
Abstract
Congenital disorders are an important cause of pregnancy loss, premature death and life-long disability. A range of interventions can greatly reduce their burden, but the absence of local epidemiological data on their prevalence and the impact of interventions impede policy and service development in many countries. In an attempt to overcome these deficiencies, we have developed a tool—The Modell Global Database of Congenital Disorders (MGDb) that combines general biological principles and available observational data with demographic data, to generate estimates of the birth prevalence and effects of interventions on mortality and disability due to congenital disorders. MGDb aims to support policy development by generating country, regional and global epidemiological estimates. Here we provide an overview of the concepts and methodological approach used to develop MGDb.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
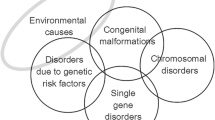
Congenital disorders, often also called birth defects, include “any potential pathological conditions arising before birth, whether they are evident at birth or become manifest later in life” (World Health Organization 1985a; World Health Organization 2000; World Health Organization 2010). Risk factors for congenital disorders include genetic, environmental and wider societal factors. The occurrence and severity of specific congenital disorders are differentially influenced by these risk factors, with some disorders influenced more by genes (e.g. single gene disorders) and others by environmental agents (e.g. those caused by infections). They are an important cause of premature death or life-long disability; however, the absence of local epidemiological data on their birth prevalence and outcomes impedes policy and service development in many countries.
To make up for this deficiency, the Modell Global Database of Congenital Disorders (MGDb) was created to meet the information needs of health policy-makers. It does so through the generation of country, regional and global estimates for selected congenital disorders, by combining general biological principles and available observational data with demographic data. Here we describe the scope of MGDb and the general principles followed in its implementation; more detailed methodology can be found in other articles within this journal supplement and in the UCL repository (Modell et al. 2016).
Terminology
MGDb uses a set of defined methods to relate demographic data to the known birth prevalence of selected congenital disorders, in order to generate estimates relevant to public health, policy-making and clinical practice. The creation of this tool requires precise definitions of all input data and outputs. Table 1 provides a list of some of the key terms along with their definitions within the context of this tool; a comprehensive glossary can be found in the UCL repository (Modell et al. 2016).
Scope of conditions
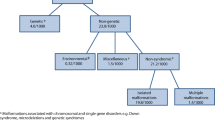
Only severe, early-onset congenital disorders that cause early death and/or life-long disability in the absence of care and present before 20 years of age are included in MGDb. The disorders currently modelled by MGDb are shown in Table 2. The rationale for modelling these disorders, where data are unavailable, is that their birth prevalence in the absence of access to diagnosis and care (baseline birth prevalence) is relatively constant in any given population. Estimation of the birth prevalence of these conditions can therefore be undertaken assuming that this will vary only with identified factors, such as interventions (e.g. folic acid fortification, prenatal diagnosis, surgical repair). Modelled estimates can be generated on a country-specific annual basis of potential (in the absence of interventions) and actual affected births. Thus, the tool can also be used to estimate the actual effects of any proposed future interventions. Although the same method can be applied to congenital disorders that arise as a result of external risk factors, such as maternal exposure to infection, malnutrition, or teratogens, at present there are insufficient input data at the global level to define a baseline birth prevalence and outcomes of these disorders. This is due to lack of precise estimates of risk associated with these exposures and the fact that risk varies more widely with place and time, requiring country-specific data.
The congenital malformation group includes the International Classification of Disease (ICD 10) (World Health Organization 2016) system groups used in most congenital anomaly registries and focuses on isolated malformations (i.e. not those associated with chromosomal disorders or genetic syndromes. Disorders associated with other malformations contribute to the multiple malformations group. Neural tube defects, orofacial clefts and congenital heart defects, are treated separately, as data relating to these malformations are more readily available. The chromosomal disorder group includes all chromosomal disorders that cause substantial disability for the affected person. In MGDb, Edwards and Patau syndrome are treated together as “other trisomies” because their outcomes are very similar. Inherited disorders have been bundled into four broad groups. Rare single gene disorders include dominant, X-linked and recessive disorders that are expected to have a similar collective birth prevalence worldwide, because their gene frequency is mainly determined by the balance between new mutation rate and natural selection, neither of which is thought to vary greatly between populations. The common recessive disorders group includes three conditions (sickle cell disorder, thalassaemia and oculocutaneous albinism) for which there is known global variation in carrier prevalence. Consanguinity-associated disorders refer to the increment of recessive disorders that are associated with parental consanguinity (Corry 2014). Disorders due to genetic risk factors are caused by the interaction of (often very common) DNA variants with other genetic and environmental factors. MGDb includes two early-onset examples (rhesus haemolytic disease of the newborn and neonatal jaundice due to G6PD deficiency) because (a) the underlying mechanisms are exceptionally well understood and (b) they are potentially lethal but can be effectively prevented and/or treated.
Overview of the methodological approach
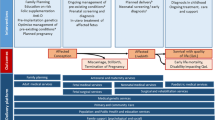
The objective of MGDb is to estimate numbers of births affected by one or more congenital disorders, and outcomes in the no-care situation and with current care. Figure 1 shows the range of possible outcomes and their modification by a variety of diagnostic and therapeutic interventions. The effect of some interventions is quantifiable as data relating to their impact are available, for example, folic acid food fortification, the identification of genetic risk and the option of termination of pregnancy. In addition to the impact of particular interventions, their availability and ease of access must be taken into account (Blencowe et al. 2017). Estimates were derived in a stepwise manner, beginning with baseline birth prevalence and outcomes in the absence of interventions (Fig. 1). Once an estimate is available for baseline birth prevalence, outcomes may be calculated based on estimated actual live birth prevalence, estimated access to services and survival with optimal or very limited care.

