
Abstract
The prevalence of non-alcoholic fatty pancreas disease (NAFPD) is increasing in parallel with obesity rates. Stress-related alterations in endoplasmic reticulum (ER), such as the unfolded protein response (UPR), are associated with obesity. The aim of this study was to investigate ER imbalance in the pancreas of a mice model of adult and perinatal diet-induced obesity. Twenty female C57BL/6J mice were assigned to control (Con) or obesogenic (Ob) diets prior to and during pregnancy and lactation. Their offspring were weaned onto Con or Ob diets up to 6 months post-partum. Then, after sacrifice, plasma biochemical analyses, gene expression, and protein concentrations were measured in pancreata. Offspring of Ob-fed mice had significantly increased body weight (p < 0.001) and plasma leptin (p < 0.001) and decreased insulin (p < 0.01) levels. Maternal obesogenic diet decreased the total and phosphorylated Eif2α and increased spliced X-box binding protein 1 (XBP1). Pancreatic gene expression of downstream regulators of UPR (EDEM, homocysteine-responsive endoplasmic reticulum-resident (HERP), activating transcription factor 4 (ATF4), and C/EBP homologous protein (CHOP)) and autophagy-related proteins (LC3BI/LC3BII) were differently disrupted by obesogenic feeding in both mothers and offspring (from p < 0.1 to p < 0.001). Maternal obesity and Ob feeding in their offspring alter UPR in NAFPD, with involvement of proapoptotic and autophagy-related markers. Upstream and downstream regulators of PERK, IRE1α, and ATF6 pathways were affected differently following the obesogenic insults.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity-associated non-alcoholic fatty pancreas disease (NAFPD) is a pathological condition characterized by ectopic fat deposition in the pancreas. In NAFPD, pancreatic stellate cells are abnormally activated, leading to excess collagen production and therefore pancreatic fibrosis [39]. This can potentially lead to pancreatic cancer [25], and currently, pancreatic adenocarcinoma is one of the deadliest malignancies in developed countries, with a prevalence increasing in parallel with obesity rates [7].
A possible cause of NAFPD is therefore obesity. Experimental models have demonstrated that maternal nutrition status during the perinatal period can directly modulate the risk of developing obesity [2], as well as obesity-associated metabolic diseases in offspring, such as non-alcoholic fatty liver disease (NAFLD) [3, 22], glucose intolerance [6], and hypertension [34]. Additionally, our research group has recently demonstrated that maternal obesity and subsequent obesogenic feeding in offspring can increase their triglyceride content and injury-related gene expressions in the pancreas [1].
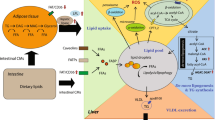
Body and organ fat accumulation promotes the release of proinflammatory cytokines and increased oxidative stress, processes which might underlie the pathogenesis of both NAFLD [24] and NAFPD [5]. However, further studies focusing on other metabolic and molecular pathways are needed. It has been described that endoplasmic reticulum (ER) stress, a mechanism responsible for protein folding and maturation, is altered in obesity-related diseases [8]. External influences, such as nutrient supply, could cause disruption of ER homeostasis, resulting in an altered pattern of protein synthesis, also termed the unfolded protein response (UPR) [27]. ER stress-induced UPR activates three chief pathways, which are regulated by the following ER transmembrane proteins: the kinase RNA (PKR)-like ER kinase (PERK), the inositol-requiring 1 alpha protein (IRE1a), and the activating transcription factor 6 (ATF6). Activation of these proteins by phosphorylation or splicing initiates a cascade of reactions; consequently, they act as transcription modulators, activating downstream effectors and regulating the gene expressions [30]. UPR is primarily a protective pathway, but an unresolved UPR could lead to increased apoptotic-related mediators and cell death. Obesity is known to trigger UPR in the liver [12], and disruption of ER stress homeostasis is thought to contribute to apoptosis of hepatocytes, a feature in many end-stage liver diseases [18]. Since the pancreas is derived from the same embryonic origin as the liver [4], it is probable that NAFPD shares similar pathogenic mechanisms with NAFLD, involving ER stress. Therefore, by utilizing a developmental programming model, we studied if maternal obesity and a postweaning obesogenic diet could promote ER stress, thus affecting molecular pathways and cellular apoptosis in the pancreas, which in turn promote the pathogenesis of NAFPD.
