
Abstract
Accurate species delimitation is of primary importance in biodiversity assessments and in reconstructing patterns and processes in the diversification of life. However, the discovery of cryptic species in virtually all taxonomic groups unveiled significant gaps in our knowledge of biodiversity. Mimicry complexes are good candidates to source for cryptic species. Indeed, members of mimicry complexes undergo selective pressures on their habitus, which results in strong resemblance even between distantly related species. In this study, we used a multi-locus genetic approach to investigate the presence of cryptic diversity within a group of mimetic day-flying moths whose systematics has long been controversial, the Euro-Anatolian Syntomis. Results showed incongruence between species boundaries and the currently accepted taxonomy of this group. Both mitochondrial and nuclear markers indicate the presence of four, well-distinct genetic lineages. The genetic distance and time of divergence between the Balkan and Italian populations of S. marjana are the same as those found between S. phegea and S. ragazzii, the last two being well-distinct, broadly sympatrically occurring species. The divergence between the two lineages of S. marjana dates back to the Early Pleistocene, which coincided with substantial changes in climatic conditions and vegetation cover in Southern Europe that have likely induced geographic and ecological vicariance. Syntomis populations belonging to the taxa kruegeri (s. str.), albionica and quercii are now considered a separate species from marjana s. str. and are thus distinguished as Syntomis quercii Verity, 1914, bona sp., stat. nov. Our results show that the species richness of mimicry complexes inhabiting temperate regions might still be severely underestimated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accurate species delimitation is of primary importance for understanding tempo and modes of species diversification (Wiens 2007). Historically, new species have mainly been recognized using morphological trait variation (Wiens and Servedio 2000). However, species can also evolve in absence of conspicuous morphological trait divergence, as shown by the discovery of the so-called cryptic species (Bickford et al. 2007). These had originally been spotted based on subtle ecological or behavioural features (Mayr 1963), though the use of genetic markers has nowadays boosted their discovery (Ayala and Powell 1972; Knowlton 1993; Beheregaray and Caccone 2007; Carstens et al. 2013). The recognition of cryptic species virtually across all taxonomic groups (Pfenninger and Schwenk 2007), including many well-studied taxa (Roca et al. 2001; Fennessy et al. 2016), unveiled significant gaps in our knowledge of biological diversity (Beheregaray and Caccone 2007; Struck et al. 2018). These gaps, in turn, constrain our understanding of the mechanisms involved in biological diversification and in the establishment of interactions at the community level, limiting the deployment of measures in many areas of biodiversity management, from conservation biology to pest or disease control (Beheregaray and Caccone 2007; Bálint et al. 2011; Robuchon et al. 2019).
Although cryptic species are widespread throughout the tree of life, they are over-reported in some groups, such as freshwater fishes, deep-sea clams, polychaetes, frogs, mites, parasitic insects and nematodes (reviewed in Pérez-Ponce de León and Poulin 2016). Cryptic species seem to be commoner in animals using non-visual mating signals (e.g. many frogs) or under selection promoting morphological stasis or convergent evolution (e.g. parasites) (Bickford et al. 2007; Struck et al. 2018). They are also common in cases of recent and/or rapid speciation when short divergence time did not allow the accumulation of detectable phenotypic differences (Reidenbach et al. 2012; Gustafsson et al. 2014). However, numerous taxa characterized by life-history traits likely encouraging cryptic diversification have been poorly investigated, and many of them are still analysed with a traditional morphological approach (Bickford et al. 2007; Struck et al. 2018). As a consequence, a clear picture of the distribution of cryptic species across the tree of life is still lacking.
Mimicry complexes are good sources of cryptic species. In particular, cryptic species are expected among closely related species involved in a same Müllerian mimicry ring (Ruxton et al. 2004, cf. Boppré et al. 2017), because phenotypic divergence is constrained by stabilizing selection on shared warning signals (but see Lawrence et al. 2019). Interestingly, it was shown that look-alikeness among co-mimics could also be achieved via introgressive hybridization of pattern-determining genes from related species (Giraldo et al. 2008; Pardo-Diaz et al. 2012; Jiggins 2017). The imbalance between species divergence and phenotypic divergence is a prerequisite for the evolution of cryptic species (Struck et al. 2018). Thus, species involved in mimicry complexes provide a good testing ground to study the contrasting effects of divergence and stasis in the evolution of cryptic species. However, the systematics of these species often results inaccurate or partial, especially if only based on the external morphology. As a matter of fact, the presence of cryptic species within mimicry complexes has predominantly been reported in some well-studied tropical taxa, which turned out as excellent test groups to evaluate how mimicry affects patterns of diversification and speciation (Pfennig 2012, and references therein; Jiggins 2017). Yet mimicry complexes are not limited to the tropical regions.
In this study, we investigated the presence of cryptic species within a group of mimetic moths occurring in temperate regions whose systematics has long been controversial, the Euro-Anatolian Syntomis. These are distasteful, day-flying moths showing aposematic colourations and belonging to a Müllerian mimicry complex that include also several unrelated co-mimics, e.g. yellow melanic morphs of Zygaena ephialtes, Z. transalpina (Zygaenidae) and Callimorpha dominula (Erebidae) (Bullini et al. 1969; Sbordoni et al. 1979; Zilli 1996). Being mainly based on features of the habitus and genital morphology (Obraztsov 1966), the taxonomy of Syntomis moths appears outdated and confused, evidently flawed by the incorrect delimitation of real species boundaries. Disputes also exist on whether the genus Syntomis should be distinguished from the genus Amata—here we treat Syntomis as a distinct genus, in agreement with part of the recent literature (e.g. Schneider et al. 1999; Igniatyev and Zolotuhin 2005; Anikin et al. 2017; de Freina and de Prins 2018), and with the evidence that Amata is polyphyletic (Przybyłowicz et al. 2019: 629–630) whereas Syntomis include a monophyletic set of Palaearctic and Oriental species characterized by synapomorphies and genetic differentiation (see Schneider et al. 1999). In particular, we focused on S. marjana, a taxon ranging from Provence, Sicily and mainland Italy across the Balkan Peninsula, Ukraine and Southern European Russia (de Freina 2008; Fibiger et al. 2011). Based on their disjunct distribution, and slight differences in size, male genitalia, wing shape and spotting, some populations have occasionally been considered distinct species, i.e. albionica (Provence), quercii (Apennines), kruegeri (Sicily), marjana proper (Balkans) and sheljuzhkoi (Caucasus) (Verity 1914; Turati 1917; Stauder 1928-1929; Obraztsov 1966; Dufay 1970; Igniatyev and Zolotuhin 2005). Preliminary genetic studies showed a substantial divergence between two populations of the taxa kruegeri (Mt Pellegrino, Palermo, Sicily) and marjana s. str. (Stari Grad, Croatia), which were proposed to be distinct species (Cianchi et al. 1980; Bullini et al. 1981). Nevertheless, this genetic evidence was not considered anymore in recent taxonomic reviews, which recognized only a single widely distributed species with more subspecies (de Freina and Witt 1987; de Freina 2008; Fibiger et al. 2011). Such discordance on the systematics of the group claims for comprehensive genetic investigations.
Here we provide the first extensive assessment of the genetic variation across some Euro-Anatolian Syntomis taxa. Firstly, we assessed the genetic variation at both nuclear and mitochondrial markers within broad sense ‘S. marjana’ and in two closely related, sympatric species, S. phegea and S. ragazzii; then, we used ordination-based clustering analyses and species delimitation methods to circumscribe putative species; finally, we compared the estimated time of divergence among sympatric and allopatric taxa. The aim of this paper is thence to clarify the taxonomy and systematics of Western Palaearctic Syntomis and disclose the extent of cryptic diversity within a mimicry complex occurring in temperate areas.
