 2014). The Japanese population of Kyushu segregates only a single haplotype and was artificially introduced from Korea in the late sixteenth century (Eguchi & Kubo, 1992) which may indicate that a founder effect resulted in complete homogeneity. The other population of Hokkaido Island was introduced within the last 40 years (Horimoto, 2004) by hitchhiking on ships from Primorye in Far East Russia (Kryukov et al., 2017), rather than from Korea or Kyushu, as shown by microsatellite analysis (Mori et al., 2014).
Comparison of magpie population history in two peninsulas: Iberia and Kamchatka
The subspecies melanotos is widely distributed throughout the Iberian Peninsula up to altitudes of 2400 m (Martinez, 2016). It is mainly sedentary, but may move seasonally (Martinez et al., 2003; Herrando & Quesada, 2012). Field observations indicate a population size expansion in the last century, which is supported by our BSP and mismatch analyses (Figs. 6 and 7a, b). This may have been facilitated by its high ecological plasticity, which resulted in recent range expansions to previously uninhabited peripheral regions (Herrando & Quesada, 2012). Due to mountainous and complex environments, resulting in fragmented habitats, the refugium might have consisted of several smaller refugia, which has also been found in other animals and plants in Iberia (“Refugia within refugia” hypothesis; Gomez & Lunt, 2007; Neto et al., 2012; Abellan & Svenning, 2014). For example, Hewitt (2011) recognized seven refugial areas in Iberia for several species. In one of such cases, six geographically isolated haplogroups of the lizard Lacerta lepida were found in Iberia, corresponding to presumed smaller refugia (Miraldo et al., 2011).
When comparing the results of the Iberian populations to those from Kamchatka, the other peninsula in our survey, we find that both haplogroups have similarly distinct positions in the network (Fig. 4) and phylogenetic tree (Fig. 5). Kamchatka’s population is genetically homogeneous, and its genetic distance from the European–Siberian haplogroup is even larger compared to that of melanotos. This distinctness is surprising given Kamchatka’s presumably young fauna. Despite the closer proximity of the eastern subspecies jankowskii (groups #5 and 6), our genetic results support an origin of camtschatica from the Western Clade. Assuming a postglacial colonization of Kamchatka, we would have expected that the colonizing birds carry haplotypes of the source population. Yet, the three haplotypes in Kamchatka are separated by at least 20 mutational steps (~ 1.5%) from the remaining haplotypes. This implies a longer lasting survival in Kamchatka, e.g., in the central depression where trees and bushes probably remained in the LGM (Melekescev et al., 1974) or in coastal refugia in the south of the peninsula (de Lattin, 1957). Low levels of genetic variation indicate severe bottlenecks, followed by spatial expansion (Table 1, Fig. 6).
Population dynamics and divergence time estimations
Our Bayesian time-calibrated tree indicated that the Clades East and West diverged as early as ~ 1.68 Ma ago (95% highest posterior density (HPD) interval 1.26–2.16) in the early Pleistocene, which agrees with the dating of Song et al. (2018). All groups #1–4 from Clade West diverged earlier, ~ 460 (319–612) kyr ago, than groups 5 and 6, which split slightly later, about 330 (208–464) kyr ago (Fig. 5).
The BSP analyses show the longest tMRCA in group 2 (which includes part of leucoptera) and the shortest in group 4 (camtschatica) (Fig. 7a). Among subspecies, much longer tMRCA were estimated for jankowskii, serica, and leucoptera (106–130 kyr, Fig. 7b). This is putatively due to the presence of each of these three subspecies in two distinct haplogroups implying incomplete panmixia within a subspecies and may suggest that this estimation could be an artifact. Zhang et al. (2012) estimated tMRCA for the eastern clade of serica as 250 kyr and the onset of expansion at 100 kyr ago. Conversely, Song et al. (2018) estimated tMRCA’s as 540 kyr for the Eurasian clade and 370 kyr for the East Asian clade (comprised of serica and anderssoni). Both estimates are higher than ours. A longer tMRCA was estimated for melanotos compared to group #1 (Fig. 7a, Table 1), which supports the hypothesis that this subspecies survived the LGM in the Iberian refugium.
With the exception of the Mongolian population of leucoptera, all plots indicate a population growth at different periods in the recent past. Group #5 started to grow much earlier than group #6 (Fig. 7a). Rather fast population growth of three haplogroups #1, 5, and 6 began approximately 38–30 kyr ago and occurred before the LGM (Fig. 7a). This agrees with estimates of Song et al. (2018) and supports their hypotheses about longer tMRCA for haplogroups of the West versus East lineages.
As we have shown earlier (Goroshko et al., 2018; Kryukov et al., 2017), leucoptera and jankowskii are still expanding year by year towards each other, and jankowskii is moving evidently faster than leucoptera. Based on field observations and BSP modeling, this study provides further evidence for these demographic dynamics. Considering the steepness of the BSP curves, the Amur river population of jankowskii is growing faster than any population of the two contacting subspecies (Fig. 7c, d). Moreover, it started to expand earlier than all others (Fig. 7b). This fits well with ornithological observations of a fast, westward range expansion of jankowskii in the Amur river basin where it currently reaches the range of leucoptera along the Trans-Siberian railway and approaches leucoptera at Argun’ river basin near the border between Mongolia and China (this study). The expansion in the last decades is not necessarily connected with anthropogenic factors, at least in the unpopulated regions of South Siberia and Mongolia. Londei (2018) hypothesized that magpies were initially associated with wild grazing ungulates, which can be still observed in some areas such as the Mongolian steppes or on islands in the Mediterranean Sea. Note, however, that magpies are very plastic in their ecological niche and may use a variety of habitats. Currently, they mostly occupy anthropogenic landscapes, villages, and big cities, but have retained some ancient foraging habits. They are adapted to semi-open landscapes such as the edge of the forest and forest-steppes, and might expand their ranges along intrazonal landscapes, such as river valleys, trees along the roads, and high-power lines used for nesting. Such a plasticity makes hypotheses on their historical ranges rather speculative. The declining population of Mongolian leucoptera during the last ~ 10 kyr (Fig. 6d) is difficult to interpret. These changes may be connected to dry conditions in the Late Glacial and predominantly dry conditions during most of the Holocene in eastern and northern Mongolia (Klinge & Sauer, 2019).
Population analysis, hybridization, and gene flow
The present study provides evidence for hybridization and gene flow between leucoptera and jankowskii, with 15.8% (3 out of 9) and 23.8% (5/21) samples with mixed ancestry in eastern Mongolia and central Transbaikalia, respectively (Figs. 3 and 8). Accordingly, we observed a number of mixed breeding pairs. Three northernmost samples (Fig. 3, #9–11) may represent descendants of long-distance migrants from the range of jankowskii that traveled to the west along the species’ northern distribution border and entered the nesting range of leucoptera where mating with a consubspecific partner was difficult or impossible. Together with two additional samples (#14–15) from further south, these five genetically close samples from Transbaikalia may represent F1 hybrids. Since they possessed leucoptera mt haplotypes, we assume that they originated from crosses between jankowskii males and leucoptera females.
We found quite the opposite genetic structure in the eastern Mongolian population. Among three admixed samples from Kerulen river valley, two were genetically similar while the third possessed a higher proportion of jankowskii alleles (#58, 61, 62, Fig. 3) indicating different orders of backcrossing. Both subspecies co-occur in the Argun’ river basin, but are generally separated by 40 km or more during the nesting period. Thus, we speculate that these species are not fully parapatric, but connected by sporadic gene flow. Moreover, the borders may fluctuate across time: we found several uninhabited nests from previous years in the zone between both ranges. Young birds are prone to vagrancy and we found flocks of jankowskii in the postbreeding period migrating for rather long distance into the range of leucoptera. Importantly, also admixed groups of individuals from both subspecies were met by us during the non-breeding time.