The sequence of events covered in MGDb and stepwise modelling process used to derive estimates for specific congenital disorders. The outcomes that are impacted by interventions are shown in italics
Sources of data
Demographic data
Demographic data not only provide the basis for quantitative epidemiological estimates and the assessment of service needs, they offer an overall picture of many aspects of a population. It is particularly important to recognise the speed of demographic change; all epidemiological estimates need to take account of this dynamic background. The principal source of demographic data is the UN World Population Prospects (UN WPP) (United Nations Population Division 2015), with other data sources used for specific indicators (Bittles and Black 2015; Blencowe et al. 2017; Institute for Health Metrics and Evaluation (IHME) 2015; Inter-agency Group for Child Mortality Estimation (UN-IGME) (2015); UNAIDS 2016). Table 3 provides a summary of the demographic indicators and their sources used to generate estimates for the prevalence and outcomes of congenital disorders.
Baseline birth prevalence
The frequency of a disorder in a population is usually described in terms of incidence (number of new cases arising in a defined time-period) and prevalence (number of cases actually present in a given unit of population, e.g. per thousand or per million). However, the incidence of congenital disorders is usually expressed as birth prevalence because although many congenital disorders arise at or around the time of conception, many affected embryos either fail to implant or miscarry in early pregnancy, and so never come to the attention of health services. Therefore, for practical purposes, their prevalence at birth is counted as their incidence, and expressed as rate per 1000 live births. MGDb follows the European Surveillance of Congenital Anomalies and Twins (EUROCAT) convention in counting all pregnancy outcomes after 20 weeks gestation as births. In keeping with ICD-10, all births with any signs of life following separation from the mother, regardless of gestation, are counted as live births.
Baseline, or potential, birth prevalence is the prevalence that would apply in the absence of any intervention. It is the foundation of the MGDb and provides the “envelope” into which all outcomes must fit. It includes stillbirths and live births but excludes miscarriages, and uses “foetal death” (death in utero after 20 weeks’ gestation) as a proxy for stillbirth. Once baseline birth prevalence is known, when appropriate observational data are available baseline births can be allocated to each potential outcome (Fig. 1) thus generating an overall epidemiological picture of the current status of each disorder group.
The baseline birth prevalence for most countries worldwide can be calculated from available data or estimated for many congenital disorders (Table 2). The methodology used to obtain these estimates is described in detail elsewhere (Modell et al. 2016). Briefly, the birth prevalence of maternal age-related chromosomal disorders can be calculated from maternal age distributions for which global data are available (Moorthie et al. 2017). The Hardy-Weinberg (H-W) equation can be used to calculate the birth prevalence of haemoglobin disorders, consanguinity-related disorders, rhesus haemolytic disease and susceptibility to neonatal jaundice due to G6PD deficiency, from available global data on carrier prevalences (Abbas and Yunis 2014; Bittles and Black 2015; Piel et al. 2013a, b). With regard to congenital malformations, there are sufficient observational data available in the literature and from registries on neural tube defects and orofacial clefts to make estimates for countries in every region (Modell et al. 2016; Blencowe et al. 2017), since these severe malformations are evident at birth.
However, data on all other congenital malformations and rare single gene disorders are only available in high-income settings. To generate estimates for congenital malformations for countries outside high-income settings, we have utilised publicly accessible data from the EUROCAT website to obtain average European rates for birth prevalence (Moorthie et al. 2017; Modell et al. 2016). Studies examining ethnic differences have only shown variation for polydactyly, congenital hypothyroidism, orofacial clefts and neural tube defects (Chitty and Winter 1989; Petrini et al. 1997; Terry et al. 1985; Yang et al. 2004). In addition, data from the International Clearinghouse for Birth Defects Surveillance and Research (ICBDSR) indicate little inter-ethnic variation for other malformations. As a result we have assumed that the baseline birth prevalence for these conditions is little affected by ethnicity; therefore, average European rates have been used to generate estimates for countries with no or insufficient observational data (Modell et al. 2016). In the absence of more recent data on the birth prevalence of rare single gene disorders, we have utilised the rates of Baird et al. (Baird et al. 1988) to represent global collective baseline birth prevalences. However, the contribution of specific single gene disorders to collective birth prevalence is known to vary in countries, especially where there are founder effects.
Data on outcomes in the absence of diagnosis and care, and with optimal care (i.e. with the interventions generally available in high-income settings), can be obtained for most groups of congenital disorders. In any setting, a proportion of the population has access to optimal care; however, there is no routinely collected indicator from all countries that provides this information. We have therefore developed a method for estimating the proportion of access to optimal care using infant mortality as a proxy indicator (Modell et al. 2016). Country-specific estimates of outcomes can then be calculated, based on outcomes in the absence of care and with optimal care, and the proportion of the population with access to care (Modell et al. 2016).
There are only three possible outcomes for early-onset congenital disorders in the absence of interventions–foetal death, life with some degree of disability and premature death (Fig. 1). The types and availability of specific interventions have evolved over time, impacting on birth prevalence for specific disorders in different time-periods and the subsequent number of survivors (Modell et al. 2016). Increased access to prenatal diagnosis and termination of pregnancy can act to reduce birth prevalence whilst improvements in diagnosis and care impact on outcomes. Provision of early diagnosis and care can greatly reduce annual numbers surviving with disability for disorders with effective cures. For children with incurable disorders it can both prolong survival and ameliorate the levels of disability. As a result, there may be a steady annual increase in the cumulative number of individuals living with these disorders and requiring appropriate care. The evolution of this effect needs to be quantified in order to assess current and future patient numbers and service needs. A full description of the sources of data on the impact of interventions is available elsewhere (Blencowe et al. 2017; Modell et al. 2016).
