
Abstract
Platelet-activating factor (PAF) accumulates during cerebral ischemia, and inhibition of this process plays a critical role in neuronal survival. Recently, we demonstrated that LAU-0901, a novel PAF receptor antagonist, is neuroprotective in experimental stroke. We used magnetic resonance imaging in conjunction with behavior and immunohistopathology to expand our understanding of this novel therapeutic approach. Sprague–Dawley rats received 2 h middle cerebral artery occlusion (MCAo) and were treated with LAU-0901 (60 mg/kg) or vehicle 2 h from MCAo onset. Behavioral function, T2-weighted imaging (T2WI), and apparent diffusion coefficients were performed on days 1, 3, and 7 after MCAo. Infarct volume and number of GFAP, ED-1, and NeuN-positive cells were conducted on day 7. Behavioral deficit was significantly improved by LAU-0901 treatment compared to vehicle on days 1, 3, and 7. Total lesion volumes computed from T2WI were significantly reduced by LAU-0901 on days 1, 3, and 7 (by 83%, 90%, and 96%, respectively), which was consistent with decreased edema formation. Histopathology revealed that LAU-0901 treatment resulted in significant reduction of cortical and subcortical infarct volumes, attenuated microglial infiltration, and promoted astrocytic and neuronal survival. These findings suggest LAU-0901 is a promising neuroprotectant and provide the basis for future therapeutics in patients suffering ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is a leading cause of death and disability worldwide. Conventional therapies for ischemic stroke include thrombolytic therapy, prevention of inappropriate coagulation and thrombosis, and surgery to repair vascular abnormalities. Only one FDA-approved therapy exists for treatment of acute ischemic stroke, the thrombolytic tissue plasminogen activator (tPA), but due to comorbid conditions and contraindications, only 5–8% of all ischemic stroke patients are eligible for treatment with tPA [1]. Thus, there is an urgent need for good therapeutic strategies to reduce the devastating deficits resulting from stroke.
Platelet-activating factor (PAF, 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) is a potent, short-lived phospholipid mediator of leukocyte functions, platelet aggregation, and proinflammatory signaling. PAF accumulates in the brain after cerebral ischemia, and, in conjunction with nonspecific glutamate release and glutamate reuptake inhibition, leads to intracellular Ca2+ overload, mitochondrial dysfunction, generation of reactive oxygen species, and inflammation-mediated excitotoxicity [2]. In order to attenuate PAF’s proinflammatory and excitotoxic effects during reperfusion injury, we used LAU-0901, a novel PAF inhibitor, to promote neuronal survival and prevent ischemic brain injury.
LAU-0901 (2,4,6-trimethyl-1, 4-dihydro-pyridine-3, 5-dicarboxylic acid) is a highly potent and selective PAF receptor antagonist, named as such to recognize the collaboration between Louisiana State University Health Sciences Center, New Orleans, and the Universidad de Alcala, Spain, universities (Fig. 1). LAU-0901 has been used as an anti-inflammatory, highly protective treatment in a variety of inflammation models [3–6]. Recently, we showed that LAU-0901 has potent neuroprotective activity when used as a treatment for ischemia–reperfusion injury in rats and mice [7, 8]. In a dose–response study, we found that a 60-mg/kg dose of LAU-0901 resulted in improved behavior and reduced brain infarction after focal cerebral ischemia [7]. Thus, we choose this dose in the current study. The effect of LAU-0901 treatment was investigated using multimodal magnetic resonance imaging (MRI), neurobehavioral assays, and end point immunohistochemistry. No prior studies have used MRI to evaluate the temporal effects of LAU-0901 treatment on brain ischemia.

Chemical structure of LAU-0901 (Nicolas G. Bazan, Carlos Sunkel, Victor Marcheselli and Julio Alvarez-Builla Gomez. “2,4,6-trimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid esters as neuroprotective drugs.” United States Patent No. 6,566,359)
Materials and Methods
Animal Preparation
All studies were approved by the Institutional Animal Care and Use Committees of the Louisiana State University Health Sciences Center, New Orleans, and Loma Linda University. Male Sprague–Dawley rats (Charles River Lab., Wilmington, MA) weighing 260–349 g were fasted overnight but allowed free access to water. Anesthesia was induced with 3% isoflurane in a mixture of 70% nitrous oxide and 30% oxygen. All rats were orally intubated and mechanically ventilated. Temperature probes were inserted into the rectum and the left temporalis muscles to maintain temperatures at 36–37°C during surgical procedures. The right femoral artery and vein were catheterized for blood sampling for arterial gases, pH, and plasma glucose.