In northern China and the Amur river basin, only traces of hybridization were recorded. Although field observations showed that breeding success in mixed pairs of the east Mongolian population dropped drastically due to failed fertilization or embryonic mortality, we nevertheless identified some levels of introgression by SNP analyses (Fig. 8). Several birds with weak signs of admixture (in southern Transbaikalia, northern China, Argun’ river basin, and middle Amur) indicate several generations of backcrossing. In total, these complex patterns along the mosaic contact zone may be due to varying time points of secondary contact. In eastern Mongolia, contact might have started at least 100 years ago (Tugarinov, 1932), whereas in the Argun’ river basin, based on interviewing local residents and our own observations, contact started only 20–30 years ago. In northern China and the Amur river basin, contact is still absent as only single backcrossed hybrids were found.
Role of vocalization in species recognition and hybridization
Acoustic signals, including calls, play an important role in the speciation of birds and serve as an important prezygotic isolating barrier (Panov, 1989; Wilkins et al., 2013). Magpies have a rich vocalization repertoire: 15 types of songs and calls were reported for adults (Goodwin, 1986; Cramp & Perrins, 1994). Bird calls are often believed to be highly heritable, a view that was based mainly on observations of birds raised in captivity. However, also learned calls have been reported previously (Edwards et al., 2005; Marler, 2004). In admixed magpie populations, both alternatives may be considered. The intermediate pattern of the alarm call, representing a kind of convergence, may be genetically determined in hybrids (reflecting a similar inheritance mode as in other cases of complex quantitative traits). Alternatively, it may be developed ontogenetically in the progeny of a mixed pair as a result of learning from both parents with their specific call types.
Differences in chatter calls between western and eastern Pica pica subspecies groups were already noted before (Ebels, 2003; Goodwin, 1986). In a previous study, we presented spectrograms and quantitatively analyzed the geographic variation of this character in most subspecies of magpies (Kryukov et al., 2017). In the present paper, we confirmed that averaged chattering rates for pure populations of both subspecies differ significantly (Fig. 12, Table 2). For the first time, intermediate calls of hybrid magpies regarding chattering rates were discovered. Moreover, we detected bilingual birds alternating both specific kinds of calls: fast and slow (Fig. 10c–d).
In the Argun’ river population, magpies with intermediate calls were absent, which agrees with the genetic data, implying that the population should be treated as admixed but not hybridogeneous. Among the records obtained in the hybridogeneous population of Kerulen, only the recordings of three birds potentially stem from hybrids. Two of these performed intermediate chattering, and one was bilingual (Fig. 10c–f). Our bioacoustic data implies that 18.7% (3 out of 16) are hybrids, which is close to the proportion estimated by nuclear SNPs (15.8%; 3 out of 19). Despite the low sample size, the similarity of these estimates points to a pronounced genetic basis of magpie calls.
Mutual learning from different passerine species in their zones of sympatry is a rather common phenomenon and was called “mixed singing” (Helb et al., 1985). Both intermediate types of songs and bilingual singers were reported for the hybrid zones of the leaf warblers Phylloscopus collybita abietinus and Ph. c. tristis (Marova et al., 2017). A mainly genetic control of the song dialects was evidenced both by analysis of mitochondrial DNA (Marova et al., 2017) and by nuclear SNP analysis (Shipilina et al., 2017), whereas in the nightingales Luscinia luscinia and L. megarhynchos, the mixed calls in the hybrid zone reflected cultural transmission (heterospecific learning) rather than introgression (Vokurková et al., 2013). In sympatric flycatcher populations of Ficedula hypoleuca and F. albicollis, mixed songs lead to hybridization (Haavie et al., 2004). In contrast, due to suspected hybridization, intermediate vocalization was reported for the zone of sympatry of the doves Streptopelia vinacea and S. capicola (de Kort et al., 2002). In the latter two cases, songs are believed to be inherited.
The convergence of the acoustic signaling in a contact zone might lead to elevated levels of hybridization. In magpies, such a convergence may be advantageous for a pair’s territorial defense against heterosubspecific neighbors. But this would also ease finding a mate and thus would promote backcrossing. Yet, it would be non-adaptive because of reduced fitness of the hybrid progeny as shown above. Overall, our results show that bioacoustic analysis in the contact zones of magpies (and any other birds with innate calls) may help to reveal hybrid individuals even in the field, preliminary by ear and subsequently confirmed by spectrograms. Targeted studies are required to discriminating between several reasons for mixed calls (song convergence) and bilinguality, and understanding their evolutionary consequences.
Evolutionary aspects of introgression in the case of magpies
Introgression is the most important outcome of hybridization. Levels of gene flow across a secondary contact zone may vary, from restricted gene flow in a steep cline as found, e.g., with the Picus viridis sharpei/P. viridis viridis zone (Pons et al., 2019), to moderate introgression (e.g., voles Microtus californicus; Lin et al., 2018) and massive introgression (e.g., hares Lepus timidus/L. mandshuricus; Kinoshita et al., 2019). The extreme is “mitochondrial capture”—the complete mt genome replacement (e.g., between buntings Emberiza citrinella and E. leucocephalos; Irwin et al., 2009). In the present paper, we describe a rather rare case of introgression without evident direct secondary contact of the breeding ranges.
During several years of field observations, we identified only a few putative hybrids between leucoptera and jankowskii based on intermediate phenotypic features. In eastern Mongolia, hybrids always mated with pure leucoptera. Only once we identified a putative intersubspecific pair of pure leucoptera and jankowskii, whereas pure pairs of leucoptera and jankowskii were never found nesting in close contact, when both subspecies live in neighboring territories. The spatial distribution of mixed pairs and nests with solely unfertilized eggs increases towards the east in eastern Mongolia, which is consistent with the results of SNP-based analyses of hybridization. Intermediate and mixed vocalization of hybrids, as well as their expected intermediate appearance, may facilitate hybridization between the two subspecies. Nonetheless, a certain degree of reproductive isolation, i.e., incomplete female hybrid sterility, should act against frequent introgression. Such a balance between selection against hybrids and dispersal of parental forms into the zone was predicted in the “tension model” of hybrid zone (Barton & Hewitt, 1985; Key, 1968). Note, however, that the stability of this zone is not yet established, and its fate remains unclear; it seems that postzygotic isolation would prevent merging of both forms, but only quite strong negative selection would be able to reinforce the prezygotic isolation and establish semi-sympatry (Liou & Price, 1994). Further invasion of jankowskii is expected, potentially extending the introgression range. On the invading forefront, asymmetry in the number of both forms may cause difficulties in mating with conspecifics, supporting hybridization (Irwin, 2020). Nonetheless, taking into account the very low population size and density in the contact zone, the situation may smolder a long time. The boundaries of species will remain semi-permeable, but the distinctness of both taxa is expected to be preserved.
Usually, introgression due to range expansion is considered to be asymmetric, resulting in gene flow from the local population to the invader (Currat et al., 2008; Drovetski et al., 2015; Excoffier et al., 2009; Levanen et al., 2018). Since all eight genetically confirmed hybrid magpies in Transbaikalia and eastern Mongolia bear mtDNA of leucoptera (Figs. 2, 3 and 8), the introgression of nuclear genes seems to occur only due to invading males of jankowskii to the west entering the range of leucoptera (the resident form), but not from leucoptera to the east. Thus, asymmetric sex-biased introgression apparently occurs from invaders, into the “wrong” direction. Our genetic data are thus consistent with observation by Goroshko et al. (2018) that during the last decades, jankowskii has been expanding its range faster to the west than leucoptera to the east, sometimes mating deep within the leucoptera range with residential leucoptera individuals.
mtDNA is usually much more prone to be maintained in a population after introgression than nuclear genes (Kinoshita et al., 2019; Lin et al., 2018; Pons et al., 2014; Renoult et al., 2009). In our case, however, introgression was detected based on nuclear genes, rather than mitochondrial markers, strongly indicating predominantly male-biased dispersal in magpies. A similar case was previously reported for Locustella warblers (Drovetski et al., 2015). Our study thus documents a rather rare case of mitonuclear discordance accompanying introgression (Sloan et al., 2017; Toews & Brelsford, 2012).