Materials and methods
Sampling and laboratory procedures
The sampling strategy was designed to include most of the geographic variation from Syntomis marjana, S. ragazzii and S. phegea. We analysed 1150 specimens from 34 localities representing most of the described subspecies (we did not analyse S. m. odessana, S. m. sheljuzhkoi and S. p. phegea). Sampling localities and sample sizes are given in Table 1 and Fig. 1. Specimens were preliminarily identified following the characters described in Obraztsov (1966), de Freina and Witt (1987) and Fibiger et al. (2011). Biological tissue was stored at − 80° until subsequent analyses. We also obtained two dry specimens from the related species S. nigricornis and S. aequipunta for use in the phylogenetic analysis (see below).

Geographical distribution of Syntomis phegea, S. ragazzii, S. marjana and S. quercii in the Western Palaearctic region, and geographical locations of the 36 populations sampled and analysed. Locations are numbered as in Table 1. Dotted lines outline the approximate range of the different subspecies named in the text. The map was drawn using the software Canvas 11 (ACD Systems of America, Inc.)
Standard horizontal starch gel (10%) electrophoresis was performed to analyse the genetic variation at 22 allozyme loci: α-glycerol phosphate dehydrogenase (α-Gpdh), malate dehydrogenase (Mdh-1 and Mdh-2), isocitrate dehydrogenase (Idh-1 and Idh-2), 6-phosphogluconate dehydrogenase (6Pgdh), glyceraldehyde-3-phosphate dehydrogenase (Gapdh), superoxide dismutase (Sod-1 and Sod-3), xanthine dehydrogenase (Xdh), hexokinase (Hk-1 and Hk-2), adenylate kinase (Adk), esterase (Est-1, Est-2 and Est-5), acid phosphatase (Acph), aldolase (Ald), aconitase (Aco), triosephosphate isomerase (Tpi), mannose phosphate isomerase (Mpi), and phosphoglucomutase (Pgm).
Electrophoretic techniques and staining procedures followed Selander (1971), Ayala et al. (1972) and Harris and Hopkinson (1976), with minor modifications. Alleles were numbered according to their mobility (in mm) with respect to the most common allele (named 100) in a reference population (S. phegea from population 6, Camerata Nuova).
DNA was extracted from the legs of a subsample of available specimens (Table 1) following the standard cetyltrimethylammonium-bromide (CTAB) protocol (Doyle and Doyle 1990). A fragment from the mitochondrial Cytochrome Oxidase I gene (COI) was amplified and sequenced. The polymerase chain reaction (PCR) primers were REVNANCY and PAT2K837 from Simon et al. (1994). Amplifications were performed in a 10-μL reaction volume containing MgCl2 (2 mM), four dNTPs (0.2 mM each), two primers (0.2 μM each), the enzyme Taq polymerase (0.5 U, Promega), its reaction buffer (1X, Promega) and 10–100 ng of DNA template. PCRs were conducted with an initial step at 95 °C for 5 min, 32 cycles at 94 °C for 1 min, 45 s at 57 °C (COI) and 1 min at 72 °C, followed by a single final step at 72 °C for 5 min. Purification and sequencing of PCR products were conducted on both strands by Macrogen Inc. (http://www.macrogen.com), using an ABI PRISM® 3730 sequencing system (Applied Biosystems). All sequences have been deposited in GenBank (accession numbers: MW343496-MW343511).
Allozyme data analysis
The effective number of alleles per locus was calculated with GENALEX (Peakall and Smouse 2006). Allele frequencies, mean observed and expected heterozygosity and proportion of polymorphic loci were computed for each sampling site (population) using BIOSYS-2 (Swofford and Selander 1999). BIOSYS-2 was also used to evaluate departures from the expected Hardy-Weinberg equilibrium for each locus at each sampling site, and the linkage equilibrium between each pair of loci after application of the Bonferroni correction for multiple tests. As no departures from Hardy-Weinberg equilibria were observed, indicating panmixia in all populations, we conducted a population-based assessment of genetic affinities.
A pairwise matrix of unbiased Nei’s genetic distances (Nei 1978) among all populations was computed by BIOSYS-2. Clustering of populations was assessed via principal coordinate analysis (PCoA) of Nei’s distances among populations, as implemented in GENALEX. PCoA-defined clusters were then compared with candidate taxa, and a matrix of fixed differences (i.e. fully diagnostic loci) among them was generated.
Sequence data analysis, species delimitation and phylogenetic reconstruction
Electropherograms of sequences were visually checked using FinchTV 1.4.0 (Geospiza Inc.) and aligned using Clustal X 2.0 (Larkin et al. 2007). Nucleotide variation was assessed using MEGA 6.0 (Tamura et al. 2013); haplotype and nucleotide diversity were estimated using DnaSP 5.10 (Librado and Rozas 2009).
We applied three different species delimitation methods traditionally employed to delimit groups of sequences that potentially correspond to distinct species: (i) Automatic Barcode Gap Discovery (ABGD; Puillandre et al. 2012a) that detects gaps in the distribution of pairwise genetic distances, assuming that it corresponds to a threshold between intra- and inter-specific distances; (ii) General Mixed Yule Coalescent model (GMYC; Pons et al. 2006), which starts from an ultrametric tree and tests whether the branching rates fit better with a coalescent model or a speciation model, using the transition point between speciation and coalescence to delimit species; and (iii) Bayesian implementation of the Poisson Tree Processes (bPTP; Zhang et al. 2013), which also compares speciation and coalescent models but relies on the substitution rates calculated for each node instead of the branching rates. These three methods were chosen because they proved to be effective in recognizing cryptic species either in large and small groups of sequences, and are among the most widely used with mtDNA data (Fujita et al. 2012; Puillandre et al. 2012a, b; Zhang et al. 2013; Schwarzfeld and Sperling 2015).
ABGD analysis was performed on the ABGD web server platform (http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html), using the default parameters (gap width X = 1.5, prior intraspecific divergences from P = 0.001 to P = 0.1, with 20 steps) and the Kimura-2-parameter (K80) model to compute a pairwise genetic distance matrix. The phylogenetic trees used as input for the GMYC and bPTP analyses were generated using the Bayesian method implemented in BEAST 1.8.1 (Bayesian evolutionary analysis by sampling trees; Drummond et al. 2012). We selected the Yule pure-birth speciation model as tree prior, a strict clock model and the best-fit model of molecular evolution estimated by Jmodeltest 2.1.3 (Darriba et al. 2012) under the Bayesian Information Criterion, i.e. the TIM2 transitional model with a proportion of invariant sites (+I). The Markov chain Monte Carlo (MCMC) length was 10 million generations, sampling trees every 1000 generations. The independence of the effective sample size (ESS values »200) for the estimated parameters was evaluated using Tracer 1.6, after removing the first 10% of samples as burn-in. The consensus tree was then generated by TreeAnnotator 1.8.1 (BEAST package), using the maximum clade credibility criterion, after removing the first 1000 sampled trees as burn-in. We included in the analysis also the sequences of S. nigricornis and S. aequipuncta. The GMYC analysis was then run using the splits R package (Ezard et al. 2015), applying both the single threshold and the multiple threshold methods. bPTP analysis was conducted on the web server platform (https://species.h-its.org/ptp/), with a 100,000 MCMC length and a 10% burn-in.