Under-5 mortality
For most congenital disorders mortality is highest within the first 5 years of life, the period for which survival data with no care and with optimal care are most complete and most reliable. To avoid double counting, it is important to allow for the overlaps inherent in multiple causes of death. For assessing total deaths of people with a given disorder, all deaths of affected individuals must be included, whatever the cause. However, when the aim is to assess attributable deaths–deaths that are specifically due to a defined disorder, the background mortality should also be considered. This is because some affected individuals who would have died of their disorder in fact die earlier from unrelated causes, and the proportion of such deaths varies with place and time.
To obtain attributable early deaths, numbers are adjusted for deaths from other causes using country rates for neonatal, infant and under-5 deaths (Modell et al. 2016). The adjustment makes relatively little difference in countries where early background mortality is low, but when background mortality is high a sizeable difference is observed.
Long-term survival
Table 4 shows the main sources used for estimating survival with limited care, and with optimal care, defined as the best care available around the time the patients were born. The upper age limit of the observational data is also shown. This allows the construction of survival curves in a no-care and an optimal care situation which were then used to estimate number of survivors.
Lifetime survival curves with optimal care are only available for some congenital disorders—Down syndrome, spina bifida, orofacial clefts, and haemoglobin disorders. For most other disorders, observational survival data are only available to age 20 or 30 years. Lifetime survival curves were completed for these disorder groups by extrapolating the observed rate of attrition in the last full 5-year interval recorded to 80 years of age. Normal survival is expected for correctable congenital malformations, congenital hypothyroidism and disorders due to genetic risk factors, as optimal care should allow for effective cure or appropriate management of these conditions. Long-term survival in the absence of care is based on literature available from high-income settings in the 1950s for spina bifida, Down syndrome, congenital heart disease and haemoglobin disorders and on expert opinion for most other disorders (Table 4).
Lifetime survival curves are used to project future patient numbers, but they need adjustment for use in calculating current patient numbers, since mortality is usually higher in the early years. The current number of survivors in each age group is largely determined by the services available around their year of birth. Retrospective survival curves were derived from prospective curves, taking into consideration the historical evolution of services, for use in calculating the number and age distribution of patients living at any given time. For this purpose, survival data for each phase in the evolution of care were obtained from the literature (Modell et al. 2016).
Disability and cure with optimal care and with no care
Definitive cure of conditions is limited to operable congenital malformations. Survivors with most other disorders live with some disability. The survival outcome can range from well (e.g. congenital hypothyroidism with regular replacement treatment) to the very severe (e.g. associated severe physical and mental disability). Due to this diversity, quantification of physical disability is limited to estimates of the proportion of survivors at age 5 with severe disability (including reduced life expectancy), less severe disability (with lesser or no effect on life expectancy) and effectively cured.
Data regarding long-term disability outcomes, depending on the levels of access to care, were obtained through literature review. The proportion of each population without optimal care is assumed to have no access to care. Although we have used the term no-care, this does not refer to the absolute lack of care as this is unlikely to be the case in reality, with supportive care at home or in a facility available in many settings. In addition, we have used estimates for survival with no care based on available data, which in large part is based on survival in the 1950s in “high income” settings (Modell et al. 2016); consequently the survival that we have estimated is likely to be an overestimate for a total absence of care.
Examples of MGDb outputs by WHO region
The aim of the present exercise is to support health policy makers and practising clinicians at the country level. Outputs are therefore generated to provide a basis for assessing non-financial and financial costs and benefits, and to allow critical comparison with other estimates. MGDb output data are generated for each disorder group by country; these can be aggregated by WHO region. All outputs are expressed in terms of annual numbers and rate per 1000 live births.
Baseline birth prevalence and actual birth prevalence
Figure 2 shows the estimated baseline birth prevalence of the groups of congenital disorders by WHO region. All charts also include rates for Western Europe because most currently available interventions are deployed in this sub-region near-equitably at high coverage, and surveillance data are available. Observed outcome data may therefore be used to describe the “power” of each intervention when fully deployed at the population level. There is little inter-regional difference in the baseline prevalence of chromosomal disorders, congenital malformations and baseline single-gene disorders. Most of the inter-region difference is due to genetic disorders where there are inter-country differences in carrier prevalence, or according to the prevalence of consanguinity.

Estimated baseline total birth prevalences of congenital disorder by WHO region. AFR African, AMR American, EMR Eastern Mediterranean, SEAR South-East Asian, WPR Western Pacific Region, W Europe Western Europe
Figure 3 shows the corresponding distribution of three possible outcomes in a situation with limited access to care, providing a baseline for assessing the effects of interventions. The estimated effect of pre-birth interventions on affected birth prevalence in 2010–2014 by type of intervention can be seen in Fig. 4. Globally, the largest contributions were from anti-D for rhesus negative mothers and termination of pregnancy for foetal impairment. The effect of pre-pregnancy genetic counselling is relatively small because in most cases risk is only detected retrospectively, i.e. after the diagnosis of an affected child (Modell et al. 2016).