Taxonomic considerations
The level of reproductive isolation is an important criterion of taxonomic rank in many species concepts. The dilemma of treating closely related allopatric populations as either species or subspecies remains unresolved. A similar issue arises in the cases of hybridizing forms. Under the biological species concept, the situation in the contact zones is the deciding factor. The paradigm of isolating mechanisms was the key point in the framework of the synthetic theory of evolution (Mayr, 1963). It is impossible to solve this dilemma without detailed data on gene flow between populations that have come into contact and its dynamics.
In birds, closely related species are mainly isolated by prezygotic mechanisms, which prevent heterogeneous mating due to differences in plumage, vocalization, and behavioral displays (Edwards et al., 2005). For the two groups of magpie subspecies coming into contact in Transbaikalia and Mongolia, we demonstrated that their reproductive isolation is incomplete, despite likely prezygotic isolation by evident differences in phenotypes and calls. In addition, we found evidence for postzygotic isolation in the form of increased embryonic mortality in hybridogeneous populations. The reasons for the embryonic mortality, e.g., chromosomal rearrangements resulting in an excess of unviable gametes due to heterokaryotypic recombination (Feulner & De-Kayne, 2017), remain to be investigated.
In the earlier studies, the high levels of genetic divergence in the mt markers between Clades West and East were reported (Haring et al., 2007; Kryukov et al., 2004, 2017; Lee et al., 2003; Song et al., 2018). Recently published data on complete mt genomes of five magpie subspecies showed a mean distance of 0.039 between representatives of the same two subspecies groups (Kryukov et al., 2020) and their differentiation was further confirmed by the present nuclear SNP analysis (Figs. 8 and 9). Besides genetic differentiation as described above, we also found striking morphological differences, such as bigger body size in leucoptera than in jankowskii (Table S2). These data, together with differences in plumages and vocalizations, allopatric distribution, and partly genetic incompatibilities (hybrid infertility) in the contact zone, are a reasonable basis to consider leucoptera and jankowskii as belonging to distinct species. Their species rank is compatible with most species’ concepts.
These species correspond to the two mitochondrial Clades East and West. The eastern forms, under the priority rule of the Zoological Nomenclature Codex, should be named Pica serica Gould, 1845. Overall, the proposed full species rank of Pica serica is consistent with the current status in the latest editions of the Bird Checklists (Gill et al., 2021; Madge et al., 2020). Concerning the subspecies of P. serica, some comments are necessary. The nomenclature of the East Asian forms of magpie was based on outdated information on their distribution (Rustamov, 1954), which proved to be incorrect. Some authors treated P. p. jankowskii as a synonym of P. p. anderssoni Lönnberg, 1923 (Del Hoyo & Collar, 2016; Madge, 2009; Song et al., 2018). A later, detailed analysis of eight morphometric characters and coloration demonstrated that magpies from the terra typica of P. p. anderssoni (in north-eastern China, see Fig. 1) do not differ from P. p. serica living to the south. Accordingly, P. p. anderssoni was considered as a junior synonym for P. p. serica (Red’kin et al., 2021), a view we follow here, as there is no support for this taxon at all. The same study and the data presented above (Table S2), however, reported clear morphometric differences between P. p. jankowskii, P. p. alashanica Stegmann, 1928, and P. p. serica (that study included the type specimens of jankowskii and alashanica). P. p. jankowskii nests mainly in the Amur river basin, P. p. alashanica inhabits the arid landscapes of the Gobi and Alashan deserts, and P. p. serica lives farther south (Fig. 1). Consequently, Pica serica should comprise P. s. serica, P. s. jankowskii, and P. s. alashanica (Red’kin et al., 2021).
Based on the close relationships in mtDNA among the seven subspecies of Clade West—P. p. melanotos, P. p. pica, P. p. fennorum, P. p. hemileucoptera, P. p. bactriana, P. p. leucoptera, and P. p. camtschatica, which are altogether very distant from the subspecies of Clade East (Figs. 4 and 5), we propose to retain the former group under Pica pica (Linnaeus, 1758). P. p. camtschatica is geographically well isolated and differs in ecology, plumages, and vocalization (Kryukov et al., 2017), and its status might be elevated to distinct species, but this should be evaluated by further analyses. Concerning the other species, the monotypic P. mauritanica, P. asirensis, and P. bottanensis as well as P. hudsonia and P. nuttalli, we agree with their species rank suggested by Song et al. (2018) and Birds of the World (Madge et al., 2020). In particular, for the latter two Nearctic species, lack of sufficient genetic data still leaves their status rather preliminary.
Conclusions
In this paper, we conducted integrative genetic and bioacoustic analyses of closely related forms of the widely distributed Eurasian magpie Pica pica s.l. that were densely sampled from within and around a secondary contact zone in southern Siberia and eastern Mongolia. We combined genome-wide SNP analysis with detailed field observation and bioacoustics. Finally, we carried out a phylogeographic study covering most of the range of Pica pica s.l. in the Palearctic. This comprehensive data set and in-depth investigation provided insights into the current state and structure of the secondary contact zone in Asia. Because of reciprocal expansions into regions previously uninhabited by magpies (with predominating invasion by the eastern populations), hybridization sporadically occurs due to partial collapse of both pre- and postzygotic isolation. We found that during several rounds of backcrossing, asymmetric male-biased introgression of nuclear, but not mitochondrial markers, takes place, even over long distances from the actual zone of contact and that introgression occurs from the more actively invading eastern form into the western populations. Intermediate and mixed vocalization of hybrids might play an important role in the dynamics of hybridization and demography. The observed hybrid infertility and the fact that hybridization is very sporadic justifies the elevation of the two subspecies to the species level. Our study confirms the importance of integrative studies of initial stages of natural secondary contact zones as an approach to better understand the complex dynamics of evolutionary processes. Generally, magpies might serve as a useful model system for more thorough analyses of speciation genetics and genomics. Continued extensive ecological and genetic monitoring of magpie contact zones are needed to shed light on the development of species’ borders and on the mechanisms by which species evolve as entities in spite of ongoing gene flow.
Data availability
The mitochondrion sequences obtained in the current study are deposited in the GenBank (https://www.ncbi.nlm.nih.gov/nucleotide/) under accession numbers OK317735–OK317879 (https://www.ncbi.nlm.nih.gov/popset?DbFrom=nuccore&Cmd=Link&LinkName=nuccore_popset&IdsFromResult=2156341295) and OL539421–OL539424 (https://www.ncbi.nlm.nih.gov/popset?DbFrom=nuccore&Cmd=Link&LinkName=nuccore_popset&IdsFromResult=2183463245). SNP data files are available at https://github.com/nhmvienna/PicaPica_Introgression.
References
Abbott, R., Albach, D., Ansell, S., Arntzen, J. W., Baird, S. J. E., Bierne, N., Boughman, J., Brelsford, A., Buerkle, C. A., Buggs, R., Butlin, R. K., Dieckmann, U., Eroukhmanoff, F., Grill, A., Cahan, S. H., Hermansen, J. S., Hewitt, G., Hudson, A. G., Jiggins, C., & Zinner, D. (2013). Hybridization and speciation. Journal of Evolutionary Biology, 26(2), 229–246. https://doi.org/10.1111/j.1420-9101.2012.02599.x
Abbott, R. J., Barton, N. H., & Good, J. M. (2016). Genomics of hybridization and its evolutionary consequences. Molecular Ecology, 25, 2325–2332. https://doi.org/10.1111/mec.13685
Abellán, P., & Svenning, J. -C. (2014). Refugia within refugia – patterns in endemism and genetic divergence are linked to Late Quaternary climate stability in the Iberian Peninsula. Biological Journal of the Linnean Society, 113, 13–28. https://doi.org/10.1111/bij.12309
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., Selker, E. U., Cresko, W. A., & Johnson, E. A. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE, 3(10), e3376. https://doi.org/10.1371/journal.pone.0003376
Bandelt, H. -J., Forster, P., & Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Barton, N. H., & Hewitt, G. M. (1985). Analysis of hybrid zones. Annual Review of Ecology and Systematics, 16, 113–148. https://doi.org/10.1146/annurev.es.16.110185.000553
Birkhead, T. R. (1991). The magpies: The ecology and behaviour of black-billed and yellow-billed magpies. T & AD Poyser.