The phylogenetic relationships among the candidate species were inferred using maximum likelihood (ML) and Bayesian methods. The ML-tree was generated using the algorithm implemented in IQTREE (Nguyen et al. 2014) applying the TIM2+I model of substitution; the robustness of the inferred tree topology was assessed using the non-parametric bootstrap method with 1000 pseudo-replicates, and the SH-like approximate likelihood ratio test (SH-aLRT), also with 1000 bootstrap replicates. The Bayesian phylogenetic analysis was performed using the time-calibrated procedure implemented in BEAST 1.8.1 (Drummond and Rambaut 2007), to get a comparative framework of the times of divergence among the candidate species and to get an approximate historical contextualization of the speciation events. In the absence of internal calibration points derived from fossil data or geologic events directly linked to the evolutionary history of Syntomis, to time-calibrate the tree we used an uncorrelated relaxed clock model and a fixed substitution rate of 0.0015 per site per million of years, i.e. the mean COI substitution rate estimated for arthropods by Brower (1994). Although the use of non-fossil-based calibrations is usually discouraged (Schenk 2016), this option is often the only available for most of the soft-bodied invertebrates, and it continues to be extensively used in dating divergences in insects, including Lepidoptera (e.g. Hinojosa et al. 2018). We chose the substitution rate by Brower (1994) among the several available ones (Papadopoulou et al. 2010) since it falls in the middle of the ranges estimated for this gene fragment in insects, and it is so far the most used, facilitating comparative inferences. The Yule pure-birth speciation model was chosen as tree prior, and the TIM2+I as substitution model. Two independent runs were performed, each with a Markov chain Monte Carlo (MCMC) length of 10 million generations, with sampling every 1000 generations. The independence of the estimated parameters (ESS values > 200) and the convergence between runs were evaluated using Tracer 1.6, after removing the first 10% of samples as burn-in. The two runs were combined using LogCombiner 1.8.1 (BEAST package), and an annotated maximum clade credibility tree was computed with TreeAnnotator 1.8.1.
Results
Allozyme data analysis
Among the 22 loci analysed, four (Sod-1, Sod-3, α-Gpdh, Gapdh) resulted monomorphic in all studied populations. The allele frequencies at the 18 polymorphic loci are given in the Supplementary Table 1. The overall number of alleles observed at each polymorphic locus varied from two to nine (Pgm). No departures from both the Hardy-Weinberg and linkage equilibria were observed (no significant value after the Bonferroni correction). The mean sample size per locus, the effective number of alleles and the observed and expected heterozygosity for each population are shown in Table 1. The mean sample size ranged from 3.5 (pop. 28, S. m. kruegeri, from Rocca Busambra) to 38.1 (pop. 6, S. phegea from Camerata Nuova); the average number of alleles per locus varied from 1.1 (several samples) to 1.5 (pop. 8, S. phegea from Monte Faito, and pop. 15, S. ragazzii from Monte Faito); the mean observed heterozygosity ranged from 0.006 (pop. 2, S. phegea from Capriana) to 0.097 (pop. 34, S. m. marjana from Stari Grad); the mean expected heterozygosity ranged from 0.006 (pop. 2, S. phegea from Capriana) to 0.095 (pop. 16, S. ragazzii from Roccamandolfi).
The pairwise matrix of population genetic distances D (Nei 1978) is given in Supplementary Tab. 2, whereas a heatmap of the D values is shown in Fig. 2a. The inspection of the heatmap revealed four main clusters of populations with lower values of genetic distance, which coincided with populations of S. phegea, S. ragazzii, S. m. marjana, and S. m. kruegeri + S. m. quercii + S. m. albionica. The mean genetic distances observed within these groups are 0.002, 0.013, 0.013 and 0.017, respectively, which are values commonly observed at the intraspecific level (Bullini and Sbordoni 1980). The mean genetic distances among these groups range from 0.338 (S. phegea vs S. m. marjana) to 0.629 (S. ragazzii vs S. m. kruegeri + S. m. quercii + S. m. albionica); the genetic distance between S. m. marjana and S. m. kruegeri + S. m. quercii + S. m. albionica was 0.432; these values agree with those commonly observed among closely related species (Bullini and Sbordoni 1980). The highest values of genetic distance observed within groups are 0.076, between southern (pop. 34, Stari Grad) and northern (pop. 31, Sgonico) populations of S. m. marjana; 0.075, between S. m. kruegeri (pop. 28, Rocca Busambra) and S. m. quercii (pop. 24, Aschi Alto); and 0.063, between S. r. asperomontana (pop. 21, S. Stefano in Aspromonte) and S. r. ragazzii (pop. 16, Roccamandolfi); these values agree with those commonly observed among subspecies (Bullini and Sbordoni 1980). A summarized matrix of the mean genetic distances observed within and among these groups is given in Table 2.

a Heatmap representing the pairwise matrix of population genetic distance (D Nei 78) among the Western Palaearctic Syntomis populations analysed in this study, based on 22 allozyme loci; warmer colours indicate higher genetic identity. b Principal coordinate analysis of the 31 analysed populations, based on the unbiased Nei genetic distance (D Nei 78), calculated on 22 allozyme loci; number in parentheses refers to the proportion of variance explained by the first two principal coordinates. The graph was drawn using the software Canvas 11 (ACD Systems of America, Inc.)
The ordination-based clustering analysis resulting from PCoA also defined four main clusters of populations (Fig. 2b). The first and second PCoA axes explained 40.48% and 34.10% of the total variance, respectively. Populations of S. m. kruegeri + S. m. quercii + S. m. albionica were grouped in a distinct and well-defined cluster with respect to populations of S. m. marjana, which were also combined into a single cluster; the other two clusters coincided with populations of S. phegea and S. ragazzii.
The four clusters were also defined by some fully diagnostic loci, resumed in Table 2. We found four 100% diagnostic loci between S. phegea and S. m. marjana, five between S. marjana and S. m. kruegeri + S. m. quercii + S. m. albionica, five between S. m. marjana and S. ragazzii, seven between S. ragazzii and S. m. kruegeri + S. m. quercii + S. m. albionica, seven between S. ragazzii and S. phegea, and nine between S. phegea and S. m. kruegeri + S. m. quercii + S. m. albionica.
Sequence data
We amplified and sequenced a 781-bp fragment from the final section of the mitochondrial COI gene from 38 Syntomis individuals. No indels, stop codons or nonsense codons were observed. We found 16 different haplotypes defined by 81 (10.3%) variable positions, of which 68 (8.7%) were parsimony informative. The mean haplotype diversity (h) and nucleotide diversity (π) values for this dataset were 0.950 (± 0.021 SD) and 0.032 (± 0.008 SD), respectively. A full list of the haplotype found within each studied population is presented in Table 1.
The results from the species delimitation analyses are resumed in Table 3. ABGD recognized a gap in the pairwise distance distribution between 0.013 and 0.035. Taking the prior maximal distance (Pmax) threshold within this gap, ABGD computed a primary partition with six groups, coinciding with S. phegea, S. ragazzii, S m. marjana, S. m. kruegeri + S. m. quercii + S. m. albionica, S. nigricornis and S. aequipuncta. The secondary partition, achieved by the recursive analysis, recognized the same groupings when using Pmax values included in the gap; further splits were recognized for lower values: northern S. m. marjana from southern S. m. marjana (for Pmax = 0.0077), and S. m. kruegeri from S. m. albionica + quercii (for Pmax = 0.0046). GMYC single-threshold analysis rejected the null hypothesis of a coalescent model (likelihood ratio 6.415; LR test: 0.04, significant) and recognized six candidate species, which coincided with those recovered from ABGD analyses. The multiple-threshold analysis also rejected the coalescent model (likelihood ratio 6.873; LR test: 0.03, significant), but suggested further splits between northern and southern S. m. marjana and between S. m. kruegeri and S. m. albionica + quercii (as in ABGD analysis). Finally, bPTP returned eight putative species, coinciding with S. nigricornis (support = 1.0), S. aequipuncta (support = 1.0), S. phegea (support = 1.0), S. ragazzii (support = 0.79), northern S. m. marjana (support = 0.87), southern S. m. marjana (support = 0.94), S. m. kruegeri (not supported) and S. m. albionica + S. m. quercii (not supported).