Middle Cerebral Artery Occlusion
Rats underwent 2 h of right middle cerebral artery occlusion (MCAo) by an intraluminal filament, as previously described [9]. In brief, the right common carotid artery bifurcation was exposed through a midline neck incision and the occipital artery branches of the external carotid artery were isolated, ligated, and dissected. After careful isolation of the internal carotid artery (ICA), a 3–0 monofilament coated with poly-l-lysine was advanced through the ICA to the MCA until mild resistance was felt. The neck incision was closed with a silk suture, and the animals were then allowed to recover. After 2 h of MCAo, rats were reanesthetized with the same anesthetic combination and intraluminal sutures were carefully removed.
Behavioral Tests
The animals underwent neurobehavioral assays by an observer blinded to the treatment groups at 60 min (during MCAo) and then on days 1, 3, and 7 after MCAo. The battery consisted of the postural reflex test and the forelimb placement test to visual, tactile, and proprioceptive stimuli. Neurological function was graded on a scale of 0–12 (normal score—0; maximum score—12) as previously described [9]. All behavioral tests were performed on the same days as the MRI scans, prior to the administration of anesthesia.
Treatment
The agents (LAU-0901; 60 mg/kg; n = 5) or vehicle (45% cyclodextran, 1 ml/kg; n = 6) were administered i.p. at the time of reperfusion, i.e., 2 h from onset of MCAo.
Magnetic Resonance Imaging
MRI was performed at 1, 3, and 7 days following MCAo. Rats were lightly anesthetized using isoflurane (1.0%) and imaged on a Bruker Avance 4.7 T MRI (Bruker Biospin, Billerica MA) as previously described [10]. A thermostat-controlled heated water cushion maintained body temperature at 35–37 ± 1°C. Two imaging data sets were acquired: (1) a six echo T2 and (2) a DWI sequence where each sequence collected 20 coronal slices (1 mm thickness and interleaved by a 1 mm). The T2 sequence had the following parameters: TR/TE = 4,600/10.2 ms, matrix = 1282, field of view (FOV) = 3 cm, and two averages. The DWI sequence had the following parameters: TR/TE = 1,000/25 ms, two b values (0.72, 1,855.65 s/mm2), matrix = 1282, FOV = 3 cm, and two averages. Total imaging time was 40 min. The apparent diffusion coefficient (ADC) maps were computed using an in-house MatLab routine, and ADC values were calculated for each pixel using the equation ADC = ln((So/Sn)/b), where Sn is the pixel intensity for a DWI, and So is the pixel intensity for the corresponding unweighted image. The T2-weighted maps were also computed on a pixel-by-pixel basis using a nonlinear least square curve fit to the data by using the equation, M(t) = Mo(1 − e −t/T2), where Mo is the initial magnetization before decay, t is the echo time (in milliseconds), and T2 is the spin–spin relaxation time. ADC and T2 maps were transferred to an analysis program, Cheshire™ (Hayden Image/PARAXEL International Corp.) for region of interest (ROI) analysis. Three-dimensional images were obtained to delineate the volumetric development of ischemic injury. Analysis included extraction of lesion, nonlesioned brain, and total brain volumes. ROI was drawn semiautomatically on each slice and manually checked. From these ROIs, the lesion volume was calculated over all slices (volume of lesion/total brain volume) and expressed as a percentage. T2-weighted imaging (T2WI) was used to assess the lesion volume estimation and to obtain tissue level T2 values. The T2 values provide a quantitative measure of the water “content” and hence edema. Conversely, DWI was acquired to estimate tissue water mobility quantitatively from the ADC. The tissue level water mobility is thought to reflect the level of tissue integrity [10].