Bonfield, J. K., Smith, K. F., & Staden, R. (1995). A new DNA sequence assembly program. Nucleic Acids Research, 23(24), 4992–4999. https://doi.org/10.1093/nar/23.24.4992
Bouckaert, R., Heled, J., Kuhnert, D., Vaughan, T., Wu, C. -H., Xie, D., Suchard, M. A., Rambaut, A., & Drummond, A. J. (2014). BEAST 2: A software platform for Bayesian evolutionary analysis. PLOS Computational Biology, 10(4), e1003537. https://doi.org/10.1371/journal.pcbi.1003537
Brelsford, A., Toews, D. P. L., & Irwin, D. E. (2017). Admixture mapping in a hybrid zone reveals loci associated with avian feather coloration. Proceedings of the Royal Society B, 284(1866). https://doi.org/10.1098/rspb.2017.1106
Burt, J. (2006). Syrinx, Version 2.6h. University of Washington, Seattle, USA. Accessed November 2015, from http://SyrinxPC.com/
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A., & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular. Ecology, 22(11), 3124–3140. https://doi.org/10.1111/mec.12354
Charif, R. A., Waack, A. M., & Strickman, L. M. (2008). Raven Pro 1.3 User’s Manual. Cornell Lab of Ornithology, Ithaca, NY.
Cheng, T. -H. (1987). A synopsis of the avifauna of China. Science Press.
Chung, M. Y., Chung, M. G., López-Pujol, J., Ren, M. X., Zhang, Z. Y., & Park, S. J. (2014). Were the main mountain ranges in the Korean Peninsula a glacial refugium for plants? Insights from the congeneric pair Lilium cernuum – Lilium amabile. Biochemical Systematics and Ecology, 53, 36–45. https://doi.org/10.1016/j.bse.2013.12.019
Coyne, J., & Orr, H. A. (2004). Speciation. Sinauer Associates.
Cramp, S., & Perrins, C. M. (Eds.) (1994). Handbook of the birds of Europe the Middle East and North Africa. The birds of the Western Palearctic. Volume VIII. Crows to Finches. Oxford University Press, Oxford.
Crispo, E., Moore, J. -S., Lee-Yaw, J. A., Gray, S. M., & Haller, B. C. (2011). Broken barriers: Human-induced changes to gene flow and introgression in animals. BioEssays, 33, 508–518. https://doi.org/10.1002/bies.201000154
Currat, M., Ruedi, M., Petit, R. J., & Excoffier, L. (2008). The hidden side of invasions: Massive introgression by local genes. Evolution, 62, 1908–1920. https://doi.org/10.1111/j.1558-5646.2008.00413.x
de Kort, S. R., den Hartog, P. M., & ten Cate, C. (2002). Diverge or merge? The effect of sympatric occurrence on the territorial vocalizations of the vinaceous dove Streptopelia vinacea and the ring-necked dove S. capicola. Journal of Avian Biology, 33, 150–158. https://doi.org/10.1034/j.1600-048X.2002.330205.x
de Lattin, G. (1957). Die Ausbreitungszentren der holarktischen Landtierwelt. In O. Pflugfelder (Ed.), Verhandlungen der Deutschen Zoologischen Gesellschaft, vom 21. bis 26. Mai 1956 in Hamburg. Zoologischer Anzeiger, 20. Supplementband, 380–410.
del Hoyo, J., & Collar, N. J. (2016). HBW and BirdLife International Illustrated Checklist of the Birds of the World. 2. Passerines. Barcelona.
Dobzhansky, T. (1940). Speciation as a stage in evolutionary divergence. The American Naturalist, 74(753), 312–321. https://doi.org/10.1086/280899
Drovetski, S. V., Semenov, G., Red’kin, Y. A., Sotnikov, V. N., Fadeev, I. V., & Koblik, E. A. (2015). Effects of asymmetric nuclear introgression, introgressive mitochondrial sweep, and purifying selection on phylogenetic reconstruction and divergence estimates in the pacific clade of Locustella warblers. PLoS ONE, 10(4), e0122590. https://doi.org/10.1371/journal.pone.0122590
Drummond, A. J., Rambaut, A., Shapiro, B., & Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution, 22(5), 1185–1192. https://doi.org/10.1093/molbev/msi103
Drummond, A. J., Suchard, M. A., Xie, D., & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29(8), 1969–1973. https://doi.org/10.1093/molbev/mss075
Dufresnes, C., & Martínez-Solano, I. (2020). Hybrid zone genomics supports candidate species in Iberian Alytes obstetricans. Amphibia-Reptilia, 41(1), 105–112. https://doi.org/10.1163/15685381-20191312
Earl, D. A., & von Holdt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4, 359–361. https://doi.org/10.1007/s12686-011-9548-7
Ebels, E. B. (2003). Trends in systematics. Speciation in Pica Magpies. Dutch Birding, 25, 103–116.
Edwards, S. V., Kingan, S. B., Calkins, J. D., Balakrishnan, C. N., Jennings, W. B., Swanson, W. J., & Sorenson, M. D. (2005). Speciation in birds: Genes, geography, and sexual selection. Proceedings of the National Academy of Sciences, 102(1), 6550–6557. https://doi.org/10.1073/pnas.0501846102
Edwards, S. V., Shultz, A. J., & Campbell-Staton, S. C. (2015). Next-generation sequencing and the expanding domain of phylogeography. Folia Zoologica, 64(3), 187–206. https://doi.org/10.25225/fozo.v64.i3.a2.2015
Eguchi, K., & Kubo, H. (1992). The origin of the magpie Pica pica sericea in Japan – an investigation of historical records. Journal of the Yamashina Institute for Ornithology, 24, 32–39 (in Japanese with English abstract). https://doi.org/10.3312/jyio1952.24.32
Elgvin, T. O., Trier, C. N., Tørresen, O. K., Hagen, I. J., Lien, S., Nederbragt, A. J., Ravinet, M., Jensen, H., & Sætre, G. P. (2017). The genomic mosaicism of hybrid speciation. Science Advances, 3(6), e1602996. https://doi.org/10.1126/sciadv.1602996
Ellegren, H., Smeds, L., Burri, R., Olason, P. I., Backstrom, N., Kawakami, T., Kunstner, A., Makinen, H., Nadachowska-Brzyska, K., Qvarnstrom, A., Uebbing, S., & Wolf, J. B. W. (2012). The genomic landscape of species divergence in Ficedula flycatchers. Nature, 491, 756–760. https://doi.org/10.1038/nature11584
Excoffier, L., & Lischer, H. E. L. (2010). Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier, L., Foll, M., & Petit, R. J. (2009). Genetic consequences of range expansions. Annual Review of Ecology, Evolution, and Systematics, 40, 481–501. https://doi.org/10.1146/annurev.ecolsys.39.110707.173414
Feulner, P. G. D., & De-Kayne, R. (2017). Genome evolution, structural rearrangements and speciation. Journal of Evolutionary Biology, 30(8), 1488–1490. https://doi.org/10.1111/jeb.13101
Fok, K. W., Wade, C. M., & Parkin, D. T. (2002). Inferring the phylogeny of disjunct populations of the azure-winged magpie Cyanopica cyanus from mitochondrial control region sequences. Proceedings of the Royal Society B, 269(1501), 1671–1679. https://doi.org/10.1098/rspb.2002.2057
Freeland, J. R., & Boag, P. T. (1999). Phylogenetics of Darwin’s finches: Paraphyly in the tree-finches, and two divergent lineages in the warbler finch. The Auk, 116(3), 577–588. https://doi.org/10.2307/4089320
Fu, Y. -X. (1997). Statistical test of neutrality against population growth, hitchhiking and background selection. Genetics, 147(2), 915–925.