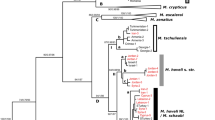
The ML-tree retrieved by IQTREE (log-likelihood score: − 1608.30, s.e. 61.93) recognized six main branches, which were consistent with results from the species delimitation methods (Fig. 3a). The sequences of the taxa kruegeri, quercii and albionica clustered together (high support = 96.4/98%), and form the sister clade (high support = 87.4/88%) of S. m. marjana clade (high support = 98/100%). The sequences of S. ragazzii form a unique supported cluster (high support = 96.5/79%), which resulted more related to S. nigricornis (moderate support = 80/66%) than S. phegea, whereas S. phegea resulted more related to S. aequipuncta albeit with weak support (77/54%).

Phylogenetic relationships among the 16 mtDNA haplotypes observed. a Maximum likelihood tree retrieved by the analysis in IQTREE; support values at the relevant nodes are SH-aLRT support (%) and standard bootstrap support (%) based on 1000 replicates. b Maximum clade credibility tree recovered by the Bayesian analysis in BEAST, showing the divergence time from the most recent common ancestor (TMRCA) for the major clades; node bars (in blue) represent 95% highest posterior density (HPD) intervals for node ages; posterior probabilities for each node are also shown; MYA: million years ago. The graph was drawn using the software Canvas 11 (ACD Systems of America, Inc.)
The time-calibrated phylogenetic analysis conducted in BEAST returned two congruent runs that fully converged on a stationary distribution with satisfactory ESS values (>>200) for all the parameters of interest. A chronogram based on the maximum clade credibility (MCC) tree is presented in Fig. 3b. The tree topology was consistent with both the ML-tree and the species delimitation analyses, as it showed six main branches corresponding with S. phegea, S. aequipuncta, S. nigricornis, S. ragazzii, S. m. kruegeri + S. m. quercii + S. m. albionica and S. m. marjana. The time to the most recent common ancestor (TMRCA) for all the analysed taxa was estimated at 2.87 million years ago (mya) (95% HPD:1.87–4.12); TMRCA between S. m. kruegeri + S. m. quercii + S. m. albionica and S. m. marjana at 1.99 mya (95% HPD: 1.11–2.96), roughly the same as that between S. phegea and S. aequipuncta (mean: 1.92 mya, 95% HPD: 0.93–3.05), and higher than that estimated for S. ragazzii and S. nigricornis (mean: 1.29, 95% HPD: 0.66–2.07). The estimated TMRCA for the clades coinciding with S. m. marjana, S. m. kruegeri + S. m. quercii + S. m. albionica and S. ragazzii were 0.61 mya (95% HPD: 0.26–1.1), 0.37 mya (95% HPD: 0.10–0.76) and 0.36 mya (95% HPD: 0.08–0.67), respectively.
Discussion
Both mitochondrial and nuclear markers consistently supported the existence of two distinct, highly differentiated, genetic clusters within S. marjana, distributed West and East of the Adriatic Sea, respectively. The extent of the genetic differentiation between these two clusters and the estimated time of their divergence are of the same magnitude of those occurring between other well-recognized species of this complex. Furthermore, all species delimitation methods clearly point to these two genetic clusters as distinct species. These results are consistent with preliminary evidence showing the genetic divergence between the taxa kruegeri and marjana s. str. (Cianchi et al. 1980; Bullini et al. 1981), and suggest considering the populations of the lineage kruegeri + quercii + albionica as a distinct species, under the oldest available name having priority, i.e. S. quercii Verity 1914 (see below). Our data also highlight further genetic sub-structuring at the intraspecific level, suggesting a role for recent Pleistocene events in triggering population-level genetic differentiation.
The taxa kruegeri, marjana and quercii were originally described as aberrations or subspecies of other species, the first two of S. phegea (Ragusa 1904; Stauder 1913), the last one of S. mestralii (a species from the Middle East). Subsequently, kruegeri and marjana were considered valid species by Turati (1917), who ranked quercii as a subspecies of marjana. However, the substantial similarity of these taxa in habitus, habitat preferences (dry grasslands), and phenology led most authors to combine them into a unique polytypic species (Obraztsov 1966; Bertaccini et al. 1997; Igniatyev and Zolotuhin 2005; Freina 2008; Fibiger et al. 2011). Despite this, we detected substantial genetic differentiation between S. marjana s. str. and S. quercii (the latter redefined here to comprise also the taxa albionica and kruegeri) that can be attributed to a speciation event occurred at some point in the Early Pleistocene or before. Our estimate of the divergence time between these two lineages is probably biased by the use of a non-fossil-based calibration of the molecular clock (Papadopoulou et al. 2010; Schenk 2016), but it provides an approximate timing of their split. Indeed, the great changes in bioclimatic conditions characterizing the Plio-Pleistocene transition prompted speciation in several Mediterranean taxa (Hewitt 1996, 2011), including Lepidoptera (Schmitt 2007), and likely affected the evolutionary history of European Syntomis. These changes consisted of a generalized increase of drought and a reduction of the forest habitats in Southern Europe (Hewitt 2011), followed by the beginning of glacial cycles, that resulted in alternate expansion and retreat of xeric and forest habitats (Fauquette et al. 1999; Suc and Popescu 2005; Abrantes et al. 2010). Most European Syntomis are thermophilous (with the partial exception of S. phegea), occur in dry habitats, and are distributed in South-Eastern Europe with several relatives in steppe areas from the Middle-East to Central Asia, that likely represents the centre of origin of this group (“phegea-Gruppe” of Obraztsov 1966). The common ancestor of S. marjana and S. quercii might have benefited from the increase of aridity at the end of Pliocene (Malatesta 1985; Fauquette et al. 1999) by spreading across South-Eastern Europe. Then, with the onset of the first glacial cycles, the cooler conditions and the spreading of broad-leaved forests in the Italian and Balkan Peninsulas (Abrantes et al. 2010) could have trapped the ancestor in the south of both peninsulas, where populations would have found relatively hot and dry refugia. This vicariance process has been claimed as the most likely explanation for the existence of several pairs of sibling species with allo-parapatric distribution in the Italian and Balkan peninsulas (Racheli and Zilli 1985). The Isonzo river in NE Italy and Slovenia, which virtually separates the distribution of these species, has been considered a suture zone and biogeographical boundary for several vertebrate and invertebrate species (Taberlet et al. 1998; Hewitt 1999). However, in our case it is hard to define a boundary between these two species because most of the populations west of the Isonzo river have not been found anymore over the last 50 years and could not be genotyped. Further genetic assays on museum specimens are therefore required.