Histopathology and Immunostaining
Animals were perfused with 4% paraformaldehyde on day 7, and brains were removed and embedded in a gelatin matrix using MultiBrain™ Technology (NeuroScience Associates, Knoxville, TN) as previously described [11]. Coronal sections were stained with thionine (Nissl), and immunohistochemical procedures were performed on the adjacent sections. To quantitate infarct volume, histological sections were digitized at nine standardized coronal levels and analyzed (MCIDTM Core imaging software, Linton, Cambridge, UK) as previously described [9]. Infarct volume was calculated as the integrated product of cross-sectional area and intersection distance and corrected for brain swelling. The following antibodies were used: glial fibrillary acid protein (GFAP; Santa Cruz, SDS Biosciences, Sweden) to label reactive astrocytes and Cd68/ED-1 (Serotec, Raleigh, NC) for activated microglia/microphages, and neuron-specific nuclear protein (NeuN; Chemicon/Millipore, Billerica, MA). The number of GFAP, ED-1, and NeuN-positive cells was counted (Zeiss Axio Imager 4.6.3) in the cortex and striatum at the level of the central lesion (bregma level −0.3 mm; magnification ×40). Data were expressed as numbers of positive cells per high-power microscopic field. Brain slices were imaged on a Zeiss LSM-510 Meta laser confocal microscope with a ×10 objective (Zeiss Plan-NEOFLUAR 10×/0.3). Fluorophore visualization (excitation/emission capture) was achieved as follows: GFAP: DyLight 488 (488 nm/505–530 nm, green) and ED1: DyLight 594 (594 nm/603–636 nm, red). The image resolution was set to 2.26 μm/pixel, and the cubic voxel dimension was 129.5 μm. Computer-generated MosaiX-processed images of NeuN, GFAP, ED-1, and GFAP/ED-1 double staining from vehicle and LAU-0901 rats were generated.
Statistical Analysis
Data are presented as mean values ± SEM. Repeated measures analysis of variance (ANOVA) followed by Bonferroni procedures to correct for multiple comparisons were used for intergroup comparisons. Two-tailed Student’s t tests were used for two-group comparisons. Differences at P < 0.05 were considered statistically significant.
Results
Physiological Variables
Rectal and cranial (temporalis muscle) temperatures, arterial blood gases, and plasma glucose showed no significant differences among groups (Table 1). LAU-0901-treated rats significantly increased body weight on day 7 compared to vehicle group (Table 1). There were no adverse behavioral side effects observed after LAU-0901 administration to rats in the present study.
Neurobehavioral Assessment
LAU-0901 treatment significantly improved behavioral scores compared to vehicle on days 1, 3, and 7 (Fig 2a). Significant improvement of postural reflex, visual, tactile, and proprioceptive placing reactions were observed at different times in LAU-0901-treated rats compared to the vehicle group (Fig. 2b–g).

a Total neurological score (normal score—0; maximal deficit—12) during MCAo (60 min) and at various times after treatment. At 60 min of MCAo, all animals had a score of 11 (of a possible 12). LAU-0901-treated rats had significantly improved neurological score on days 1, 3, and 7 compared to vehicle group. b–g Time course of recovery of visual, tactile, proprioceptive contralateral forelimb placing reactions, and postural reflex (normal score—0; maximal deficit—2) following MCAo in rats. Bar graphs show improvement of placing reactions in LAU-0901-treated rats vs. vehicle group. Values shown are mean ± SEM (vehicle group, n = 6 and LAU-0901 group, n = 5). Asterisk significantly different from corresponding vehicle group (P < 0.05; repeated measures ANOVA followed by Bonferroni tests)
MRI Lesion Volume and Brain Edema
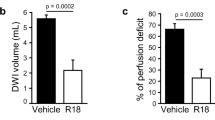
T2WI revealed large cortical and subcortical lesions and enlarged ventricules in vehicle rats (Fig. 3a). In contrast, LAU-0901-treated rats showed initial smaller infarcts that were only localized to the subcortex on days 1 and 3 and were almost indistinguishable from normal tissues by day 7. In addition, signficantly decreased cystic lesion development and decreased ventricular enlargement were observed in LAU-0901-treated rats (Fig. 3a). Total lesion volumes computed from T2WI were significantly reduced by LAU-0901 treatment on day 1 (by 83%), day 3 (by 90%), and day 7 (by 96%, Fig. 3b). When the cortical and subcortical regions were analyzed separately, LAU-0901 significantly reduced subcortical lesion volumes on day 1 (by 60%), day 3 (by 69%), and day 7 (by 86%), and cortical lesion volumes were practically nonexistent on days 1, 3, and 7 as compared to vehicle (Fig. 3b). Three-dimensional infarct volumes were computed from T2WI on days 1, 3, and 7 after MCAo (Fig. 3c). Lesion volume was dramatically reduced with LAU-0901 treatment and was mostly localized only in the subcortical areas of the brain. When the cortical and subcortical edema was quantified, LAU-0901 reduced cortical edema on days 1, 3, and 7 (by 36%, 27%, and 34%, respectively) and also subcortical edema on days 3 and 7 (by 20% and 22%, respectively) compared to vehicle-treated group (Fig. 4a). LAU-0901 increased the ADC in the cortex (by 153%) and striatum (by 132%) on day 1, consistent with increased water motility and cellular survival (Fig. 4b). These dynamic processes reflect repair mechanisms associated with LAU-0901 treatment of the brain lesion.