Games, P. A., & Howell, J. F. (1976). Pairwise multiple comparison procedures with unequal N’s and/or variances: A Monte Carlo study. Journal of Educational Statistics, 1(2), 113–125. https://doi.org/10.2307/1164979
Garcia-Elfring, A., Barrett, R. D. H., Combs, M., Davies, T. J., Munshi-South, J., & Millien, V. (2017). Admixture on the northern front: Population genomics of range expansion in the white-footed mouse (Peromyscus leucopus) and secondary contact with the deer mouse (Peromyscus maniculatus). Heredity, 119, 447–458. https://doi.org/10.1038/hdy.2017.57
Gill, F., Donsker, D. & Rasmussen, P. (Eds.). (2021). IOC World Bird List (v11.2). https://doi.org/10.14344/IOC.ML.11.2
Gómez, A., & Lunt, D. H. (2007). Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula. In Weiss S., Ferrand N. (Eds.), Phylogeography of southern European refugia (pp. 155–188). Springer, Dordrecht. https://doi.org/10.1007/1-4020-4904-8_5
Goodwin, D. (1986). Crows of the world. Seattle, WA.
Goroshko, O. A., Kryukov, A. P., Songtao, L., Huashan, D., & Bazyr-ool, B. K. (2018). On distribution, subspecies and taxonomic rank of the magpie (Pica pica) in the Hailar-Argun’ river basin (north-east China and Transbaikalia, Russia). Baikal Zoological Journal, 2(23), 38–45. (In Russian, English summary).
Grummer, J., & A., Avila, L. J., Morando, M. M. & Leach, A. D. (2021). Four species linked by three hybrid zones: Two instances of repeated hybridization in one species group (genus Liolaemus). Frontiers in Ecology and Evolution, 9, 624109. https://doi.org/10.3389/fevo.2021.624109
Gurgul, A., Miksza-Cybulska, A., Szmatoła, T., Semik-Gurgul, E., Jasielczuk, I., Bugno-Poniewierska, M., Figarski, T., & Kajtoch, L. (2019). Evaluation of genotyping by sequencing for population genetics of sibling and hybridizing birds: An example using Syrian and Great Spotted Woodpeckers. Journal of Ornithology., 160, 287–294. https://doi.org/10.1007/s10336-018-1601-9
Haavie, J., Borge, T., Bures, S., Garamszegi, L. Z., Lampe, H. M., Moreno, J., Qvarnstrom, A., Torok, J., & Saetre, G. P. (2004). Flycatcher song in allopatry and sympatry – convergence, divergence and reinforcement. Journal of Evolutionary Biology, 17(2), 227–237. https://doi.org/10.1111/j.1420-9101.2003.00682.x
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium 41, 95–98. https://doi.org/10.14601/Phytopathol_Mediterr-14998u1.29
Haring, E., Gamauf, A., & Kryukov, A. (2007). Phylogeographic patterns in widespread corvid birds. Molecular Phylogenetics and Evolution, 45(3), 840–862. https://doi.org/10.1016/j.ympev.2007.06.016
Harpending, H. C. (1994). Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biology., 66(4), 591–600.
Harrison, R. G., & Larson, E. L. (2014). Hybridization, introgression, and the nature of species boundaries. Journal of Heredity, 105(S1), 795–809. https://doi.org/10.1093/jhered/esu033
Harrison, R. G., & Larson, E. L. (2016). Heterogeneous genome divergence, differential introgression, and the origin and structure of hybrid zones. Molecular Ecology, 25(11), 2454–2466. https://doi.org/10.1111/mec.13582
Harvey, M. G., Smith, B. T., Glenn, T. C., Baircloth, B. C., & Rumfield, R. T. (2016). Sequence capture versus restriction site associated DNA sequencing for shallow systematics. Systematic Biology, 65(5), 910–924. https://doi.org/10.1093/sysbio/syw036
Helb, H. W., Dowsett-Lemaire, F., Bergmann, H. H., & Condrads, K. (1985). Mixed singing in the European songbirds – a review. Zeitschrift Für Tierpsychologie, 69(1), 27–41.
Herrando, S., & Quesada, J. (2012). Urraca común, Pica pica. En, SEO/BirdLife. Atlas de las aves en invierno en España 2007–2010. Ministerio de Agricultura, Alimentación y Medio Ambiente-SEO/BirdLife. Madrid.
Hewitt, G. M. (1999). Post-glacial recolonization of European Biota. Biological Journal of the Linnean Society, 68(1–2), 87–112. https://doi.org/10.1006/bijl.1999.0332
Hewitt, G. M. (2001). Speciation, hybrid zones and phylogeography – or seeing genes in space and time. Molecular Ecology, 10(3), 537–549. https://doi.org/10.1046/j.1365-294x.2001.01202.x
Hewitt, G. M. (2011). Quaternary phylogeography: The roots of hybrid zones. Genetica, 139(5), 617–638. https://doi.org/10.1007/s10709-011-9547-3
Horimoto, T. (2004). Records of magpie Pica pica in Iburi district, south-western Hokkaido. Journal of the Yamashina Institute for Ornithology, 36(1), 87–90 (in Japanese with English abstract). https://doi.org/10.3312/jyio.36.87
Howard, D. J. (1993). Reinforcement: Origin, dynamics, and fate of an evolutionary hypothesis. In R. G. Harrison (Ed.), Hybrid zones and the evolutionary process (pp. 46–69). Oxford Univ. Press.