Pleistocene climatic changes also support the origin of intraspecific genetic sub-structuring. Although our data do not allow fine-scale phylogeographic inferences, they reveal sub-structuring in S. quercii, S. ragazzii and S. marjana s. str., which can be explained by the interaction between climate fluctuations and the physiographic heterogeneity of the Italian and Balkan peninsulas. In particular, we found genetic differentiation between the Sicilian and Apennine populations of S. quercii, between the central and southernmost Apennine populations of S. ragazzii, and between the northern and southern populations of S. marjana (Fig. 3 and Supplementary Tab. 2). Substantial intraspecific differentiation between Sicilian and mainland populations, and between the southernmost and central Apennine populations, is commonly observed in phylogeographic studies on Italian species (e.g. Canestrelli and Nascetti 2008; Canestrelli et al. 2010; Chiocchio et al. 2019; Scalercio et al. 2020). These patterns can be related to the presence of several geographic discontinuities that acted as ecological and physiographic barriers at some point during the Pleistocene. In fact, during Pleistocene, this area has been affected by several glacio-eustatic oscillations of the sea level (Bonfiglio et al. 2002) that induced repeated fragmentations of populations in distinct sub-refugia. The genetic isolation within distinct sub-refugia led populations toward genetic differentiation in more distinct genetic lineages, a process that boosted the evolution of biodiversity hotspots in southern Italy as well as in the other Mediterranean peninsulas (Hewitt 2011). This scenario, commonly named as “refugia-within-refugia” scenario (Gómez and Lunt 2007), fits well with the geographic pattern of genetic structure observed in S. ragazzii and S. quercii. In contrast, no apparent paleogeographic evidence supports the genetic differentiation observed within S. marjana in a relatively restricted area along the NE Adriatic, with no evident orographic discontinuity. However, this area is characterized by geological discontinuities (Cvetkovic et al. 2016) that affect groundwater availability and, as a consequence, vegetation and bioclimatic conditions (DMEER 2017). These bioclimatic discontinuities likely represented ecological barriers that could have trapped S. marjana populations in distinct climatic refugia, at least during the last part of the Pleistocene. A similar pattern has been outlined in some other species (Hewitt 2011; Pabijan et al. 2015). Further investigations will then be required to track the evolutionary history of S. marjana in the Balkan peninsula during the Pleistocene.
We found a general low level of intra-population genetic diversity in all of the analysed Syntomis species, a pattern quite uncommon in insect populations (Schmitt and Seitz 2004). Such low values may reflect the effects of recent bottlenecks, perhaps induced by the last glaciation, in combination with rapid postglacial range expansions. This seems to be the case of S. phegea. Although we did not analyse populations from the northern and the eastern part of its range, the observed low level of population genetic diversity and genetic differentiation at both nuclear and mitochondrial markers support for the hypothesis of a (very) recent and quick range expansion by this species. On the other hand, we found a latitudinal gradient of population genetic diversity in S. ragazzii, with the southernmost populations showing the highest levels of diversity. This pattern may be ascribed to repeated cycles of fragmentation within the southern sub-refugia (see above) followed by gene admixture, a model used to explain similar patterns of genetic diversity observed in several other taxa with sub-Apennine distribution (Canestrelli et al. 2010). However, as other causes may explain these patterns in Lepidoptera (e.g. Wolbachia infections, Duplouy et al. 2010) we postpone any speculation to further investigations.
In the phylogenetic relationships outlined by mitochondrial DNA, noteworthy is the recovery of S. ragazzii as more closely related to the Anatolian-Caucasian S. nigricornis than S. phegea, despite S. ragazzii and S. phegea being long considered sibling species and can still hybridize (althought limited to F1, Sbordoni et al. 1982). Vicariance between Southern Apennine and Anatolian taxa is common in Lepidoptera and can be ascribed to several biogeographic processes (Racheli and Zilli 1985) other than a direct geological connection between the two regions, the last of which estimated in the late Miocene (Rögl 1999). Interestingly, the estimated divergence between these two species is more recent than that between the other sister pairs. However, more samples of S. nigricornis and other middle-eastern taxa are needed to resolve the biogeographic history of this connection. Finally, the weak support values recovered for the relationship of S. phegea and S. aequipuncta with the lineages of quercii-marjana and ragazzii-nigricornis stress the need to broaden the analysis to more genetic markers and species to fully resolve the evolutionary history of Western Palaearctic Syntomis (see also Przybyłowicz et al. 2019).
Our results highlight that in Euro-Anatolian Syntomis the degree of genetic divergence is not strictly related with that of phenotypic divergence, as expected for species involved into mimicry complexes. These moths happen thus to be at the crossroad between independent evolutionary divergence and constraints limiting their phenotypic diversification (Leimar et al. 2012). Indeed, species belonging to the same Müllerian mimicry ring share the costs of predation, but they have to be perceived very similar by predators. As a consequence, the external appearance of these species is expected to be under strong selection to maintain a common signal. A different set of characters (e.g. morphological, genetic, physiological and behavioural) will reveal their interplay with the various evolutionary forces, which is in turn affected by the allopatric/sympatric occurrence of populations of these moths with other co-mimics. Many Syntomis species are sympatric and coexist in the same on very close biotopes, at least to the scale of bird predators’ home ranges. S. quercii and S. marjana have allo-parapatric distribution, but they both overlap with S. phegea (except in Sicily, where the latter is absent, all records hitherto being misidentifications), which is hugely abundant and has already been shown to be a model also for some distantly related co-mimics (Sbordoni et al. 1979). In this context, the departure from a successful aposematic signal would be strongly counter-selected during and after speciation. Strong ecological constraints would then be responsible for morphological stasis, in the face of substantial genetic divergence, thus eventually leading to the evolution of so-called cryptic diversity (Bickford et al. 2007; Struck et al. 2018). In Syntomis, such stasis also extends to the configuration of the genitalia, all fairly homogeneous across the group, which cannot be modelled by predation or other external agents. This circumstance contrasts with the striking diversification in genitalia seen in other almost indistinguishable moths (e.g. the sympatric species pairs Grammodes geometrica-G. occulta, Dysgonia algira-D. torrida, Noctua fimbriata-N. tirrenica and Cilix glaucata-C. hispanica, to name just a few), suggesting that Syntomis genitalia could have maintained stable configurations due of phylogenetic inertia, as they perhaps do not play any species isolation role.
Mechanical compatibility of genitalia between different Syntomis species has been proved by the detection of rare hybrids S. phegea x S. ragazzii (Sbordoni et al. 1982) and heterospecific fertile matings S. phegea x S. marjana reared from the wild (Rupinpiccolo, NE Italy, A. Zilli, unpublished). Mate recognition and species integrity in Syntomis have, therefore, to rely on other systems than mechanical compatibility. As seen above, visual stimuli are unfit candidates, as any departure from the shared pattern would weaken the efficacy of their aposematic signalling. Bickford et al. (2007) recognize that cryptic diversity is particularly common in taxa not using visual mating signals. Sheer evidence that Syntomis are unlikely to use visual mate recognition systems is based on field observations of pseudo-copulae with distantly related species belonging to an alternative mimicry ring based on different colours (red and black), i.e. some burnet moth species of the genus Zygaena (Zygaenidae). Such mismatings are not uncommon during the peaks of abundance of these moths, when they cluster together on flower heads and get confused by congestion of pheromonal plumes all around and tactile abdominal stimuli that lead them to clasp almost anything within their reach (Lees and Zilli 2019).
The occurrence of temporal displacement as reproductive isolating mechanism among Syntomis has been suggested by Sbordoni et al. (1982), who reported partial temporal displacement between S. phegea and S. ragazzii in Central Italy, and a complete displacement between S. phegea and S. marjana in the Karst Plateau. However, during the fieldwork campaigns run by us in the Karst in different years, S. phegea and S. marjana were often found flying simultaneously, occasionally indulging in heterospecific copulae. Furthermore, S. phegea, S. ragazzii and S. quercii can be found flying together in a same biotope in the Apennines. Therefore, the issue of temporal isolation between sympatric Syntomis is worth of further investigation, as a trade-off situation is expected between selective pressures promoting synchrony—Müllerian mimicry being most effective under such circumstance—and opposite ones enhancing temporal displacement as a way of ethological reproductive isolation.