a Representative T2WI from vehicle and LAU-0901-treated rats. Enlarged ventricules and T2 hyperintensites were observed in the cortex and striatum of vehicle-treated rats out to 7 days, consistent with edema formation. In contrast, LAU-0901-treated animals had smaller lesion size, with only subcortical involvement and decreased ventricular size on days 1, 3, and 7 after MCAo. b Total, cortical, and subcortical lesion volumes, computed from T2WI, were significantly reduced by LAU-0901 treatment compared to vehicle group. Values shown are mean ± SEM (vehicle group, n = 6 and LAU-0901 group, n = 5). Asterisk significantly different from corresponding vehicle group (P < 0.05; repeated measures ANOVA followed by Bonferroni tests). Yellow arrows indicate brain hyperintensites and red arrows ventricle size. c 3D infarct volumes were computed from T2WI on days 1, 3, and 7 after MCAo. Vehicle-treated rats showed large cortical and subcortical infarct volumes that slowly decreased over the course of 7 days. By contrast, infarct volume was dramatically reduced in rats treated with LAU-0901 and was mostly localized in the subcortical areas. 3D reconstructions are from the same animal in each group over the 7-day time course

a Brain edema was measured from T2WI. Coronal brain diagram showing ROI for T2WI brain edema measurements. LAU-0901 reduced T2 values (decreased edema) within cortical and subcortical lesions. No differences in edema were detected on the contralateral side. b LAU-0901 increased the apparent diffusion coefficient (ADC) in cortex and striatum on day 1 consistent with water motility and cellular survival. Data are means ± SEM (vehicle group, n = 6 and LAU-0901 group, n = 5). Asterisk significantly different from vehicle-treated group (P < 0.05; repeated measures ANOVA followed by Bonferroni tests)
Histopathology
All animals survived uneventfully. Cortical and subcortical infarct areas were significantly smaller on multiple bregma levels compared to vehicle-treated rats (Fig. 5a). In fact, by day 7, no cortical infarct was observed in LAU-0901-treated rats. In addition, total, cortical, and subcortical infarct volumes were reduced by LAU-0901 compared with vehicle-treated rats (Fig. 5b). Representative images of Nissl, GFAP (marker for astrocytes), ED-1 (marker for microglia), and GFAP/ED-1 double staining are presented in Fig. 5c. Brains from vehicle-treated rats exhibited a pannecrotic lesion involving both cortical and subcortical regions of the right hemisphere (Fig. 5c). In addition, neuronal loss, GFAP-positive reactive astrocytes outlining the lesion territory, and massive ED-1-positive microglia/macrophage infiltration were observed in vehicle rats (Fig 5c). In contrast, LAU-0901 treatment attenuated damage as well as decreased ED-1-positive microglia/macrophages and increased GFAP-positive astrocytes. In order to determine the effect of LAU-0901 treatment on specific cell types, NeuN, GFAP, and ED-1-positive cells were quantified and compared to the vehicle group in the cortex (A, B, C) and subcortex (S; see brain diagram for ROI, Fig. 5d). LAU-0901 treatment significantly increased neuronal survival in the subcortex (by 95%) and cortical region B (by 114%), increased GFAP-positive reactive astrocytes in the subcortex (by 103%) and cortical regions B and C (116% and 48%), and decreased ED-1-positive microglia/macrophage infiltration in the subcortex (by 71%) and cortical regions B and C (by 83% and 90%) 7 days after MCAo (Fig. 5d).