Materials and methods
Animal models
Twenty female C57BL/6J outbred mice (Charles River Laboratories, Margate, UK) were randomly allocated to either a standard control diet (Con) (RM1, Special Dietary Services, Essex, UK) or an obesogenic high-fat-sucrose diet (Ob) (824053, Special Dietary Services) sweetened with condensed milk (Nestle, Vevey, Switzerland) as previously described [21]. The mice were kept on their respective diets 6 weeks before mating, during pregnancy and during lactation. Ob diet animals reached a 30 % increase in body weight before entering the mating cycle. The female animals were mated with male mice from the same litter in order to minimize genetic variability. Conception was determined by the formation of a vaginal plug. A total of four to five animals per experimental group gave birth (litters with at least five pups). Litters with five to nine pups were included into the study. Each litter was reduced to six pups within 48 h of birth. At day 21 postpartum, offspring were weaned onto either the control diet (Con) or the obesogenic diet (Ob) creating the following experimental groups: offspring of control weaned onto control diet (Con-Con), offspring of control weaned onto obesogenic diet (Con-Ob), offspring of obese weaned onto control diet (Ob-Con), and offspring of obese weaned onto obesogenic diet (Ob-Ob). At 6 months postpartum, female offspring were weighed, sacrificed by rising CO2, and the pancreata were extracted and appropriately weighed and stored for further analysis.
All the animals had access to food and water ad libitum and were kept in a thermostatically controlled environment (22 °C) in a 12-h light/dark cycle. The animals were treated in accordance with The Animal Scientific Procedures Act 1986 guidelines and UK Home Office. All the studies were approved by Local University College London Ethics Committee.
Plasma biochemical analysis
Plasma samples were assayed for leptin (R&D 1301, BioVendor, Brno, Czech Republic) and insulin (90080, Crystal Chem, Downers Grove, IL, USA) using ELISAs, according to the manufacturers’ instructions.
Western blotting
Western blotting analyses were performed as previously established [35]. Briefly, protein was isolated from murine whole pancreas tissue using RIPA lysis and extraction buffer (Thermo Scientific, Waltham, MA, USA) and quantified by the Pierce BCA Protein Assay Kit (Thermo Scientific). Thirty micrograms of protein per sample was loaded onto NuPAGE Bis-Tris Mini Gels (Life Technologies, Carlsbad, CA, USA) (n = 3–4 per experimental group) for electrophoresis and subsequently transferred to 0.45-μm Invitrolon PVDF membranes (Invitrogen, Life Technologies Corporation, UK) by electroblotting. The membranes were incubated in blocking buffer followed by overnight incubation with primary antibodies at 4 °C (Table 1); the membranes were then incubated with the secondary antibodies (Table 1). Finally, Pierce ECL Western Blotting Substrate (Thermo Scientific) was used for revelation procedures. The protein bands were visualized using FlourChem imager (ProteinSimple, CA, USA), and densitometry measurements were calculated using AlphaView SA software (version 3.4.0). The resulting bands were normalized against β-actin from the same samples.
Quantitative real-time PCR
Pancreas samples were homogenized using TRIzol reagent (Invitrogen, CA, USA) by following the suppliers’ protocol. Sample concentrations were measured using a NanoDrop ND-1000 Spectrometer (Thermo Scientific). DNase treatment and retrotranscription to cDNA were carried out using the Qiagen QuantiTect Reverse Transcriptase kit (Qiagen). Quantitative real-time PCR (qPCR) was performed as previously described [28] (n = 3–4 per experimental group). GAPDH was used as a control housekeeping gene for all reactions. All primers were obtained from Sigma-Aldrich (San Luis, MO, USA) (Table 2). Ct values were calculated using the 2−ΔΔCt method according to GAPDH as the internal control.
Statistical analysis
All data are shown as mean± standard error of the mean (SEM). Two-way ANOVA was applied for studying the effect of maternal obesogenic feeding and offspring obesogenic feeding. Comparison of the means was carried out by Tukey post hoc test. A statistically significant result was determined with a p value of less than 0.05. SPSS 14 software (SPSS, Chicago, IL, USA) was used for the statistical analysis.