Estimated outcomes of congenital disorders if no intervention, by type of outcome, 2010–2014. Note: The small discrepancy between total affected birth prevalence in the previous chart and total outcomes represents estimated under-5 deaths due to other causes (omitted for the sake of clarity). AFR African, AMR American, EMR Eastern Mediterranean, SEAR South-East Asian, WPR Western Pacific Region, W Europe Western Europe

Estimated reduction in affected births per 1000 due to pre-birth interventions, by WHO region, 2010–2014. AFR African, AMR American, EMR Eastern Mediterranean, SEAR South-East Asian, WPR Western Pacific Region, W Europe Western Europe, FA Folic acid
Birth outcomes, outcome at 5 years of age and effect of interventions
The estimated distribution of actual outcomes in 2010–2014, taking account of interventions and charted within the envelope of potential birth prevalence, can be seen in Fig. 5. By definition, the sum of all outcomes fits into the envelope of baseline birth prevalence. Rates for Western Europe indicate that all available interventions, when deployed at the population level, reduce mortality due to congenital malformations, chromosomal disorders, the two genetic risk factors and haemoglobin disorders by over 80%, but there is far less effect on mortality due to other single-gene disorders. Effective cure is possible for many congenital malformations by paediatric surgery (online resources Fig. 1). Table 5 shows the estimated per cent reduction in unfavourable outcomes due to interventions at 5 years of age, in 2010–2014. The greatest proportional reduction is in under-5 deaths. There is much less reduction in disability—in fact, disability at age 5 due to congenital disorders was increasing in the Eastern Mediterranean, African and South-East Asian regions.

Estimated actual outcomes of congenital disorders by type of outcome, 2010–14. AFR African, AMR American, EMR Eastern Mediterranean, SEAR South-East Asian, WPR Western Pacific Region, W Europe Western Europe
Mean life expectancy
Table 6 shows disorder-specific mean life expectancy with no care and optimal care, calculated from the full survival curves. The difference between life expectancy with no care and with optimal care measures the survival benefit of care. Mean life expectancy is used to calculate costs of the disorder and the benefits of interventions, in terms of years of life lost, lived with disability or lived cured. The rates in Table 6 represent life expectancy with the disorder in the absence of any other cause of death. These “ideal” rates are adjusted by multiplying local life expectancy divided by a notional optimal life expectancy of 80 years.
Years affected per person born is the same as local mean life expectancy. This provides the envelope for all estimates of outcomes. The world average is 2.5 years affected per person born (range 3.25 for the EMR to 2.2 for WPR). In the absence of intervention, they would be responsible for 2 years loss of life per person born (range 1.7–2.7 years). Figure 6 shows estimated outcomes in terms of years of life lost, lived with disability or lived cured per person born in 2010–2014. The striking reduction in death and disability in Western Europe demonstrates the potential of global implementation of interventions for congenital disorders.

Estimated actual outcomes for total congenital disorders, expressed as years of life lost, lived with disability or lived cured per person in the relevant birth cohort, 2010–2014, by WHO region
Estimated number and age distribution of living patients
To plan for current service needs and project future changes, policy-makers need to know the approximate numbers of persons presently living with specific disorders. This information can only be reliably obtained using a patient register. However, few such registers exist even in high-income settings, even for readily diagnosable disorders such as Down syndrome. The method we have developed extends the principles described above (i.e. consideration of availability of interventions, their impact and evolution along with demographic data) to estimate the number and age distribution of patients currently receiving care. As WPP provides age distribution data from 1950 onwards, it is possible to generate estimates in any year from 1950 onwards, allowing a comprehensive picture of the history of selected disorders up to a chosen year. As an example, Fig. 7 shows global estimates for spina bifida by 5-year age intervals in 2010.

Estimated distribution of outcomes for spina bifida in 2010. The total outline shows the number of individuals who would be living with spina bifida if survival equalled the population norm, according to age in 2010 (6.98 million). The outline reflects the age distribution of the world population in 2010. The green fill shows cases avoided by folic acid food fortification (only 56,000 in 2010 because fortification only started to become policy in the late 1990s). The orange fill shows cases avoided by termination of pregnancy (196,000 in 2010). The grey fill shows deaths that would have occurred even with best care available when the affected person was born (unavoidable deaths). The great majority of deaths occurred soon after birth, but they appear in all age groups because the chart shows all outcomes. The black fill shows the numbers of deaths that occurred because of lack of access to available care (avoidable deaths) (total losses from the current world population due to spina bifida = 6.35 million). The blue fill shows estimated numbers living with spina bifida in 2010 (399,000)
Future projections
Future projections of the likely effects of implementing interventions, such as folic acid food fortification or prenatal diagnosis with the option of termination of pregnancy are of particular interest to policy-makers. They can be calculated as follows: (a) assuming no change in present policies and (b) assuming worldwide spread of available interventions. Table 7 summarises the estimated long-term effects of different policy decisions for spina bifida at a global level. All of the estimates take account of change in access to care during the time period involved.
Discussion
The absence of epidemiological data on congenital disorders has led to the creation of MGDb. However, as with all modelling initiatives, the estimates produced can only be as accurate as the input data, and they are approximations only. We emphasise that our aim is only to produce order of magnitude estimates that can be used as a starting point for service planning. They must not be considered as definitive, and require continuous improvement and refinement as better information becomes available. In undertaking this task, it proved necessary to clarify some concepts and develop some new methods to address gaps in available observational data. Articles within this journal describe some methodological aspects and further details can be accessed via the UCL repository (Modell et al. 2016).