a Cortical and subcortical infarct areas measured at nine coronal levels and b total infarct volume in rats on day 7 of survival. c Computer- generated MosaiX-processed images of GFAP (green), ED-1 (red), and GFAP/ED-1 double staining (overlay) on day 7 at a magnification ×10. d Coronal brain diagram showing locations of regions for cell counts in cortex (A, B, and C) and striatum (S). e Number of NeuN-positive neurons, GFAP-positive astrocytes, and ED-1-positive microglia cells and on day 7 after 2 h of MCAo. LAU-0901 treatment increased NeuN, GFAP, and decreased ED-1-positive cell counts relative to vehicle. Data are mean ± SEM (vehicle group, n = 6 and LAU-0901 group, n = 5). Asterisk significantly different from saline (P < 0.05; repeated measures ANOVA followed by Bonferroni tests)
Discussion
We have shown here that the novel PAF receptor antagonist, LAU-0901, provides neurobehavioral recovery, reduces brain infarction and brain edema, increases the apparent diffusion coefficient, attenuates microglial infiltration, and promotes astrocytic and neuronal survival when administered 2 h after onset of stroke in rats. Activation of signaling cascades occurs after stroke, which leads to the inflammatory modulation of metabolic, hemodynamic, and permeability properties; resolution is varied and complicated. One fundamental element in all of these signaling cascades, occurring early after the onset of brain ischemia, is the rapid release of unesterified essential fatty acids (arachidonic and docosahexaenoic acid) as well as lysophospholipids from membranes via phospholipase A2 activity [12]. Arachidonic acid is metabolized into leukotrienes, LTC4, D4, E4, prostaglandins, and prostacyclins, while lysophospholipids are converted into PAF. Under normal conditions, PAF is produced in nontoxic quantities in response to specific stimuli by a variety of cell types, including platelets, neutrophils, monocytes, eosinophils, and endothelial cells, and promotes memory formation via increased synaptic plasticity and long-term potentiation [13, 14]. During ischemia, elevated levels of PAF accumulate and have been shown to be neurotoxic via excessive glutamate release and the induction of proinflammatory cyclooxygenase 2 [15]. Excessive PAF also contributes to cerebral damage by increasing intracellular calcium, disrupting the blood–brain barrier, reducing cerebral blood flow, and stimulating leucocytes [2].
PAF antagonists have been shown to have many beneficial effects in various animal models of cerebral ischemia. They reduce brain injury after neonatal hypoxic ischemia in rats [16], global cerebral ischemia in gerbils [17, 18], and focal cerebral ischemia in rats [19] and mice [20]. PAF antagonists also have beneficial effects in other models of ischemia and reperfusion, including the reduction of myocardial reperfusion injury in swines [21], lung ischemia–reperfusion injury in canines [22], intestinal ischemia–reperfusion injury in mice [23], and acute hepatic injury [24] and renal ischemia injury in rats [25].
Recently, we have shown that LAU-0901, the novel PAF inhibitor, results in marked infarct volume reduction and neurobehavioral improvement in focal cerebral ischemia in rats and mice [7]. LAU-0901 significantly improved the neurological score compared with vehicle-treated rats at 1, 2, 3, and 7 days. In addition, all doses of LAU-0901 treatment (30, 60, and 90 mg/kg) significantly reduced total infarct volume compared to vehicle rats by 76%, 88%, and 90%, respectively. Mice treated with LAU-0901 (30 and 60 mg/kg) reduced total infarction by 29% and 66%, respectively. Furthermore, local cerebral blood flow (LCBF) continually increased in mice treated with LAU-0901 by 77% of baseline at 6 h as compared to an LCBF of 41% relative to baseline observed in the vehicle-treated rats [7]. In a follow-up long-term survival study, 60 mg/kg of LAU-0901, administered 2 h after onset of stroke, not only yielded short-term improvements (as observed on days 1, 2, 3, and 7) but also resulted in beneficial effects on behavioral outcome that persisted 2, 3, and 4 weeks after MCAo [8]. The histological results in LAU-0901-treated animals were concordant with the neurological outcome: namely, a 20% increase in preserved brain tissue at 30 days, a remarkable reduction in the incidence of extensive cystic–necrotic lesions, and markedly increased GFAP and Nissl-positive cell count [8]. The exact mechanisms of LAU-0901 against a multitude of pathogenic PAF actions are still not completely understood. However, recent studies showed that LAU-0901 reduces cell infiltration and expression of inflammatory mediators in a model of epithelial injury in mice [26] and induces hippocampal neuroprotection and limits somatostatin interneuronal cell loss and inflammation in experimental epilepsy in rats [27].