Results
Effect of maternal and offspring obesogenic feeding in phenotypical and plasma biochemical parameters
At 6 months old, offspring weaned onto an obesogenic diet had higher body weight (78 %, p < 0.001) (Fig. 1a) and pancreas weight (20 %, p < 0.05) (Fig. 1b), as compared with those weaned onto normal diets. Plasma leptin concentration had also increased due to offspring obesogenic diet (1275 %, p < 0.001) (Fig. 1c), while insulin concentrations decreased in comparison to control-fed offspring (−40 %, p < 0.01) (Fig. 1d).

Phenotypic and biochemical parameters. Effects of maternal and offspring obesogenic feeding on a body weight, b pancreas weight, and plasma biochemical concentrations of c leptin and d insulin determined by ELISAs. *p < 0.05; **p < 0.01; ***p < 0.001; n = 4–5 animals per group
We have previously described in this experimental model that maternal obesogenic feeding alone also predisposed offspring to a NAFPD phenotype, characterized by increased macrovesicular fat infiltration, worsened oral glucose tolerance test, and increased fibrogenic-related gene expression [1].
Effect of maternal and offspring obesogenic feeding in pancreatic protein levels of UPR mainstream regulators
UPR upstream regulator, PERK, did not show changes in protein level pattern by maternal or offspring obesogenic feeding, with an elevated intersample variability (Fig. 2a, b). However, UPR immediate activator eukaryotic initiation factor 2 alpha (EIF2α) total protein levels decreased in the offspring as an effect of maternal obesogenic feeding (−13 %, p < 0.05) (Fig. 2e), but also due to the direct obesogenic feeding in the offspring (−22 %, p < 0.01). Interestingly, the phosphorylation of EIF2α was significantly reduced by maternal obesity (−50 %, p < 0.001) (Fig. 2d), causing a significant decrease in the ratio between p-EIF2 α and t-EIF2 α (−42 %, p < 0.01) (Fig. 2f).

Pancreas protein concentration of PERK UPR pathway. Effect of maternal and offspring obesogenic feeding on a phosphorylated and b total PERK and c their ratio, as well as on d phosphorylated and e total EIF2α and f their ratio determined by Western blot. *p < 0.05; **p < 0.01; ***p < 0.001; n = 3–4 animals per group
According to the IRE1α branch, there was no statistical change observed in IRE1α protein concentrations, with no notable difference in phosphorylated or total hepatic protein levels, nor their ratio (Fig. 3a–c). However, maternal obesogenic feeding led to an increase in the levels of total unspliced and spliced X-box binding protein 1 (XBP1) (62 %, p < 0.1, and 87 %, p < 0.05, respectively) (Fig. 3d, e).

Pancreas protein concentration of IRE1α UPR pathway. Effect of maternal and obesogenic feeding on a phosphorylated IRE1α, b total IRE1α and c their ratio, as well as on d spliced XBP1 and e unspliced XBP1 and f their ratio determined by Western blot. *p < 0.05; n = 3–4 animals per group
For the ATF6 pathway, we observed an opposite tendency of interaction between maternal and offspring obesity in the whole protein (p90) and after splicing (p50) (Fig. 4a, b). This tendency was also reflected by the p50/p90-ATF6 ratio, which showed Ob-Ob animals with the highest ratio (from 1558 to 3437 %, p < 0.1) (Fig. 4c).

Pancreas protein concentration of ATF6 UPR pathway. Effect of maternal and offspring obesogenic feeding on a fragmented p50-ATF6, b p90-ATF6, and c their ratio determined by Western blot; n = 3–4 animals per group
Effect of maternal and offspring obesogenic feeding on pancreatic mRNA expression of other ER stress mediators
Activating transcription factor 4 (ATF4) and the proapoptotic transcription factor C/EBP homologous protein (CHOP) are mainly regulated by the PERK-eIF2α UPR pathway. Both maternal and offspring obesogenic feeding significantly increased their hepatic gene expression (p < 0.05 and <0.01, respectively) (Fig. 5a, b). Indeed, the “double hit” of obesogenic feeding in mother and offspring resulted in significantly increased Ob-Ob liver ATF4 and CHOP messenger RNA (mRNA) expressions in comparison with all other experimental groups (p from <0.05 to <0.01 and from 157 to 217 % for ATF4 and p from <0.01 to <0.001 and from 99 to 147 % for CHOP).