Most estimates generated by MGDb are likely to be underestimates. This is because it is difficult to diagnose many congenital disorders, especially when they cause stillbirth or early death, or in settings with limited diagnostic capacity, which can lead to death and disability being attributed to other causes and a lack of reliable prevalence data. Reliable cause of death data can only be obtained in high-income settings where, by definition, prevention and care are also available. Therefore, if medically certified death rates are assumed to apply globally, early deaths due to congenital disorders will be greatly underestimated. In addition, wherever possible attempts were made to avoid overestimation; some examples are given below. The estimates for single gene disorders are based on older literature and hence only cover conditions diagnosable at that time; but with advances in DNA technology new single gene disorders are being steadily identified (Boycott et al. 2013). The rates for effect of parental consanguinity are largely based on the work of Alan Bittles; but this largely estimates mortality rather than mortality plus disability (Bittles 2003; Bittles and Black 2010), and rates for Rhesus haemolytic disease are lower than those published elsewhere (Bhutani et al. 2013).
Strengths
The strength of this approach lies in beginning with baseline birth prevalence, which provides the most robust estimates. It provides the envelope for all possible outcomes; consequently, overestimation of one outcome leads to underestimation of another, for example overestimation of mortality leads to underestimation of disability. We have endeavoured to address factors that can lead to overestimation of mortality and prevalence as detailed by Liu et al.(Liu et al. 2012, 2002) and described below.
-
1.
Potential bias towards high prevalence populations, which is a possibility when rates are based on the literature. However, we have endeavoured to use rates based on international registry data. For high-income settings the rates come from EUROCAT, which contains population-based data. Most data for lower-income settings has been obtained from ICBDSR, and most participating registries are hospital-based. This could result in bias due to selective referral of high risk pregnancies, but is unlikely as this often requires routine foetal anomaly scanning. Gene frequency data for haemoglobinopathies, G6PD deficiency and rhesus negativity are based on many large-scale population surveys. Where data were from selected populations, they were excluded.
-
2.
The uncertainty in applying prevalence estimates derived from largely European populations to populations with different fertility patterns, or specific genetic and environmental contexts. For congenital malformations, comparative studies by ethnic group in high-income settings yield comparable rates with two exceptions: (a) a lower birth prevalence of neural tube defects and orofacial clefts among people of Sub-Saharan African origin, and (b) an increased prevalence of congenital malformation syndromes in groups in which consanguineous marriage is customary. Allowances have been made for differences in maternal age distribution, total fertility rate and observed ethnic differences in the birth prevalence of NTD, OFC, CHT and the effect of parental consanguinity by using country-specific data for these conditions and estimating the impact of consanguinity.
-
3.
The fact that some estimates include stillbirths and terminations as well as live births. Due to differences in the definition of stillbirths we have used foetal deaths as an approximation for stillbirths. As many terminations occur before 20 weeks, we have adjusted TOP rates to those that would apply from 20 weeks onwards. In addition, when observed data applied only to live births we added estimates for foetal deaths based on data from registries and the literature.
-
4.
Double counting of infants with more than one congenital disorder. We ensured throughout that numbers and rates apply for affected individuals within the groups specified in Table 1 and created a separate category for cases with more than one congenital malformation.
-
5.
Overestimating mortality due to congenital disorders in lower income settings. Estimates for mortality in a no-care situation are based on historical data from high-income settings and are likely to underestimate a truly no-care situation.
Weaknesses
As improved data become available from other regions, it will be important to test the assumptions made and to modify them as necessary. The biggest assumptions relate to our estimate of access to care and long-term survival, due to the paucity of data in these areas. A further limitation of this work is a paucity of population-based, cause-specific fatality rate data for varying care settings, necessitating reliance on historical data or expert opinion. There is an urgent need for improved outcome data to verify or amend these estimates. This is an important area for future research, to fully document the long-term impact of living with these conditions and improving the estimates available to more fully quantify the benefits arising from interventions.
Differences from other estimates
Currently, the only available national and regional estimates for mortality and disability due to congenital disorders were undertaken as part of the Global Burden of Diseases (GBD) study. GBD estimates are limited to congenital anomalies, defined as disorders included in ICD10 chapter VII (the Q chapter)–“Congenital malformations, deformations and chromosomal abnormalities”; that is, they deal only with developmental structural anomalies. Congenital anomaly groups specified in the GBD are Down syndrome, unbalanced chromosomal rearrangements, neural tube defects, orofacial clefts, congenital heart anomalies and other congenital anomalies. These exclude single-gene disorders or genetic risk factors modelled in MGDb; consequently, they were excluded from the comparison.
Table 8 summarises the results of a comparison of estimates of under-5 deaths produced by the GBD (Lopez et al. 2006b) and MGDb. There is reasonable correspondence between estimates for high-income regions and for East and Central Europe where vital registration data are available, but with wide divergences in other regions. The MGDb estimates are consistently higher for most other regions; however, they do fall within the uncertainty range of the GBD data for North Africa and the Middle East and most of sub-Saharan Africa (see online resources Fig. 2).
In existing global estimates, confusion about terminology leads to under estimation of the true burden of congenital disorders. For example, estimates of the “congenital” contribution to under-5 mortality published by the GBD study (Lopez et al. 2006a, b) and WHO (Liu et al. 2012) cover only congenital anomalies. They do not include genetically determined disorders (single-gene disorders and disorders due to genetic risk factors), although estimates from MGDb indicate that they contribute to around 50% of under-5 deaths due to congenital disorders. This important point underlines the need for a clearly agreed terminology for community genetics.