Preliminary breeding experiments run by us (not shown) are fully compatible with chemically mediated interactions between sexes in Syntomis moths, with females typically resting in a calling posture and attracting males. Bendib and Minet (1998) demonstrated the presence of sex pheromone glands in female S. phegea. In contrast, Schneider et al. (1999) highlighted that males of all Syntomis species bear androconial organs at the base of forelegs. Thus, all evidence points to chemically mediated interactions as the main driver of mate recognition in Syntomis moths.
Taxonomic remarks and conclusions
Our research confirms that in Syntomis, one of the taxonomically most controversial groups of European moths, one additional cryptic species is involved. This is the case of a group of populations living in the Italian peninsula, Sicily and Provence that has been noted by different authors as quercii, kruegeri and albionica, respectively. Genetic information points out that these populations have to be combined in a distinct species under the name Syntomis quercii Verity 1914 (bona sp., stat. nov.). In fact, when first named by Ragusa (1904), the oldest name kruegeri was infrasubspecific and before it was made available for nomenclature by Turati (1917), Verity’s (1914) description intervened.
Further genetic sub-structuring has been detected in S. quercii, S. ragazzii and S. marjana. Automatically defining subspecific rank to genetically differentiated populations is considered a common mistake (see Zilli 1996, and references therein). However, it is worth noting that at least in S. ragazzii, diversification in southern Italy (Calabrian Apennine) had already been pointed out with the description of two subspecies (silaensis Obraztsov 1966 and asperomontana [Stauder & Turati], 1917). Furthermore, partially distinct genetic make-ups also characterize the Sicilian populations of S. quercii, which coincide with the original concept of nominotypical kruegeri.
Research on the Euro-Anatolian taxa of Syntomis highlights how the Palaearctic species of this group provide an interesting example to study trade-offs between morphological stasis and genetic divergence in complexes involved into mimicry relationships of Müllerian type. New studies have to be focused on understanding the genomic basis of speciation and origin of cryptic diversity within this group of moths.
Data availability
The dataset generated during the current study is available in the GenBank repository (see corresponding GenBank numbers in the “Material and Methods” section)
References
Abrantes, F., Voelker, A., Sierro Sanchez, F., Naughton, F., Rodrigues, T., Cacho, I., et al. (2010). Paleoclimate variability in the Mediterranean region. In P. Lionello (Ed.), The climate of the Mediterranean region (pp. 1–86). London: Elsevier.
Anikin, V. V., Sachkov, S. A., & Zolotuhin, V. V. (2017). "Fauna lepidopterologica Volgo-Uralensis": from P. Pallas to present days. Proceedings of the Museum Witt 6. Munich-Vilnius: Museum Witt & Nature Research Center.
Ayala, F. J., & Powell, J. R. (1972). Allozymes as diagnostic characters of sibling species of Drosophila. Proceedings of the National Academy of Sciences, 69(5), 1094–1096.
Ayala, F. J., Powell, J. R., Tracey, M. L., Mourão, C. A., & Pérez-Salas, S. (1972). Enzyme variability in the Drosophila willistoni group. IV. Genic variation in natural populations of Drosophila willistoni. Genetics, 70(1), 113–139.
Bálint, M., Domisch, S., Engelhardt, C. H. M., Haase, P., Lehrian, S., Sauer, J., Theissinger, K., Pauls, S. U., & Nowak, C. (2011). Cryptic biodiversity loss linked to global climate change. Nature Climate Change, 1(6), 313–318.
Beheregaray, L. B., & Caccone, A. (2007). Cryptic biodiversity in a changing world. Journal of Biology, 6(4), 9.
Bendib, A., & Minet, J. (1998). Female pheromone glands in Arctiidae (Lepidoptera). Evolution and phylogenetic significance. Comptes Rendus de l’Académie des Sciences, (III) (Sciences de la Vie), 321(12), 1007–1014.
Bertaccini, E., Fiumi, G., & Provera, P. (1997). Bombici e Sfingi d’Italia (Lepidoptera: Heterocera). Volume 2. Bologna: Natura-Giuliano Russo Editore.
Bickford, D., Lohman, D. J., Sodhi, N. S., Ng, P. K., Meier, R., Winker, K., et al. (2007). Cryptic species as a window on diversity and conservation. Trends in Ecology & Evolution, 22(3), 148–155.
Bonfiglio, L., Mangano, G., Marra, A. C., Masini, F., Pavia, M., & Petruso, D. (2002). Pleistocene Calabrian and Sicilian bioprovinces. Geobios, 35, 29–39.
Boppré, M., Vane-Wright, R. I., & Wickler, W. (2017). A hypothesis to explain accuracy of wasp resemblances. Ecology and Evolution, 7(1), 73–81.
Brower, A. V. (1994). Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proceedings of the National Academy of Sciences, 91(12), 6491–6495.
Bullini, L., & Sbordoni, V. (1980). Electrophoretic studies of gene-enzyme systems: microevolutionary processes and phylogenetic inference. Bollettino di Zoologia, 47(suppl. 1), 95–112.
Bullini, L., Sbordoni, V., & Ragazzini, P. (1969). Mimetismo mülleriano in popolazioni italiane di Zygaena ephialtes (L.) (Lepidoptera, Zygaenidae). Archivio Zoologico Italiano, 54, 181–214.
Bullini, L., Cianchi, R., Stefani, C., & Sbordoni, V. (1981). Biochemical taxonomy of the Amata phegea complex (Ctenuchidae, Syntominae). Nota lepidopterologica, 4(4), 125–127.
Canestrelli, D., & Nascetti, G. (2008). Phylogeography of the pool frog Rana (Pelophylax) lessonae in the Italian peninsula and Sicily: multiple refugia, glacial expansions and nuclear–mitochondrial discordance. Journal of Biogeography, 35(10), 1923–1936.
Canestrelli, D., Aloise, G., Cecchetti, S., & Nascetti, G. (2010). Birth of a hotspot of intraspecific genetic diversity: notes from the underground. Molecular Ecology, 19(24), 5432–5451.
Carstens, B. C., Pelletier, T. A., Reid, N. M., & Satler, J. D. (2013). How to fail at species delimitation. Molecular Ecology, 22(17), 4369–4383.
Chiocchio, A., Colangelo, P., Aloise, G., Amori, G., Bertolino, S., Bisconti, R., Castiglia, R., & Canestrelli, D. (2019). Population genetic structure of the bank vole Myodes glareolus within its glacial refugium in peninsular Italy. Journal of Zoological Systematics and Evolutionary Research, 57(4), 959–969.
Cianchi, R., Stefani, C., Sbordoni, V., & Bullini, L. (1980). Ricerche elettroforetiche sulle specie italiane del complesso Amata phegea (Lepidoptera, Ctenuchidae): aspetti genetici, tassonomici ed evolutivi. Atti XII Congresso Nazionale Italiano di Entomologia, 2, 239–242.
Cvetkovic, V., Prelević, D., & Schmid, S. (2016). Geology of South-Eastern Europe. In Mineral and thermal waters of Southeastern Europe (pp. 1–29). Cham: Springer.