Brain edema is a serious complication of ischemic stroke and can lead to mechanical compression of adjacent brain structure, cerebral herniation, and death [28]. MRI is the most widely used technique for assessment of the brain edema and ischemic lesion. The present study used multimodal MRI to assess in vivo the neuroprotection conferred by LAU-0901. Multiparametric MRI (DWI, T2WI, ADC, 3D volume analysis) was performed to monitor both brain edema and lesion volume. We discovered that total lesion volumes computed from T2WI were significantly reduced by LAU-0901 treatment on days 1, 3, and 7 with decreased cystic lesion development and decreased ventricular enlargement. LAU-0901 dramatically reduced cortical edema on days 1, 3, and 7 and also subcortical edema on days 3 and 7 compared to the vehicle-treated group. LAU-0901 treatment also resulted in an increase of the ADC on day 1, consistent with increased water mobility and cellular survival, and reduced the persistence of edema at days 3 and 7 relative to vehicle-treated rats.
Inflammation can extend ischemic brain injury and adversely affect stroke outcome. Microglial activation is an early response to brain ischemia. Microglia continuously respond to changes in brain homeostasis and to specific signaling molecules released by neighboring cells. These signaling molecules, including ATP, glutamate, cytokines, prostaglandins, zinc, reactive oxygen species, and HSP60, may induce microglial proliferation and migration to the sites of injury [29]. Microglial activation requires hours to days to fully develop and thus presents a target for therapeutic intervention. Our results demonstrate that LAU-0901 treatment dramatically reduces microglial infiltration, which indicates a reduction of proliferation, activation, and subsequent release of proinflammatory mediators typical of microglia in environments with substantial cellular death and debris. In addition, LAU-0901 protects neurons directly as well as indirectly via astrocytic survival/secretion of neurotrophins and other neuronal supporting messengers. The contribution of astrocyte proliferation to reactive astrogliosis warrants specific consideration. It is now clear that reactive astrogliosis is not a simple all-or-none phenomenon but is a finely gradated variety of changes that occur in context-dependent manners regulated by specific signaling events [30]. Severe diffuse reactive astrogliosis is generally found in areas surrounding severe focal lesions. Recent experimental evidence indicates that these astrocyte scars act as neuroprotective barriers to inflammatory cells and infectious agents and that they form in particular along borders of severe tissue damage [30]. It is noteworthy that the glial scar formation is associated with substantive tissue reorganization and structural changes that are long lasting and persist long after the triggering insult may have resolved. These data taken together provide a basis by which LAU-0901 attenuates ischemia–reperfusion injury via neuronal protection, astrocytic proliferation, reductions in microglial infiltration, and the inhibition of edema formation and persistence.
Since the ultimate goal of any stroke therapy is the restoration of functions that allow for a normal daily life of patients, an objective appraisal of the behavioral deficits should be performed. Several behavioral tests have been applied to ischemia research in regards to clinical criteria. A recent report has called attention to using standardized procedures for assessing functional outcome to improve translation from bench to bedside [31]. Two tests of the sensorimotor battery appear to be particularly sensitive in detecting deficits following MCAo: the postural reflex test and the forelimb placing test [32]. LAU-0901 treatment significantly improved total neurological score and visual, tactile, and proprioceptive reactions compared to vehicle on days 1, 3, and 7.