It is widely recognised that the average baseline birth prevalence of congenital anomalies is at least 20/1000 while the baseline birth prevalence of congenital disorders (including single-gene disorders and early-onset disorders due to genetic risk factors) is over 37/1000(Baird et al. 1988; Czeizel and Sankaranarayanan 1984; World Health Organization 1985a). Their contribution to early death and disability is masked in lower-income settings by high early mortality from other causes, but it has been estimated that infant mortality can fall below 10/1000 only when interventions for the prevention and care of congenital disorders are in place (Christianson and Modell 2004; World Health Organization 1985a). Consequently, they would be expected to hold a significant place in the Sustainable Development Goals (to end preventable deaths in children) and the WHO strategy for non-communicable diseases (Darmstadt et al. 2016).
Conclusions
It is possible to overcome the current difficulties in collecting high-quality population-based epidemiological data for congenital disorders in many low- and middle-income countries. The method used here to generate estimates for countries with little or no observational data makes it possible to generate useful order of magnitude estimates by (a) starting from evidence-based estimates of baseline affected birth prevalence; (b) basing estimates for high-income settings on observational data; (c) generating estimates for a baseline no-care situation using the limited observational data supplemented by expert opinion; and (d) using an empirical equation based on infant mortality rates for estimating the proportion of each population with access to the relevant services.
Abbreviations
- EUROCAT:
-
European Surveillance of Congenital Anomalies and Twins
- ICBDSR:
-
International Clearinghouse for Birth Defects Surveillance and Research
- ICD:
-
International Classification of Disease
- MGDb:
-
Modell Global Database of Congenital Disorders
- NTD:
-
Neural tube defect
- OFC:
-
Orofacial cleft
References
Abbas HA, Yunis K (2014) The effect of consanguinity on neonatal outcomes and health. Hum Hered 77:87–92. doi:10.1159/000362125
Baird PA, Sadovnick AD (1987) Life expectancy in Down syndrome. J Pediatr 110:849–854
Baird PA, Sadovnick AD (1988) Life expectancy in Down syndrome adults. Lancet 2:1354–1356
Baird PA, Anderson TW, Newcombe HB, Lowry RB (1988) Genetic disorders in children and young adults: a population study. Am J Hum Genet 42:677–693
Baylis JH, Campbell M (1956) The course and prognosis of coarctation of the aorta. Br Heart J 18:475–495
Bhutani VK et al (2013) Neonatal hyperbilirubinemia and Rhesus disease of the newborn: incidence and impairment estimates for 2010 at regional and global levels. Pediatr Res 74(Suppl 1):86–100. doi:10.1038/pr.2013.208
Bittles AH (2003) Consanguineous marriage and childhood health. Dev Med Child Neurol 45:571–576
Bittles AH, Black ML (2010) The impact of consanguinity on neonatal and infant health. Early Hum Dev 86:737–741. doi:10.1016/j.earlhumdev.2010.08.003
Bittles AH, Black ML, (2015) Global patterns and tables of consanguinity. http://consang.net
Bittles AH, Neel JV (1994) The costs of human inbreeding and their implications for variations at the DNA level. Nat Genet 8:117–121. doi:10.1038/ng1094-117
Blencowe H, Cousens S, Jassir FB, Say L, Chou D, Mathers C, Hogan D, Shiekh S, Qureshi ZU, You D, Lawn JE, Lancet. Stillbirth Epidemiology Investigator, Group (2017) National, regional, and worldwide estimates of stillbirth rates in 2015, with trends from 2000: a systematic analysis. Lancet Glob Health 4:e98–e108. doi:10.1016/S2214-109X(15)00275-2
Bojesen A, Juul S, Birkebaek N, Gravholt CH (2004) Increased mortality in Klinefelter syndrome. J Clin Endocrinol Metab 89:3830–3834. doi:10.1210/jc.2004-0777
Bowman RM, McLone DG, Grant JA, Tomita T, Ito JA (2001) Spina bifida outcome: a 25-year prospective. Pediatr Neurosurg 34:114–120
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE (2013) Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 14:681–691. doi:10.1038/nrg3555
Bundey S, Alam H (1993) A five-year prospective study of the health of children in different ethnic groups, with particular reference to the effect of inbreeding. Eur J Hum Genet 1:206–219
Campbell M (1968) Natural history of persistent ductus arteriosus. Br Heart J 30:4–13
Campbell M, Neill C, Suzman S (1957) The prognosis of atrial septal defect. Br Med J 1:1375–1383
Carter CO (1977) Monogenic disorders. J Med Genet 14:316–320
Carter CH, Maley MC (1958) Preliminary report on treatment of mongoloids. J Fla Med Assoc 44:709–713
Chitty LS, Winter RM (1989) Perinatal mortality in different ethnic groups. Arch Dis Child 64:1036–1041
Christensen K, Juel K, Herskind AM, Murray JC (2004) Long term follow up study of survival associated with cleft lip and palate at birth. BMJ 328:1405. doi:10.1136/bmj.38106.559120.7C
Christianson A, Modell B (2004) Medical genetics in developing countries. Annu Rev Genomics Hum Genet 5:219–265. doi:10.1146/annurev.genom.5.061903.175935
Corry PC (2014) Consanguinity and prevalence patterns of inherited disease in the UK Pakistani community. Hum Hered 77:207–216. doi:10.1159/000362598
Costa T, Scriver CR, Childs B (1985) The effect of Mendelian disease on human health: a measurement. Am J Med Genet 21:231–242. doi:10.1002/ajmg.1320210205