Irwin, D. E., Rubtsov, A. G., & Panov, E. N. (2009). Mitochondrial introgression and replacement between yellowhammers (Emberiza citrinella) and pine buntings (Emberiza leucocephalos) (Aves: Passeriformes). Biological Journal of the Linnean Society, 98(2), 422–438. https://doi.org/10.1111/j.1095-8312.2009.01282.x
Irwin, D. (2020). Assortative mating in hybrid zones is remarkably ineffective in promoting speciation. American Naturalist, 195(6), E150–E167. https://doi.org/10.1086/708529
Jacobs, A., Hughes, M. R., Robinson, P. C., Adams, C. E., & Elmer, K. R. (2018). The genetic architecture underlying the evolution of a rare piscivorous life history form in brown trout after secondary contact and strong introgression. Genes, 9(6), 280. https://doi.org/10.3390/genes9060280
Key, K. H. L. (1968). The concept of stasipatric speciation. Systematic Biology, 17(1), 14–22. https://doi.org/10.1093/sysbio/17.1.14
Kinoshita, G., Nunome, M., Kryukov, A. P., Kartavtseva, I. V., Han, S.-H., Yamada, F., & Suzuki, H. (2019). Contrasting phylogeographic histories between the continent and islands of East Asia: Massive mitochondrial introgression and long-term isolation of hares (Lagomorpha: Lepus). Molecular Phylogenetics and Evolution, 136, 65–75. https://doi.org/10.1016/j.ympev.2019.04.003
Klinge, M., & Sauer, D. (2019). Spatial pattern of Late Glacial and Holocene climatic and environmental development in Western Mongolia – a critical review and synthesis. Quaternary Science Reviews, 210, 26–50. https://doi.org/10.1016/j.quascirev.2019.02.020
Kryukov, A., Iwasa, M. A., Kakizawa, R., Suzuki, H., Pinsker, W., & Haring, E. (2004). Synchronic east-west divergence in azure-winged magpies (Cyanopica cyanus) and magpies (Pica pica). Journal of Zoological Systematics and Evolutionary Research, 42(4), 342–351. https://doi.org/10.1111/j.1439-0469.2004.00287.x
Kryukov, A. P., Spiridonova, L. N., Mori, S., Arkhipov, VYu., & Red’kin, Y. A., Goroshko, O. A., Lobkov, E. G., & Haring, E. (2017). Deep phylogeographic breaks in magpie Pica pica across the Holarctic: Concordance with bioacoustics and phenotypes. Zoological Science, 34(3), 185–200. https://doi.org/10.2108/zs160119
Kryukov, A. P., Spiridonova, L. N., Tyunin, A. P., Kryukov, K. A., & Dorda, B. A. (2020). Complete mitochondrial genomes of five subspecies of the Eurasian magpie Pica pica, obtained with Oxford Nanopore MinION, and their interpretation regarding intraspecific taxonomy. Mitochondrial DNA Part B, 5(3), 3792–3793. https://doi.org/10.1080/23802359.2020.1838354
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. https://doi.org/10.1093/molbev/msy096
Latinne, A., Navascués, M., Pavlenko, M., Kartavtseva, I., Ulrich, R. G., Tiouchichine, M. -L., & Catteau, G.,·Sakka, H., Quéré, J. -P., Chelomina, G., Bogdanov, A., Stanko, M., Hang, L.,·Neumann, K., Henttonen, H. & Michaux, J. (2020). Phylogeography of the striped field mouse, Apodemus agrarius (Rodentia: Muridae), throughout its distribution range in the Palaearctic region. Mammalian Biology, 100(1), 19–31. https://doi.org/10.1007/s42991-019-00001-0
Lavrenchenko, L. A. (2014). Hybrid speciation in mammals: Illusion or reality? Biology Bulletin Reviews, 4(3), 198–209. https://doi.org/10.1134/S2079086414030050
Lee, M. Y., Lissovsky, A. A., Park, S. K., Obolenskaya, E. V., Dokuchaev, N. E., Zhang, Y. P., Yu, L., Kim, Y. J., Voloshina, I., Myslenkov, A., Choi, T. Y., Min, M. -S., & Lee, H. (2008). Mitochondrial cytochrome b sequence variation and population structure of Siberian chipmunk (Tamias sibiricus) in Northeastern Asia and population subdivision in South Korea. Molecules and Cells, 26(6), 566–575.
Lee, S. I., Parr, C. S., Hwang, Y., Mindell, D. P., & Choe, J. C. (2003). Phylogeny of magpies (genus Pica) inferred from mtDNA data. Molecular Phylogenetics and Evolution, 29(2), 250–257. https://doi.org/10.1016/s1055-7903(03)00096-4
Levanen, R., Thulin, C. -G., Spong, G., & Pohjoismaki, J. L. O. (2018). Widespread introgression of mountain hare genes into Fennoscandian brown hare populations. PLoS ONE, 13(1), e0191790. https://doi.org/10.1371/journal.pone.0191790
Librado, P., & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25(11), 1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Lin, D., Bi, K., Conroy, C. J., Lacey, E. A., Schraiber, J. G., & Bowie, R. C. K. (2018). Mito-nuclear discordance across a recent contact zone for California voles. Ecology and Evolution, 8(12), 6226–6241. https://doi.org/10.1002/ece3.4129
Liou, L. W., & Price, T. D. (1994). Speciation by reinforcement of premating isolation. Evolution, 48, 1451–1459.
Londei, T. (2018). Association of Pica magpies with grazing ungulates: A clue to the genus’ origins. Rivista Italiana Di Ornitologia, 87(2), 39–42. https://doi.org/10.4081/rio.2017.295
Madge, S., & Burn, H. (1999). Crows and jays: a guide to the crows, jays and magpies of the world. C. Helm (Ed.). London.
Madge, S. (2009). Common Magpie. In J. del Hoyo, A. Elliott, J. Sargatal, D. A. Christie, E. de Juana (Eds.), Handbook of the birds of the world alive. Lynx Edicions, Barcelona.
Madge, S., Christie, D. A., & Kirwan, G. M. (2020). Oriental Magpie (Pica serica). In S. M. Billerman, B. K. Keeney, P. G. Rodewald, & T. S. Schulenberg (Eds.), Birds of the World (version 1.0). Cornell Lab of Ornithology, Ithaca, NY, USA. https://doi.org/10.2173/bow.orimag1.01
Mallet, J. (2005). Hybridization as an invasion of the genome. TRENDS in Ecology and Evolution, 20(5), 229–237. https://doi.org/10.1016/j.tree.2005.02.010
Marler, P. (2004). Bird calls: a cornucopia for communication. In P. Marler & H. Slabbekoorn, H. (Eds.), Nature’s music. The science of birdsong (pp.132–177). Elsevier, San Diego – London.
Marova, I., Shipilina, D., Fedorov, V., Alekseev, V., & Ivanitskii, V. (2017). Interaction between common and Siberian chiffchaff in a contact zone. Ornis Fennica, 94, 66–81.
Martín-Gálvez, D., Dawson, D. A., Horsburgh, G. J., & Burke, T. (2009). Isolation, characterization and chromosome locations of polymorphic black-billed magpie Pica pica (Corvidae, Aves) microsatellite loci. Molecular Ecology Resources, 9(6), 1506–1512. https://doi.org/10.1111/j.1755-0998.2009.02703.x
Martínez, J. G. (2016). Urraca – Pica pica. In A. Salvador & M. B. Morales (Eds.), Enciclopedia Virtual de los Vertebrados Españoles. Museo Nacional de Ciencias Naturales, Madrid. https://doi.org/10.20350/digitalCSIC/8709
Martínez, J. G., Soler, M., & Soler J. J. (2003). Urraca, Pica pica. In R. Martí & J. C. Del Moral (Eds.), Atlas de las Aves Reproductoras de España. Dirección General de Conservación de la Naturaleza-Sociedad Española de Ornitología. Madrid.
Martínez, J. G., Soler, J. J., Soler, M., Moller, A. P., & Burke, T. (1999). Comparative population structure and gene flow of a brood parasite, the Great spotter cuckoo (Clamator glandarius), and its primary host, the Magpie (Pica pica). Evolution, 53(1), 269–278. https://doi.org/10.1111/j.1558-5646.1999.tb05352.x
Matyushkin, E. A. (1976). European – East Asian interrupted ranges of terrestrial vertebrates. Zoological Journal, 55(9), 1277–1291. (In Russian).
Mavárez, J., & Linares, M. (2008). Homoploid hybrid speciation in animals. Molecular Ecology, 17(19), 4181–4185. https://doi.org/10.1111/j.1365-294X.2008.03898.x
Maxwell, L. M., Walsh, J., Olsen, B. J., & Kovach, A. I. (2021). Patterns of introgression vary within an avian hybrid zone. BMC Ecology and Evolution, 21, 14. https://doi.org/10.1186/s12862-021-01749-1
Mayr, E. (1963). Animal species and evolution. Belknap Press of Harvard University Press; Cambridge.
Melekescev, I. V., Braitseva, O. A., Erlikh, E. N., Shantser, A. E., Chnelebaeva, A. I., Lupikina, E. G., Egorova, I. A., & Kozhemjaka, N. N. (1974). Kamchatka, Kurils and Komandor Islands. History of landscape development in Siberia and Far East. Nauka Press, Moscow (in Russian).
Millette, K. L., Gonzalez, A., & Cristescu, M. A. (2020). Breaking ecological barriers: Anthropogenic disturbance leads to habitat transitions, hybridization, and high genetic diversity. Science of the Total Environment, 740, 140046. https://doi.org/10.1016/j.scitotenv.2020.140046
Miraldo, A., Hewitt, G. M., Paulo, O. S., & Emerson, B. C. (2011). Phylogeography and demographic history of Lacerta lepida in the Iberian Peninsula: Multiple refugia, range expansions and secondary contact zones. BMC Evolutionary Biology, 11, 170. https://doi.org/10.1186/1471-2148-11-170
Mori, S., Hasegawa, O., Eguchi, K., Hayashi, Y., Fujioka, M., Kryukov, A., & Nishiumi, I. (2014). The origin and trend of common magpie in Japan: Microsatellite analysis of old and new introduced populations. Ornithological Science, 13, 59.