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nature Methods, 9(8), 772.
de Freina, J. J. (2008). Über die Biologie, Morphologie, Phänologie und Taxonomie von Amata (Syntomis) kruegeri (Ragusa, 1904) (Lepidoptera: Arctiidae, Syntominae, Syntomini). Nachrichten des Entomologischen Vereins Apollo, (N.F.), 28(3/4), 97–107.
de Freina, J. J., & de Prins, W. (2018). Nomenclatural and taxonomic notes on three South-East Asian species of Amata (Lepidoptera: Erebidae: Arctiinae: Syntomini). Zootaxa, 4461(1), 141–145.
de Freina, J. J., & Witt, T. J. (1987). Die Bombyces und Sphinges der Westpalaearktis. München: Forschung & Wissenschaft.
DMEER (2017). Digital Map of European Ecological Regions. Update 01 Nov 2017. European Environment Agency. http://www.eea.europa.eu/data-and-maps/data/digital-map-of-european-ecological-regions.
Doyle, J. J., & Doyle, J. L. (1990). Isolation of plant DNA from fresh tissue. Focus, 12(13), 39–40.
Drummond, A. J., & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7(1), 214.
Drummond, A. J., Suchard, M. A., Xie, D., & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29(8), 1969–1973.
Dufay, C. (1970). Amata albionica Dufay, bona species et son ethologie (Lep. Ctenuchidae). Entomops, 17, 31–40.
Duplouy, A., Hurst, G. D. D., O'Neill, S. L., & Charlat, S. (2010). Rapid spread of male-killing Wolbachia in the butterfly Hypolimnas bolina. Journal of Evolutionary Biology, 23, 231–235.
Ezard, T., Fujisawa, T. & Barraclough, T.G. (2015). SPLITS: SPecies' LImits by Threshold Statistics. R package version 1.0-18/r45.
Fauquette, S., Suc, J. P., Guiot, J., Diniz, F., Feddi, N., Zheng, Z., Bessais, E., & Drivaliari, A. (1999). Climate and biomes in the West Mediterranean area during the Pliocene. Palaeogeography, Palaeoclimatology, Palaeoecology, 152(1-2), 15–36.
Fennessy, J., Bidon, T., Reuss, F., Kumar, V., Elkan, P., Nilsson, et al. (2016). Multi-locus analyses reveal four giraffe species instead of one. Current Biology, 26(18), 2543–2549.
Fibiger, M., Laszló, G. M., Ronkay, G., Ronkay, L., Speidel, W., Varga, Z., et al. (2011). Lymantriinae and Arctiinae, including phylogeny and check list of the quadrifid Noctuoidea of Europe. Noctuidae Europaeae 13. Sorø: Entomological Press.
Fujita, M. K., Leaché, A. D., Burbrink, F. T., McGuire, J. A., & Moritz, C. (2012). Coalescent-based species delimitation in an integrative taxonomy. Trends in Ecology & Evolution, 27(9), 480–488.
Giraldo, N., Salazar, C., Jiggins, C. D., Bermingham, E., & Linares, M. (2008). Two sisters in the same dress: Heliconius cryptic species. BMC Evolutionary Biology, 8(1), 324.
Gómez, A., & Lunt, D. H. (2007). Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula. In S. Weiss & N. Ferand (Eds.), Phylogeography of southern European refugia (pp. 155–188). Netherlands: Springer.
Gustafsson, A. L. S., Skrede, I., Rowe, H. C., Gussarova, G., Borgen, L., Rieseberg, L. H., Brochmann, C., & Parisod, C. (2014). Genetics of cryptic speciation within an arctic mustard, Draba nivalis. PLoS One, 9(4), e93834.
Harris, H., & Hopkinson, D. A. (1976). Handbook of enzyme electrophoresis in human genetics. Amsterdam: North-Holland.
Hewitt, G. M. (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biological Journal of the Linnean Society, 58(3), 247–276.
Hewitt, G. M. (1999). Postglacial re-colonization of European biota. Biological Journal of the Linnean Society, 68(1-2), 87–112.
Hewitt, G. M. (2011). Mediterranean peninsulas: the evolution of hotspots. In F. E. Zachos & J. C. Habel (Eds.), Biodiversity hotspots (pp. 123–147). Heidelberg: Springer.
Hinojosa, J. C., Monasterio, Y., Escobés, R., Dincă, V., & Vila, R. (2018). Erebia epiphron and Erebia orientalis: sibling butterfly species with contrasting histories. Biological Journal of the Linnean Society, 126(2), 338–348.
Igniatyev, N. N., & Zolotuhin, V. V. (2005). A review of syntomids (Lepidoptera: Syntomidae) of Russia and adjacent territories. Part 1. Genus Syntomis Ochsenheimer 1808. Eversmannia, 3(4), 28–54.
Jiggins, C. D. (2017). The ecology & evolution of Heliconius butterflies. Oxford: Oxford University Press.
Knowlton, N. (1993). Sibling species in the sea. Annual Review of Ecology and Systematics, 24(1), 189–216.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., & Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics, 23(21), 2947–2948.
Lawrence, J. P., Rojas, B., Fouquet, A., Mappes, J., Blanchette, A., Saporito, R. A., Bosque, R. J., Courtois, E. A., & Noonan, B. P. (2019). Weak warning signals can persist in the absence of gene flow. Proceedings of the National Academy of Sciences, 116(38), 19037–19045.
Lees, D. C., & Zilli, A. (2019). Moths: their biology, diversity and evolution. London: Natural History Museum.
Leimar, O., Tullberg, B. S., & Mallet, J. (2012). Mimicry, saltational evolution, and the crossing of fitness valleys. In E. Svensson & R. Calsbeek (Eds.), The Adaptive Landscape in Evolutionary Biology (pp. 259–270). Oxford: Oxford University Press.
Librado, P., & Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25(11), 1451–1452.
Malatesta, A. (1985). Geologia e paleobiologia dell’era glaciale. Roma: NIS.
Mayr, E. (1963). Animal species and evolution. Cambridge: Belknap Press of Harvard University.
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89(3), 583–590.
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., & Minh, B. Q. (2014). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274.
Obraztsov, N. S. (1966). Die Palaearktischen Amata – Arten (Lepidoptera, Ctenuchidae). Veröffentlichungen der Zoologischen Staatssammlung München, 10, 1–383.
Pabijan, M., Zieliński, P., Dudek, K., Chloupek, M., Sotiropoulos, K., Liana, M., & Babik, W. (2015). The dissection of a Pleistocene refugium: phylogeography of the smooth newt, Lissotriton vulgaris, in the Balkans. Journal of Biogeography, 42(4), 671–683.
Papadopoulou, A., Anastasiou, I., & Vogler, A. P. (2010). Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Molecular Biology and Evolution, 27(7), 1659–1672.
Pardo-Diaz, C., Salazar, C., Baxter, S. W., Merot, C., Figueiredo-Ready, W., Joron, M., McMillan, W. O., & Jiggins, C. D. (2012). Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genetics, 8, e1002752.
Peakall, R. O. D., & Smouse, P. E. (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6(1), 288–295.
Pérez-Ponce de León, G., & Poulin, R. (2016). Taxonomic distribution of cryptic diversity among metazoans: not so homogeneous after all. Biology Letters, 12(8), 20160371.
Pfennig, D. W. (2012). Mimicry: ecology, evolution, and development. Current Zoology, 58(4), 603–606.
Pfenninger, M., & Schwenk, K. (2007). Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evolutionary Biology, 7(1), 121.
Pons, J., Barraclough, T. G., Gomez-Zurita, J., Cardoso, A., Duran, D. P., Hazell, S., Kamoun, S., Sumlin, W. D., & Vogler, A. P. (2006). Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology, 55(4), 595–609.
Przybyłowicz, Ł., Lees, D. C., Zenker, M. M., & Wahlberg, N. (2019). Molecular systematics of the arctiine tribe Syntomini (Lepidoptera, Erebidae). Systematic Entomology, 44(3), 624–637.