Czeizel A, Sankaranarayanan K (1984) The load of genetic and partially genetic disorders in man. I Congenital anomalies: estimates of detriment in terms of years of life lost and years of impaired life. Mutat Res 128:73–103
Darmstadt GL et al (2016) Prevention of congenital disorders and care of affected children: a consensus statement. JAMA Pediatr 170:790–793. doi:10.1001/jamapediatrics.2016.0388
Elwood JM, Little J, Elwood JH (1992) Epidemiology and control of neural tube defects. Oxford University Press
Fleming AF, Storey J, Molineaux L, Iroko EA, Attai ED (1979) Abnormal haemoglobins in the Sudan savanna of Nigeria. I. Prevalence of haemoglobins and relationships between sickle cell trait, malaria and survival. Ann Trop Med Parasitol 73:161–172
Frid C, Drott P, Otterblad Olausson P, Sundelin C, Anneren G (2004) Maternal and neonatal factors and mortality in children with down syndrome born in 1973-1980 and 1995-1998. Acta Paediatr 93:106–112
Howes RE et al (2012) G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatistical model-based map. PLoS Med 9:e1001339. doi:10.1371/journal.pmed.1001339
Hunt GM, Oakeshott P (2003) Outcome in people with open spina bifida at age 35: prospective community based cohort study. BMJ 326:1365–1366. doi:10.1136/bmj.326.7403.1365
Institute for Health Metrics and Evaluation (IHME) (2015) GHDX Seattle. IHME, University of Washington, WA Available from http://ghdx.healthdata.org/ (Accessed [01.09.2015])
Inter-agency Group for Child Mortality Estimation (UN-IGME) (2015) Levels and trends in child mortality. childmortalityorg
Kadir A, Mossey PA, Blencowe H, Moorthie S, Lawn JE, Mastroiacovo P, Modell B (2016) Systematic review and meta-analysis of the birth prevalence of orofacial clefts in low- and middle-income countries cleft palate. Craniofac J. doi:10.1597/15–221
Kromberg JG, Castle D, Zwane EM, Jenkins T (1989) Albinism and skin cancer in Southern Africa. Clin Genet 36:43–52
Laurence KM, Tew BJ (1971) Natural history of spina bifida cystica and cranium bifidum cysticum. Major central nervous system malformations in South Wales IV. Arch Dis Child 46:127–138
Lie RT, Wilcox AJ, Skjaerven R (2001) Survival and reproduction among males with birth defects and risk of recurrence in their children. JAMA 285:755–760
Liu S, Joseph KS, Wen SW (2002) Trends in fetal and infant deaths caused by congenital anomalies. Semin Perinatol 26:268–276
Liu L et al (2012) Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet 379:2151–2161. doi:10.1016/S0140-6736(12)60560-1
Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ (2006a) Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 367:1747–1757. doi:10.1016/S0140-6736(06)68770-9
Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL (2006b) Measuring the global burden of disease and risk factors, 1990-2001. In: Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL (eds) Global burden of disease and risk factors. Washington (DC)
Lorber J (1971) Results of treatment of myelomeningocele. An analysis of 524 unselected cases, with special reference to possible selection for treatment. Dev Med Child Neurol 13:279–303
Lund PM, Taylor JS (2008) Lack of adequate sun protection for children with oculocutaneous albinism in South Africa. BMC Public Health 8:225. doi:10.1186/1471-2458-8-225
Macmahon B, McKeown T, Record RG (1953) The incidence and life expectation of children with congenital heart disease. Br Heart J 15:121–129
Modell B, Berdoukas V (1984) The clinical approach to thalassaemia. Grune & Stratton, London
Modell B, Darlison M (2008) Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 86:480–487
Modell B, Modell M (1992) Towards a healthy baby: congenital disorders and the new genetics in primary care. Oxford University Press
Modell B, Khan M, Darlison M (2000) Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 355:2051–2052. doi:10.1016/S0140-6736(00)02357-6
Modell B, Khan M, Darlison M, Westwood MA, Ingram D, Pennell DJ (2008) Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson 10:42. doi:10.1186/1532-429X-10-42
Modell B, Darlison MW, Moorthie S, Blencowe H, Petrou M, Lawn J (2016) Epidemiological methods in community genetics and the Modell Global Database of Congenital Disorders (MGDb) downloaded from UCL Discovery. http://discovery.ucl.ac.uk/1532179/
Moorthie S et al. (2017) Chromosomal disorders: estimating baseline prevalence and pregnancy outcomes worldwide. J Commun Genet. doi:10.1007/s12687-017-0336-2
Morris JK, Alberman E, Scott C, Jacobs P (2008) Is the prevalence of Klinefelter syndrome increasing? Eur J Hum Genet 16:163–170. doi:10.1038/sj.ejhg.5201956
Mossey PA, Little J (2002) Epidemiology of oral clefts: an international perspective. In: Cleft lip and palate: from origin to treatment. Oxford University Press, Oxford
Mossey PA, Modell B (2012) Epidemiology of oral clefts 2012: an international perspective. 16:1-18. Front Oral Biol. doi:10.1159/000337464
Mourant AE, Domaniewska-Sobczak K, Kopec AC, Tills D, Tills RE (1976) The distribution of the human blood groups and other polymorphisms, 2nd ed., Suppl.1 edn. OUP, Oxford
Pedersen RN, Garne E, Loane M, Korsholm L, Husby S, Group EW (2008) Infantile hypertrophic pyloric stenosis: a comparative study of incidence and other epidemiological characteristics in seven European regions. J Matern Fetal Neonatal Med 21:599–604. doi:10.1080/14767050802214824