The beneficial effect of LAU-0901 has been shown in a well-controlled animal model of MCAo. In the current study, we used a poly-l-lysine-coated suture and have found that this method yields reliable and highly consistent results (coefficient of variation of infarct volume, 9%) [7, 9]. This infarction closely mimics the large hemispheric infarcts resulting from proximal MCA and ICA occlusion in patients. We demonstrated that LAU-0901 did not have direct effects on body and cranial temperatures or arterial blood gases because these variables were carefully controlled and did not differ among groups.
Conclusion
We have shown that LAU-0901, a novel PAF inhibitor, provides neurobehavioral recovery, reduces brain infarct lesion volume and brain edema as observed by MRI, and promotes cell survival when administered after focal cerebral ischemia in rats. These findings suggest LAU-0901 is a promising neuroprotective agent and provide support for future clinical studies to confirm whether LAU-0901 is beneficial for patients suffering ischemic stroke.
References
Armstead WM, Ganguly K, Kiessling JW, Riley J, Chen XH, Smith DH, et al. Signaling, delivery and age as emerging issues in the benefit/risk ratio outcome of tPA for treatment of CNS ischemic disorders. J Neurochem. 2010;113:303–12.
Bazan NG. Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol Neurobiol. 2005;32:89–103.
He J, Bazan HE. Synergistic effect of platelet-activating factor and tumor necrosis factor-alpha on corneal myofibroblast apoptosis. Invest Ophthalmol Vis Sci. 2006;47:883–91.
Cortina MS, Gordon WC, Lukiw WJ, Bazan NG. Oxidative stress-induced retinal damage up-regulates DNA polymerase gamma and 8-oxoguanine-DNA-glycosylase in photoreceptor synaptic mitochondria. Exp Eye Res. 2005;81:742–50.
Esquenazi S, He J, Bazan HE, Bazan NG. Prevention of experimental diffuse lamellar keratitis using a novel platelet-activating factor receptor antagonist. J Cataract Refract Surg. 2004;30:884–91.
Boetkjaer A, Boedker M, Cui JG, Zhao Y, Lukiw WJ. Synergism in the repression of COX-2- and TNFalpha-induction in platelet activating factor-stressed human neural cells. Neurosci Lett. 2007;426:59–63.
Belayev L, Marcheselli VL, Khoutorova L, Rodriguez de Turco EB, Busto R, Ginsberg MD, et al. Docosahexaenoic acid complexed to albumin elicits high-grade ischemic neuroprotection. Stroke. 2005;36:118–23.
Belayev L, Khoutorova L, Atkins K, Cherqui A, Alvarez-Builla J, Bazan NG. LAU-0901, a novel platelet-activating factor receptor antagonist, confers enduring neuroprotection in experimental focal cerebral ischemia in the rat. Brain Res. 2009;1253:184–90.
Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616–22.
Ashwal S, Tone B, Tian HR, Chong S, Obenaus A. Comparison of two neonatal ischemic injury models using magnetic resonance imaging. Pediatr Res. 2007;61:9–14.
Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–65.
Bazan NG, Birkle DL, Reddy TS. Docosahexaenoic acid (22:6, n-3) is metabolized to lipoxygenase reaction products in the retina. Biochem Biophys Res Commun. 1984;125:741–7.
Izquierdo I, Fin C, Schmitz PK, Da Silva RC, Jerusalinsky D, Quillfeldt JA, et al. Memory enhancement by intrahippocampal, intraamygdala, or intraentorhinal infusion of platelet-activating factor measured in an inhibitory avoidance task. Proc Natl Acad Sci U S A. 1995;92:5047–51.
Kato K, Clark GD, Bazan NG, Zorumski CF. Platelet-activating factor as a potential retrograde messenger in CA1 hippocampal long-term potentiation. Nature. 1994;367:175–9.
Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a platelet-activating factor antagonist. J Biol Chem. 1996;271:24794–9.
Bozlu G, Atici A, Turhan AH, Polat A, Nayci A, Okuyaz C, et al. Platelet-activating factor antagonist (ABT-491) decreases neuronal apoptosis in neonatal rat model of hypoxic ischemic brain injury. Brain Res. 2007;1143:193–8.