Puillandre, N., Lambert, A., Brouillet, S., & Achaz, G. (2012a). ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Molecular Ecology, 21(8), 1864–1877.
Puillandre, N., Modica, M. V., Zhang, Y., Sirovich, L., Boisselier, M. C., Cruaud, C., et al. (2012b). Large-scale species delimitation method for hyperdiverse groups. Molecular Ecology, 21(11), 2671–2691.
Racheli, T., & Zilli, A. (1985). Modelli di distribuzione dei Lepidotteri nell’Italia meridionale. Biogeographia, 11, 165–194.
Ragusa, E. (1904). Note Lepidotterologiche. Il Naturalista Siciliano, 17(1), 18–20.
Reidenbach, K. R., Neafsey, D. E., Costantini, C., Sagnon, N. F., Simard, F., & Ragland, et al. (2012). Patterns of genomic differentiation between ecologically differentiated M and S forms of Anopheles gambiae in West and Central Africa. Genome Biology and Evolution, 4(12), 1202–1212.
Robuchon, M., Faith, D. P., Julliard, R., Leroy, B., Pellens, R., Robert, A., Thévenin, C., Véron, S., & Pavoine, S. (2019). Species splitting increases estimates of evolutionary history at risk. Biological Conservation, 235, 27–35.
Roca, A. L., Georgiadis, N., Pecon-Slattery, J., & O'Brien, S. J. (2001). Genetic evidence for two species of elephant in Africa. Science, 293(5534), 1473–1477.
Rögl, F. (1999). Mediterranean and Paratethys. Facts and hypotheses of an Oligocene to Miocene paleogeography (short overview). Geologica Carpathica, 50(4), 339–349.
Ruxton, G. D., Sherratt, T. N., & Speed, M. P. (2004). Avoiding attack: the evolutionary ecology of crypsis, warning signals and mimicry. Oxford: Oxford University Press.
Sbordoni, V., Bullini, L., Scarpelli, G., Forestiero, S., & Rampini, M. (1979). Mimicry in the burnet moth Zygaena ephialthes: population studies and evidence of a Batesian-Müllerian situation. Ecological Entomology, 4(1), 83–93.
Sbordoni, V., Bullini, L., Bianco, P., Cianchi, R., De Matthaeis, E., & Forestiero, S. (1982). Evolutionary studies on ctenuchid moths of the genus Amata: 2. Temporal isolation and natural hybridization in sympatric populations of Amata phegea and A. ragazzii. Journal of the Lepidopterists' Society, 36(3), 185–191.
Scalercio, S., Cini, A., Menchetti, M., Vodă, R., Bonelli, S., Bordoni, A., et al. (2020). How long is 3 km for a butterfly? Ecological constraints and functional traits explain high mitochondrial genetic diversity between Sicily and the Italian Peninsula. Journal of Animal Ecology, 00, 1–14.
Schenk, J. J. (2016). Consequences of secondary calibrations on divergence time estimates. PLoS One, 11(1), e0148228.
Schmitt, T. (2007). Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Frontiers in Zoology, 4(1), 11.
Schmitt, T., & Seitz, A. (2004). Low diversity but high differentiation: the population genetics of Aglaope infausta. Journal of Biogeography, 31, 137–144.
Schneider, D., Legal, L., Dier, L. W., & Wink, M. (1999). Androconial hairbrushes of the Syntomis (Amata) phegea (L.) group (Lepidoptera, Ctenuchinae): a synapomorphic character supported by sequence data of the mitochondrial 16S rRNA gene. Zeitschrift für Naturforschung. Section C, 54, 1119–1139.
Schwarzfeld, M. D., & Sperling, F. A. (2015). Comparison of five methods for delimitating species in Ophion Fabricius, a diverse genus of parasitoid wasps (Hymenoptera, Ichneumonidae). Molecular Phylogenetics and Evolution, 93, 234–248.
Selander, R. K. (1971). Biochemical polymorphism and systematics in the genus Perumyscus. 1. Variation in the old field mouse (Peromyscus polionatus). University of Texas Publications, 7103, 49–90.
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H., & Flook, P. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America, 87(6), 651–701.
Stauder, H. (1913). Syntomis phegea L. aus dem ӧsterreichischen Litorale und Mitteldalmatien. Zeitschrift für Wissenschaftliche Insektenbiologie, 9, 236–239.
Stauder, H. (1928-1929). Genus Syntomis O. im zirkum-adriatisch-tyrrhenisch-ligurischen Gebiete. Lepidopterologische Rundschau, 2, (15) 149–154, (16/17) 160–171, (18) 173–176, (19) 187–190, (20) 200–201, (21) 207–210, (22) 215–218, (23) 227–230, (24) 239–242; Entomologische Anzeiger, 9, 10–12.
Struck, T. H., Feder, J. L., Bendiksby, M., Birkeland, S., Cerca, J., Gusarov, V. I., et al. (2018). Finding evolutionary processes hidden in cryptic species. Trends in Ecology & Evolution, 33(3), 153–163.
Suc, J. P., & Popescu, S. M. (2005). Pollen records and climatic cycles in the North Mediterranean region since 2.7 Ma. Geological Society, London, Special Publications, 247(1), 147–158.
Swofford, D.L. & Selander, R.B. (1999). BIOSYS-2: a computer program for the analysis of allelic variation in population genetics and biochemical systematics (Release 2.0). University of Illinois, Urbana-Champaign.
Taberlet, P., Fumagalli, L., Wust-Saucy, A., & Cosson, J. (1998). Comparative phylogeography and postglacial colonization routes in Europe. Molecular Ecology, 7(4), 453–464.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30(12), 2725–2729.
Turati, E. (1917). Revisione delle Syntomis Paleartiche a doppio cingolo giallo, e saggio di una classificazione delle varie forme e specie. Atti della Società Italiana di Scienze Naturali, 56, 179–232 pls 2–8.
Verity, R. (1914). Contributo allo studio della variazione nei Lepidotteri tratto principalmente da materiale di Toscana, delle Marche e di Calabria. Bollettino della Società entomologica Italiana, 45, 203–238 1 pl.
Wiens, J. J. (2007). Species delimitation: new approaches for discovering diversity. Systematic Biology, 56(6), 875–878.
Wiens, J. J., & Servedio, M. R. (2000). Species delimitation in systematics: inferring diagnostic differences between species. Proceedings of the Royal Society of London B: Biological Sciences, 267(1444), 631–636.
Zhang, J., Kapli, P., & Stamatakis, A. (2013). A general species delimitation method with applications to phylogenetic placements. Bioinformatics, 29(22), 2869–2876.
Zilli, A. (1996). Colour polymorphism of Callimorpha dominula (Linnaeus, 1758) in Italy, and the problem of polytopic subspecies (Lepidoptera, Arctiidae, Callimorphinae). Mitteilungen der Münchner entomologischen Gesellschaft, 86, 79–98.
Acknowledgements
We would like to thank Mauro Zampiglia for his help on fieldwork. AC also thanks Jadranka Rota for her helpful suggestions.
Funding
Open access funding provided by Lund University. AC was supported by the SYNTHESYS Projects GB-TAF-2574, awarded by the European Community Research Infrastructure Action under the EU FP7 Framework.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Code availability
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chiocchio, A., Arduino, P., Cianchi, R. et al. Unveiling cryptic diversity among Müllerian co-mimics: insights from the Western Palaearctic Syntomis moths (Lepidoptera: Erebidae: Arctiinae). Org Divers Evol 21, 119–133 (2021). https://doi.org/10.1007/s13127-020-00474-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-020-00474-9