Pancreas mRNA levels by real-time qPCR. Effect of maternal and offspring obesogenic feeding on a ATF4, b CHOP, c EDEM, d HERP, e WSF1, and f insulin. *p < 0.05; **p < 0.01; ***p < 0.001; n = 3–4 animals per group
sXBP1-specific downstream target genes, ER degradation-enhancing alpha-mannosidase-like 1 (EDEM1), and homocysteine-responsive endoplasmic reticulum-resident (HERP) also increased their hepatic expression levels as an effect of obesogenic diets in offspring (66 %, p < 0.05 and 73 %, p < 0.01, respectively) (Fig. 5c, d). Maternal obesogenic feeding tended to decrease HERP expression levels (−19 %, p < 0.1). On the other hand, ER stress-related Wolfram syndrome 1 (WSF1) gene expression was increased by offspring obesogenic feeding (62 %, p < 0.01) (Fig. 5e), and comparisons between groups showed a tendency of WSF1 mRNA to increase in Ob-Ob compared with Con-Con mice (56 %, p < 0.1), as well as a significant increase in Ob-Ob compared with the Ob-Con group (243 %, p < 0.01).
Finally, hepatic insulin expression was increased due to obesogenic feeding in both mother and offspring (208 %, p < 0.05 and 491 %, p < 0.001, respectively) (Fig. 5f). The highest mRNA levels were observed in the Ob-Ob group, with significantly increased levels when compared with the all other groups (202 to 1294 %, p < 0.01 to <0.01).
Effect of maternal and offspring obesogenic feeding on pancreatic protein levels of CHOP and GRP78
GRP78 is a chaperone and master regulator of ER homeostasis. It inactivates the transmembrane PERK, IREα, and ATF6 blocking the UPR [30]. GRP78 levels showed a tendency to decrease in the groups with maternal obesogenic feeding (−43 %, p < 0.1) (Fig. 6a); conversely, results for CHOP protein concentrations did not show any statistically significant differences (Fig. 6b).

Pancreas protein concentration. Effects of maternal and offspring obesogenic feeding on a GRP78 and b CHOP determined by Western blot; n = 3–4 animals per group
Effect of maternal and offspring obesogenic feeding on autophagy regulators
NAFPD and pancreatic carcinogenesis are kept going by derangement of proliferation, apoptosis, metabolism, and autophagy [33]. Autophagy markers, microtubule-associated protein 1 light chain 3 beta (LC3BI and LC3BII), were quantified as possible mechanisms that could be modified by the dysregulation of ER homeostasis (Fig. 7a, b). We observed a trend toward increased levels of LC3BI and LC3BII protein levels due to maternal obesogenic feeding when compared with Con-Con and Ob-Con (118 and 205 %, respectively, p < 0.1 for both). However, there was no statistically significant difference in LC3BI/LC3BII ratios (Fig. 7c).

Pancreas protein concentration. Effects of maternal and offspring obesogenic feeding on the autophagy-related protein a LC3BI, b LC3BII, and c their ratio. *p < 0.05; n = 3–4 animals per group
Discussion
Our programming model has previously demonstrated that a NAFPD phenotype can be induced by maternal and offspring obesogenic feeding [1]. In this study, we show that ER stress plays a role in the pathogenesis of NAFPD and that this effect is a consequence of developmentally programmed obesity. To our knowledge, this is the first study exploring ER stress as a potential mechanism of developmental programming and NAFPD. It has been suggested that inhibiting ER stress may reduce the complications of obesity [10]. If ER stress is involved in the pathogenesis of NAFPD, it could similarly serve as a potential therapeutic target for pancreatic injury.
Similar to previous rodent models, our findings demonstrate that a mouse model of high-fat-diet-induced obesity can result in a pancreatic phenotype similar to that seen in human NAFPD: fat deposition in the pancreas, increased total body fat content, an obesity-associated physiological and biochemical profile, increased cytokine production, and, in some cases, even pancreatic cirrhosis, which in turn could lead to pancreatic cancer [5, 20, 25].