Muller, H. J. (1942). Isolating mechanisms, evolution, and temperature. Biology Symposium, 6, 71–125.
Müller, N. F., & du Plessis, L. (2020). Tutorial using BEAST v2.5.x. Skyline plots.
Nazarenko, A. A. (1982). On faunistic cycles (extinction – expansion – extinction…) with special reference to the East Palearctic dendrophilous avifauna. Zhurnal Obshei Biologii, 43(6), 823–835. (in Russian).
Neto, J. M., Arroyo, J. L., Bargain, B., Monros, J. S., Matrai, N., Prochazka, P., & Zehtindjiev, P. (2012). Phylogeography of a habitat specialist with high dispersal capability: The Savi’s Warbler Locustella luscinioides. PLoS ONE, 7(6), e38497. https://doi.org/10.1371/journal.pone.0038497
Nieto, F. G., Álvarez, I., Fuertes-Aguilar, J., Heuertz, M., Marques, I., Moharrek, F., Piñeiro, R., Riina, R., Rosselló, J. A., Soltis, P. S., & Villa-Machío, I. (2017). Is homoploid hybrid speciation that rare? An Empiricist’s View. Heredity, 118(6), 513–516. https://doi.org/10.1038/hdy.2017.7
Nosil, P., Funk, D. J., & Ortíz-Barrientos, D. (2009). Divergent selection and heterogeneous genomic divergence. Molecular Ecology, 18(3), 375–402. https://doi.org/10.1111/j.1365-294X.2008.03946.x
Oliveira, R., Randi, E., Mattucci, F., Kurushima, J. D., Lyons, L. A., & Alves, P. C. (2015). Toward a genome-wide approach for detecting hybrids: Informative SNPs to detect introgression between domestic cats and European wildcats (Felis silvestris). Heredity, 115, 195–205. https://doi.org/10.1038/hdy.2015.25
Omland, K. E., Baker, J. M., & Peters, J. L. (2006). Genetic signatures of intermediate divergence: Population history of Old and New World Holarctic ravens (Corvus corax). Molecular Ecology, 15(3), 795–808. https://doi.org/10.1111/j.1365-294X.2005.02827.x
Orr, H. A. (1996). Dobzhansky, Bateson, and the genetics of speciation. Genetics, 144(4), 1331–1335.
Ottenburghs, J. (2018). Exploring the hybrid speciation continuum in birds. Ecology and Evolution, 8(24), 13027–13034. https://doi.org/10.1002/ece3.4558
Ottenburghs, J., Kraus, R. H. S., van Hooft, P., Wieren, S. E., Ydenberg, R. C., & Prins, H. H. T. (2017). Avian introgression in the genomic era. Avian Research, 8, 30. https://doi.org/10.1186/s40657-017-0088-z
Panov, E. N. (1989). Natural hybridization and ethological isolation in birds. Nauka.
Patil, I. (2021). Visualizations with statistical details: the ‘ggstatsplot’ approach. Journal of Open Source Software, 6(61), 3167. https://doi.org/10.21105/joss.03167
Patterson, N., Price, A. L., & Reich, D. (2006). Population structure and eigenanalysis. PLOS. Genetics, 2(12), e190. https://doi.org/10.1371/journal.pgen.0020190
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., & Hoekstra, H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE, 7(5), e37135. https://doi.org/10.1371/journal.pone.0037135
Pfennig, K. S., Kelly, A. L., & Pierce, A. A. (2016). Hybridization as a facilitator of species range expansion. Proceedings of the Royal Society B, 283(1839), 20161329. https://doi.org/10.1098/rspb.2016.1329
Poelstra, J. W., Vijay, N., Bossu, C. M., Lantz, H., Ryll, B., Muller, I., Baglione, V., Unneberg, P., Wikelski, M., Grabherr, M. G., & Wolf, J. B. W. (2014). The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science, 344(6190), 1410–1414. https://doi.org/10.1126/science.1253226
Pons, J. -M., Masson, C., Olioso, G., & Fuchs, J. (2019). Gene flow and genetic admixture across a secondary contact zone between two divergent lineages of the Eurasian Green Woodpecker Picus viridis. Journal of Ornithology, 160, 935–945. https://doi.org/10.1007/s10336-019-01675-6
Pons, J. -M., Sonsthagen, S., Dove, C., & Crochet, P. -A. (2014). Extensive mitochondrial introgression in North American Great Black-backed Gulls (Larus marinus) from the American Herring Gull (Larus smithsonianus) with little nuclear DNA impact. Heredity, 112, 226–239. https://doi.org/10.1038/hdy.2013.98
Poschel, J., Heltai, B., Gracia, E., Quintana, M. F., Velo-Antón, G., Arribas, O., Valdeón, A., Wink, M., Fritz, U., & Vamberger, M. (2018). Complex hybridization patterns in European pond turtles (Emys orbicularis) in the Pyrenean Region. Scientific Reports, 8, 15925. https://doi.org/10.1038/s41598-018-34178-0
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959.
Quilodrán, C. S., Tsoupas, A., & Currat, M. (2020). The spatial signature of introgression after a biological invasion with hybridization. Frontiers in Ecology and Evolution. https://doi.org/10.3389/fevo.2020.569620
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., & Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901–904. https://doi.org/10.1093/sysbio/syy032
Ramos-Onsins, S. E., & Rozas, J. (2002). Statistical properties of new neutrality tests against population growth. Molecular Biology and Evolution, 19(12), 2092–2100. https://doi.org/10.1093/oxfordjournals.molbev.a004034
Razkin, O., Sonet, G., Breugelmans, K., Madeira, M. J., Gómez-Moliner, B. J., & Backeljau, T. (2016). Species limits, interspecific hybridization and phylogeny in the cryptic land snail complex Pyramidula: The power of RADseq data. Molecular Phylogenetics and Evolution, 101, 267–278. https://doi.org/10.1016/j.ympev.2016.05.002
Red’kin, Y. A., Arkhipov, V. Yu. & Zhigir, D. R. (2021). On subspecies taxonomy and nomenclature of Far Eastern subspecies of magpie Pica pica Linnaeus, 1758 of the group “serica”. Russian Journal of Ornithology, 30(2053), 1535–1544.
Remington, C. L. (1968). Suture-zones of hybrid interaction between recently joined biotas. In T. Dobzhansky, M. K. Hecht & W. C. Steere (Eds.). Evolutionary Biology (pp 321–428). Springer, Boston, MA. https://doi.org/10.1007/978-1-4684-8094-8_8
Renoult, J. P., Geeniez, P., Bacquet, P., Benoit, L., & Crochet, P. -A. (2009). Morphology and nuclear markers reveal extensive mitochondrial introgressions in the Iberian Wall Lizard species complex. Molecular Ecology, 18(20), 4298–4315. https://doi.org/10.1111/j.1365-294X.2009.04351.x
Rius, M., & Turon, X. (2020). Phylogeography and the description of geographic patterns in invasion genomics. Frontiers in Ecology and Evolution., 8, 595711. https://doi.org/10.3389/fevo.2020.595711
Rogers, A. R., & Harpending, H. (1992). Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution, 9(3), 552–569. https://doi.org/10.1093/oxfordjournals.molbev.a040727
Rustamov, A. K. (1954). Family Corvidae. In G. P. Dementiev & N. A. Gladkov (Eds.), Birds of Soviet Union (Vol. 5, pp. 13–104). Sovetskaya Nauka.