Penrose LS (1949) The incidence of mongolism in the general population. J Ment Sci 95:685–688
Petrini J, Damus K, Johnston RB Jr (1997) An overview of infant mortality and birth defects in the United States. Teratology 56:8–10. doi:10.1002/(SICI)1096-9926(199707/08)56:1/2<8::AID-TERA3>3.0.CO;2-U
Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN (2013a) Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 10:e1001484. doi:10.1371/journal.pmed.1001484
Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI (2013b) Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381:142–151. doi:10.1016/S0140-6736(12)61229-X
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP (1994) Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 330:1639–1644. doi:10.1056/NEJM199406093302303
Price WH, Clayton JF, Collyer S, De Mey R, Wilson J (1986) Mortality ratios, life expectancy, and causes of death in patients with Turner's syndrome. J Epidemiol Community Health 40:97–102
Skjaerven R, Wilcox AJ, Lie RT (1999) A population-based study of survival and childbearing among female subjects with birth defects and the risk of recurrence in their children. N Engl J Med 340:1057–1062. doi:10.1056/NEJM199904083401401
Stevenson AC (1959) The load of hereditary defects in human populations. Radiat Res Suppl 1:306–325
Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH (2006) Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab 91:3897–3902. doi:10.1210/jc.2006-0558
Tennant PW, Pearce MS, Bythell M, Rankin J (2010) 20-year survival of children born with congenital anomalies: a population-based study. Lancet 375:649–656. doi:10.1016/S0140-6736(09)61922-X
Terry PB, Bissenden JG, Condie RG, Mathew PM (1985) Ethnic differences in congenital malformations. Arch Dis Child 60:866–868
Trimble BK, Doughty JH (1974) The amount of hereditary disease in human populations. Ann Hum Genet 38:199–223
UNAIDS (2016) Global aids update. http://www.unaids.org/sites/default/files/media_asset/global-AIDS-update-2016_en.pdf
United Nations Population Division (2015) World Population Prospects: the 2015 revision. http://esa.un.org/wpp/index.htm
Visootsak J, Graham JM Jr (2006) Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis 1:42. doi:10.1186/1750-1172-1-42
Wellesley D et al (2012) Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur J Hum Genet 20:521–526. doi:10.1038/ejhg.2011.246
World Health Organization (1985a) Community approaches to the control of hereditary diseases. Report of a WHO Advisory Group on Hereditary Diseases. Geneva 3–5 October 1985. Unpublished WHO document HMG/AG/85.10 available at http://www.who.int/genomics/publications/WHOHGNWG85.10.pdf
World Health Organization (1985b) Glucose-6-phosphate dehydrogenase deficiency. Report of a WHO Working Group. Unpublished WHO Document HDP/WP/G6PD/85.9.May be obtained free of charge from: The Hereditary Diseases Programme. WHO, Geneva
World Health Organization (2000) Primary health care approaches for the control of congenital disorders and disability. Report of a WHO meeting Cairo, 6–8 December 1999. WHO/HGN/WG/00.1
World Health Organization (2010) Sixty-third world health assembly: Geneva, 17–21 May 2010,
World Health Organization 2016 The International Classification of Diseases, Tenth Revision (ICD-10)
Wren C, O'Sullivan JJ (2001) Survival with congenital heart disease and need for follow up in adult life. Heart 85:438–443
Wren C et al (2012) Mortality in infants with cardiovascular malformations. Eur J Pediatr 171:281–287. doi:10.1007/s00431-011-1525-3
Wu J, Springett A, Morris JK (2013) Survival of trisomy 18 (Edwards syndrome) and trisomy 13 (Patau Syndrome) in England and Wales: 2004–2011. Am J Med Genet A 161A:2512–2518. doi:10.1002/ajmg.a.36127
Yang J, Carmichael SL, Kaidarova Z, Shaw GM (2004) Risks of selected congenital malformations among offspring of mixed race-ethnicity birth defects. Res A Clin Mol Teratol 70:820–824. doi:10.1002/bdra.20054
Acknowledgements
The authors would like to thank Professor Helen Dolk.
Congenital Disorder Expert Group: AH Bittles, H Blencowe, A Christianson, S Cousens, M Darlison, S Gibbons, H Hamamy, B Khoshnood, CP Howson, JE Lawn, P Mastroiacovo, B Modell, S Moorthie, JK Morris, PA Mossey, AJ Neville, M Petrou, S Povey, J Rankin, L Schuler-Faccini, C Wren, KA Yunis.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
Sowmiya Moorthie, Hannah Blencowe, Matthew Darlison, Joy Lawn, Pierpaolo Mastroiacovo, Joan K Morris, Bernadette Modell, and the Congenital Disorders Expert Group declare they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Funding
This work was supported by grants from the WHO Regional Office for the Eastern Mediterranean. The time of SM and HB was funded in part through a grant from the Bill and Melinda Gates Foundation to the Child Health Epidemiology Reference Group (CHERG).
Additional information
This article is part of the Topical Collection on Epidemiological Methods in Community Genetics
Electronic supplementary material
ESM 1
(DOCX 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Moorthie, S., Blencowe, H., W. Darlison, M. et al. An overview of concepts and approaches used in estimating the burden of congenital disorders globally. J Community Genet 9, 347–362 (2018). https://doi.org/10.1007/s12687-017-0335-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12687-017-0335-3