Novel evidence suggested that ER stress plays an important role in the pathogenesis of obesity and its related comorbidities [13]. Therefore, we initially expected to found an upregulation of ER stress in those experimental groups where offspring were obese. Our study has further discovered that the three major UPR pathways are differentially activated by maternal obesity, by post-weaning obesogenic diet, or by the combination of both factors. When PERK is activated by ER stress, eIF2α is phosphorylated which reversibly attenuates protein translation in the ER to reduce the overload of protein folding [30]. These findings are consistent with those of Wang et al. demonstrating that the reduction of PERK pathway gene expression significantly affects β-cell function and glucose homeostasis in a transgenic mouse model [36]. Similarly, Kennedy and colleagues showed that PERK and eIF2α are downregulated when serum glucose levels were high [11]. Another study involving rat pancreas damaged by ethanol also showed an increase in sXBP1, which is again similar to our findings, although unspliced protein did not show an increase in this setting [16].
The UPR pathway involving ATF6 is particularly important for regulating GRP78, XBP1, and CHOP expression [32]. After dissociating from GRP78, ATF6 as p90-ATF6 form is usually moved to the Golgi organelle to produce its active form, p50-ATF6. The active form is then translocated to the nucleus, resulting in upregulation of UPR processes, i.e., an increase in protein folding, processing, and degradation [30]. According to our results, although not statistically, maternal and offspring obesogenic feeding appeared to have a significant impact on ATF6 activation. This tendency drives to the hypothesis that ER stress increases in obesity situation. Furthermore, p50-ATF6 is known to activate CHOP transcription, which was also confirmed in the Ob-Ob group in our study.
WSF1 is an ER transmembrane protein with genetic polymorphisms that have been linked to the concurrent decrease in plasma insulin concentrations [29]. Thus, the marked WSF1 expression increase observed by the obesogenic feeding may account for the concurrent decrease in plasma insulin levels and greater pancreatic injury in the obesogenic feeding groups.
ER stress causes molecular chaperone GRP78 dissociation from the main ER stress sensors, activating the UPR [30]. Thus, transcriptional activation of GRP78 is used to determine whether there is an ER stress response. Although there was no statistically significant difference, protein expression levels of GRP78 were unexpectedly downregulated in Ob-Ob animals. Interestingly, a decrease on GRP78 liver expression has been found in db/db diabetic mice, which may be associated with pancreatic regulation of insulin resistance [37]. It is unclear why GRP78 was downregulated, even though ER stress was apparent, as shown by the upregulation of the ATF6 pathway. Liu et al. investigated the role of GRP78 in acute pancreatitis and found that downregulation of GRP78 was associated with increased apoptosis [15]. Therefore, the three pathways might regulate the action of the proapoptotic CHOP protein by upregulation during severe or chronic ER stress [14]. Furthermore, overexpression of CHOP leads to cell cycle arrest or apoptosis [23]. The implication of CHOP in the apoptotic pathway resulting from ER stress suggests that CHOP is a crucial link between hepatic fat infiltration and ER stress [19]. Another study, based on a chemically induced pancreatic ER stress rat model, had increased eIF2α phosphorylation and CHOP expression with no changes on GRP78. This pathological pancreatic condition was associated with an increased autophagy as a rescue system to maintain cytoplasmic homeostasis [9].
One possible mechanism for controlling the excess of misfolded proteins and cellular apoptosis is autophagy [17]. Autophagy controls the quality and homeostasis of the cytoplasm by eliminating unwanted proteins and damaged organelles by digestion in the lysosome [31]. Interestingly, autophagy has been suggested as a potential regulatory mechanism for insulin resistance following ER stress in diabetes [38]. Nonetheless, autophagy can also cause apoptosis [40]. Once the cytosolic form from LC3BI is cleaved by ATG4, it is conjugated with phosphatidylethanolamine and as LC3BII form act on autophagosome membrane promoting its expansion [26]. A double obesogenic hit resulted in low levels of LC3BI and LC3BII and in a trend toward an increase of Ob-Ob with respect to the control animals, with no changes in either parameter in the Ob-Con group. Surprisingly, these results were not in line with the previously described UPR activation profile. A potential explanation could be that circadian rhythmicity in the pancreas activates different metabolic regulators at different time points [1]. In support of this, preliminary results (unpublished) from our group have demonstrated that maternal obesogenic feeding and offspring feeding alter 24-h rhythmicity in NAFLD-associated ER stress homeostasis in mice, suggesting that the main regulators may impinge on different activation patterns at different times of the day.