Salinas, P., Morinha, F., Literak, I., García, J., Milá, B., & Blanco, G. (2021). Genetic diversity, differentiation and historical origin of the isolated population of rooks Corvus frugilegus in Iberia. Journal of Avian Biology, 52(3), e02689. https://doi.org/10.1111/jav.02689
Schmitt, T., & Varga, Z. (2012). Extra-Mediterranean refugia: The rule and not the exception? Frontiers in Zoology, 9, 22. https://doi.org/10.1186/1742-9994-9-22
Schumer, M., Rosenthal, G. G., & Andolfatto, P. (2014). How common is homoploid hybrid speciation? Evolution, 68, 1553–1560. https://doi.org/10.1111/evo.12399
Seehausen, O. (2004). Hybridization and adaptive radiation. Trends in Ecology and Evolution, 19(4), 198–207. https://doi.org/10.1016/j.tree.2004.01.003
Seehausen, O., Takimoto, G., Roy, D., & Jokela, J. (2008). Speciation reversal and biodiversity dynamics with hybridization in changing environments. Molecular Ecology, 17(1), 30–44. https://doi.org/10.1111/j.1365-294X.2007.03529.x
Shipilina, D., Serbyn, M., Ivanitskii, V., Marova, I., & Backstrom, N. (2017). Patterns of genetic, phenotypic, and acoustic variation across a chiffchaff (Phylloscopus collybita abietinus/tristis) hybrid zone. Ecology and Evolution, 7(7), 2169–2180. https://doi.org/10.1002/ece3.2782
Short, L. L. (1969). Taxonomic aspects of avian hybridization. The Auk, 86(1), 84–105. https://doi.org/10.2307/4083543
Slager, D. L., Epperly, K. L., Renee, R. H., Rohwer, S., Wood, C., Van Hemert, C., & Klicka, J. (2020). Cryptic and extensive hybridization between ancient lineages of American crows. Molecular Ecology, 29(5), 956–969. https://doi.org/10.1111/mec.15377
Sloan, D. B., Havird, J. C., & Sharbrough, J. (2017). The on-again, off-again relationship between mitochondrial genomes and species boundaries. Molecular Ecology, 26(8), 2212–2236. https://doi.org/10.1111/mec.13959
Sokal, R. R., & Rohlf, F. J. (2012). Biometry: The principles and practice of statistics in biological research (4th ed.). WH Freeman and Co.
Sommer, R. S., & Nadachowski, A. (2006). Glacial refugia of mammals in Europe: Evidence from fossil records. Mammal Review, 36(4), 251–265. https://doi.org/10.1111/j.1365-2907.2006.00093.x
Song, G., Zhang, R., Alström, P., Irestedt, M., Cai, T., Qu, Y., Ericson, P. G. P., Fjeldså, J., & Lei, F. (2018). Complete taxon sampling of the avian genus Pica (magpies) reveals ancient relictual populations and synchronous Late-Pleistocene demographic expansion across the Northern Hemisphere. Journal of Avian Biology, 49(2), e01612. https://doi.org/10.1111/jav.01612
Stegmann, B. K. (1932). Corvid birds. Birds of the USSR, 6. USSR Acad. Sci. Press, Leningrad (in Russian).
Stepanyan, L. S. (2003). Conspectus of the ornithological fauna of Russia and adjacent territories (within the borders of the USSR as a historic region). Academkniga, Moscow (in Russian).
Tajima, F. (1989). Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics, 123(3), 585–595.
Taylor, S. A., & Larson, E. L. (2019). Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nature Ecology & Evolution, 3(2), 170–177. https://doi.org/10.1038/s41559-018-0777-y
Toews, D. P. L., & Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology, 21(16), 3907–3930. https://doi.org/10.1111/j.1365-294X.2012.05664.x
Toews, D. P. L., Lovette, I. J., Irwin, D. E., & Brelsford, A. (2018). Similar hybrid composition among different age and sex classes in the Myrtle–Audubon’s warbler hybrid zone. The Auk, 135(4), 1133–1145. https://doi.org/10.1642/AUK-18-45.1
Tugarinov, A. (1932). Birds of East Mongolia on observations of the expedition of 1928. Acad. Sci. Press, Leningrad (in Russian).
Turner, T. L., Hahn, M. W., & Nuzhdin, S. V. (2005). Genomic islands of speciation in Anopheles gambiae. PLOS Biology, 3(9), e285. https://doi.org/10.1371/journal.pbio.0030285
Vokurková, J., Petrusková, T., Reifová, R., Kozman, A., Morkovský, L., Kipper, S.,Weiss, M., Reif, J., Dolata, P. T., & Petrusek, A. (2013). The causes and evolutionary consequences of mixed singing in two hybridizing songbird species (Luscinia spp.). PLoS One, 8(4), 60–172. https://doi.org/10.1371/journal.pone.0060172
Wilkins, M. R., Seddon, N., & Safran, R. J. (2013). Evolutionary divergence in acoustic signals: Causes and consequences. Trends in Ecology & Evolution, 28(3), 156–166. https://doi.org/10.1016/j.tree.2012.10.002
Zhang, R., Song, G., Qu, Y., Alstrom, P., Ramos, R., Xing, X., Ericson, P. G. P., Fjeldså, J., Wang, H., Yang, X., Kristin, A., Shestopalov, A. M., Choe, J. C., & Lei, F. (2012). Comparative phylogeography of two widespread magpies: Importance of habitat preference and breeding behavior on genetic structure in China. Molecular Phylogenetics and Evolution, 65(2), 562–572. https://doi.org/10.1016/j.ympev.2012.07.011
Zhang, H., Yan, J., Zhang, G., & Zhou, K. (2008). Phylogeography and demographic history of Chinese black-spotted frog populations (Pelophylax nigromaculata): Evidence for independent refugia expansion and secondary contact. BMC Evolutionary Biology, 8, 21. https://doi.org/10.1186/1471-2148-8-21
Zink, R. M., Rohwer, S., Andreev, A. V., & Dittmann, D. L. (1995). Trans-Beringia comparisons of mitochondrial DNA differentiation in birds. The Condor, 97(3), 639–649. https://doi.org/10.2307/1369173
Acknowledgements
We are thankful to the staff of the following institutions for providing samples: Zoological Museum of Moscow State University, Russia; Tissues and DNA Biobank of the National Museum of Natural Sciences, Madrid, Spain; Laboratory of Behavioral Ecology and Evolution, School of Biological Sciences, Seoul National University, Seoul, Korea; Conservation Genome Resource Bank for Korean Wildlife, South Korea; Endemic Species Research Institute, Taiwan; National Museum of Nature and Science, Tsukuba, Japan; Yamashina Institute for Ornithology, Japan; Oberösterreichisches Landesmuseum, Linz, Austria. We are grateful to A. S. Yolgin and I. N. Sheremetyeva for help in a field work, L. N. Spiridonova and B.K. Bazyr-ool in molecular experiments, and M. S. Romanov in statistical analysis of vocalization. The research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation, project no. 121031500274-4, the state research program no. 121032300105-0, and supported by the grant of the Russian Foundation for Basic Research No. 16-04-01304.
Funding
Open access funding provided by University of Vienna.
Author information
Authors and Affiliations
Contributions
AK and LH conceived the study; OG, AK, and VA arranged field trips, collected samples, and recorded calls; AK analyzed the mtDNA sequences, developed all data, prepared figures and tables, and wrote the draft; LH managed ddRAD analysis, conducted parts of the mtDNA analyses, and contributed to the draft; SL and BD provided tissue samples; YR provided morphological data and taxonomic expertise; KK, VA, and MK conducted calculations and data analysis. All authors edited and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kryukov, A.P., Goroshko, O.A., Arkhipov, V.Y. et al. Introgression at the emerging secondary contact zone of magpie Pica pica subspecies (Aves: Corvidae): integrating data on nuclear and mitochondrial markers, vocalizations, and field observations. Org Divers Evol 22, 1037–1064 (2022). https://doi.org/10.1007/s13127-022-00568-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-022-00568-6