Panetta T, Marcheselli VL, Braquet P, Bazan NG. Arachidonic acid metabolism and cerebral blood flow in the normal, ischemic, and reperfused gerbil brain. Inhibition of ischemia–reperfusion-induced cerebral injury by a platelet-activating factor antagonist (BN 52021). Ann N Y Acad Sci. 1989;559:340–51.
Panetta T, Marcheselli VL, Braquet P, Spinnewyn B, Bazan NG. Effects of a platelet activating factor antagonist (BN 52021) on free fatty acids, diacylglycerols, polyphosphoinositides and blood flow in the gerbil brain: inhibition of ischemia-reperfusion induced cerebral injury. Biochem Biophys Res Commun. 1987;149:580–7.
Aspey BS, Alp MS, Patel Y, Harrison MJ. Effects of combined glutamate and platelet-activating factor inhibition on the outcome of focal cerebral ischaemia—an initial screening study. Metab Brain Dis. 1997;12:237–49.
Subbanna PK, Tyagi MG. PAF antagonism modifies neuroprotective action of histone deacetylase and calcineurin phosphatase inhibitors in mice. Indian J Exp Biol. 2006;44:886–91.
Loucks EB, Qayumi AK, Godin DV, English JC, Lim SP, Al Mahmeed T, et al. Therapeutic potential of platelet-activating factor antagonism in the management of myocardial infarction. Can J Cardiol. 2000;16:497–504.
Iwazaki S, Takeyoshi II, Ohwada S, Sunose Y, Aiba M, Tsutsumi H, et al. FR128998 (a PAF receptor antagonist) counters the increased pulmonary vascular resistance associated with ischemia–reperfusion injury in the canine lung. Int J Angiol. 2001;10:10–4.
Souza DG, Pinho V, Soares AC, Shimizu T, Ishii S, Teixeira MM. Role of PAF receptors during intestinal ischemia and reperfusion injury. A comparative study between PAF receptor-deficient mice and PAF receptor antagonist treatment. Br J Pharmacol. 2003;139:733–40.
Grypioti AD, Kostopanagiotou G, Demopoulos CA, Roussos A, Mykoniatis M. Platelet activating factor (PAF) antagonism with ginkgolide B protects the liver against acute injury. Importance of controlling the receptor of PAF. Dig Dis Sci. 2008;53:1054–62.
Lloberas N, Cruzado JM, Torras J, Herrero-Fresneda I, Riera M, Merlos M, et al. Protective effect of UR-12670 on chronic nephropathy induced by warm ischaemia in ageing uninephrectomized rats. Nephrol Dial Transplant. 2001;16:735–41.
Esquenazi S, He J, Li N, Bazan NG, Esquenazi I, Bazan HE. A novel platelet activating factor receptor antagonist reduces cell infiltration and expression of inflammatory mediators in mice exposed to desiccating conditions after PRK. Clin Dev Immunol. 2009;2009:138513.
Musto AE, Samii M. Platelet-activating factor receptor antagonism targets neuroinflammation in experimental epilepsy. Epilepsia. 2011;52:551–61.
Balami JS, Chen RL, Grunwald IQ, Buchan AM. Neuorological complications of acute ischaemic stroke. Lancet Neurol. 2011;10:357–71.
Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7:378–91.
Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35.
Freret T, Schumann-Bard P, Boulouard M, Bouet V. On the importance of long-term functional assessment after stroke to improve translation from bench to bedside. Exp Transl Stroke Med. 2011;3:6.
Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006;3:497–504.
Acknowledgments
This investigation was supported by NIH, NINDS Grant R01 NS046741 (NGB). Tiffany N. Eady is a recipient of the Ruth L. Kirschstein National Research Service Awards for Individual Predoctoral MD/PhD and Other Dual Doctoral Degree Fellows (NIH, NIA Grant F30 AG032841). We thank Dr. Kamalakar Ambadipudi and Sonny Kim for the technical assistance, Dr. Hilary Thompson for statistical analysis, and Neuroscience Associates, Inc. the for histology service.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Belayev, L., Eady, T.N., Khoutorova, L. et al. Superior Neuroprotective Efficacy of LAU-0901, a Novel Platelet-Activating Factor Antagonist, in Experimental Stroke. Transl. Stroke Res. 3, 154–163 (2012). https://doi.org/10.1007/s12975-011-0116-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-011-0116-y