Although we tried to control all the possible confounding factors in this transgenerational model, there could be some factors that limit this study, such as the genetic background of the dams and their fertility rate. Other limitation could be the lack of data from male offspring or the limited number of samples, which affected statistical results during the analyses due to the wider dispersion of the data. Finally, it is difficult to extrapolate the results of this experimental model to humans.
In summary, we have demonstrated that maternal obesity and postnatal obesogenic diets can induce an ER stress unbalance and result in a NAFPD phenotype with more severe associated metabolic outcomes. This is likely related to upregulation of ER stress pathways with resultant inflammation and cellular apoptosis, as suggested by increased proapoptotic and autophagy markers observed. However, while some UPR pathways appeared to be upregulated, these findings were not consistent across all the ER stress pathways, suggesting that while UPR dysregulation plays a role, ER homeostasis and fatty pancreas pathogenesis are regulated by other important factors. Further research in this area is therefore warranted, given the potential of these new therapeutic targets for the management of many obesity-associated diseases, including NAFPD.
References
Carter R, Mouralidarane A, Soeda J, Ray S, Pombo J, Saraswati R et al (2014) Non-alcoholic fatty pancreas disease pathogenesis: a role for developmental programming and altered circadian rhythms. PLoS One 9:e89505
Cordero P, Gonzalez-Muniesa P, Milagro FI, Campion J, Martinez JA (2014) Perinatal maternal feeding with an energy dense diet and/or micronutrient mixture drives offspring fat distribution depending on the sex and growth stage. J Anim Physiol Anim Nutr 71:589–599
Cordero P, Milagro FI, Campion J, Martinez JA (2014) Supplementation with methyl donors during lactation to high-fat-sucrose-fed dams protects offspring against liver fat accumulation when consuming an obesogenic diet. J Dev Orig Health Dis 1–11
Deutsch G, Jung J, Zheng M, Lora J, Zaret KS (2001) A bipotential precursor population for pancreas and liver within the embryonic endoderm. Development 128:871–881
Fraulob JC, Ogg-Diamantino R, Fernandes-Santos C, Aguila MB, Mandarim-de-Lacerda CA (2010) A mouse model of metabolic syndrome: insulin resistance, fatty liver and non-alcoholic fatty pancreas disease (NAFPD) in C57BL/6 mice fed a high fat diet. J Clin Biochem Nutr 46:212–223
Graus-Nunes F, Dalla Corte Frantz E, Lannes WR, da Silva Menezes MC, Mandarim-de-Lacerda CA, Souza-Mello V (2015) Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition 31:380–387
Gumbs AA, Bessler M, Milone L, Schrope B, Chabot J (2008) Contribution of obesity to pancreatic carcinogenesis. Surg Obes Relat Dis 4:186–193
Hetz C, Chevet E, Harding HP (2013) Targeting the unfolded protein response in disease. Nat Rev Drug Discov 12:703–719
Ito M, Nakagawa H, Okada T, Miyazaki S, Matsuo S (2009) ER-stress caused by accumulated intracistanal granules activates autophagy through a different signal pathway from unfolded protein response in exocrine pancreas cells of rats exposed to fluoride. Arch Toxicol 83:151–159
Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K (2012) Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep 2:799
Kennedy J, Katsuta H, Jung MH, Marselli L, Goldfine AB, Balis UJ et al (2010) Protective unfolded protein response in human pancreatic beta cells transplanted into mice. PLoS One 5:e11211
Kim SH, Kim KH, Kim HK, Kim MJ, Back SH, Konishi M et al (2015) Fibroblast growth factor 21 participates in adaptation to endoplasmic reticulum stress and attenuates obesity-induced hepatic metabolic stress. Diabetologia 58:809–818
Lee J, Ozcan U (2014) Unfolded protein response signaling and metabolic diseases. J Biol Chem 289:1203–1211
Lenna S, Trojanowska M (2012) The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr Opin Rheumatol 24:663–668
Liu Y, Zhou ZG, Zhou B, Wang R, Yan H, Li Y (2013) Downregulation of GRP78 and XIAP is correlated with apoptosis during cerulein-induced acute pancreatitis in rats via regulation of caspase activation. Mol Med Rep 7:725–730
Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW et al (2011) Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140:987–997
Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J et al (2014) Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 124:1406–1417
Malhi H, Kaufman RJ (2011) Endoplasmic reticulum stress in liver disease. J Hepatol 54:795–809
Maris M, Overbergh L, Gysemans C, Waget A, Cardozo AK, Verdrengh E et al (2012) Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia 55:1167–1178
Matsuda A, Makino N, Tozawa T, Shirahata N, Honda T, Ikeda Y et al (2014) Pancreatic fat accumulation, fibrosis, and acinar cell injury in the Zucker diabetic fatty rat fed a chronic high-fat diet. Pancreas 43:735–743
Mouralidarane A, Soeda J, Sugden D, Bocianowska A, Carter R, Ray S et al (2015) Maternal obesity programs offspring non-alcoholic fatty liver disease through disruption of 24-h rhythms in mice. Int J Obes (Lond) 39:1339
Oben JA, Patel T, Mouralidarane A, Samuelsson AM, Matthews P, Pombo J et al (2010) Maternal obesity programmes offspring development of non-alcoholic fatty pancreas disease. Biochem Biophys Res Commun 394:24–28
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389
Podrini C, Borghesan M, Greco A, Pazienza V, Mazzoccoli G, Vinciguerra M (2013) Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr Pharm Des 19:2737–2746
Preziosi G, Oben JA, Fusai G (2014) Obesity and pancreatic cancer. Surg Oncol 23:61–71
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Ramadori G, Konstantinidou G, Venkateswaran N, Biscotti T, Morlock L, Galie M et al (2015) Diet-induced unresolved ER stress hinders KRAS-driven lung tumorigenesis. Cell Metab 21:117–125
Ripoli M, Barbano R, Balsamo T, Piccoli C, Brunetti V, Coco M et al (2011) Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res 160:74–81
Rohayem J, Ehlers C, Wiedemann B, Holl R, Oexle K, Kordonouri O et al (2011) Diabetes and neurodegeneration in Wolfram syndrome: a multicenter study of phenotype and genotype. Diabetes Care 34:1503–1510
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63
Tavano F, Pazienza V, Fontana A, Burbaci FP, Panebianco C, Saracino C et al (2015) SIRT1 and circadian gene expression in pancreatic ductal adenocarcinoma: effect of starvation. Chronobiol Int 32:497–512
Taylor PD, Samuelsson AM, Poston L (2014) Maternal obesity and the developmental programming of hypertension: a role for leptin. Acta Physiol (Oxf) 210:508–523
Veyrat-Durebex C, Montet X, Vinciguerra M, Gjinovci A, Meda P, Foti M et al (2009) The Lou/C rat: a model of spontaneous food restriction associated with improved insulin sensitivity and decreased lipid storage in adipose tissue. Am J Physiol Endocrinol Metab 296:E1120–E1132
Wang R, Munoz EE, Zhu S, McGrath BC, Cavener DR (2014) Perk gene dosage regulates glucose homeostasis by modulating pancreatic beta-cell functions. PLoS One 9:e99684
Yamagishi N, Ueda T, Mori A, Saito Y, Hatayama T (2012) Decreased expression of endoplasmic reticulum chaperone GRP78 in liver of diabetic mice. Biochem Biophys Res Commun 417:364–370
Zhang N, Cao MM, Liu H, Xie GY, Li YB (2015) Autophagy regulates insulin resistance following endoplasmic reticulum stress in diabetes. J Physiol Biochem 71:319–327
Zhang X, Cui Y, Fang L, Li F (2008) Chronic high-fat diets induce oxide injuries and fibrogenesis of pancreatic cells in rats. Pancreas 37:e31–e38
Zhang J, Morris MW Jr, Dorsett-Martin WA, Drake LC, Anderson CD (2013) Autophagy is involved in endoplasmic reticulum stress-induced cell death of rat hepatocytes. J Surg Res 183:929–935
Acknowledgments
This work was funded by the Welcome Trust, the Obesity Action Campaign, and the Fiorina Elliot Charity grant.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Soeda, J., Mouralidarane, A., Cordero, P. et al. Maternal obesity alters endoplasmic reticulum homeostasis in offspring pancreas. J Physiol Biochem 72, 281–291 (2016). https://doi.org/10.1007/s13105-016-0476-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-016-0476-6