
Abstract
Missense variants represent a significant proportion of variants identified in clinical genetic testing. In the absence of strong clinical or functional evidence, the American College of Medical Genetics recommends that these findings be classified as variants of uncertain significance (VUS). VUSs may be reclassified to better inform patient care when new evidence is available. It is critical that the methods used for reclassification are robust in order to prevent inappropriate medical management strategies and unnecessary, life-altering surgeries. In an effort to provide evidence for classification, several in silico algorithms have been developed that attempt to predict the functional impact of missense variants through amino acid sequence conservation analysis. We report an analysis comparing internally derived, evidence-based classifications with the results obtained from six commonly used algorithms. We compiled a dataset of 1118 variants in BRCA1, BRCA2, MLH1, and MSH2 previously classified by our laboratory’s evidence-based variant classification program. We compared internally derived classifications with those obtained from the following in silico tools: Align-GVGD, CONDEL, Grantham Analysis, MAPP-MMR, PolyPhen-2, and SIFT. Despite being based on similar underlying principles, all algorithms displayed marked divergence in accuracy, specificity, and sensitivity. Overall, accuracy ranged from 58.7 to 90.8% while the Matthews Correlation Coefficient ranged from 0.26–0.65. CONDEL, a weighted average of multiple algorithms, did not perform significantly better than its individual components evaluated here. These results suggest that the in silico algorithms evaluated here do not provide reliable evidence regarding the clinical significance of missense variants in genes associated with hereditary cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Accurate variant classification is of significant importance to patient care. In particular, clinically actionable variants have the potential to help guide medical management decisions. In such cases, these decisions are guided by the National Comprehensive Cancer Network (NCCN) guidelines (https://www.nccn.org/professionals/physician_gls/f_guidelines.asp). For example, due to the high risk of developing breast and/or ovarian cancer women with pathogenic variants in BRCA1 and BRCA2 are recommended to receive increased screening, chemoprevention, and prophylactic surgeries (Daly et al. 2016). Similarly, individuals with pathogenic variants in MLH1 and MSH2 are recommended to receive more frequent colonoscopies starting at an earlier age due to the risk of developing colorectal cancer (Provenzale et al. 2016).
Given the importance of accurate clinical variant classification, the American College of Medical Genetics (ACMG) have published guidelines for the interpretation of sequence variants that often require multiple lines of evidence (Richards et al. 2015). For some classes of variants, such as missense variants, obtaining this evidence can be a challenge. Missense variants result in amino acid substitutions in the protein product that may affect protein structure and/or function and, in rare cases, RNA splicing. However, commonly used criteria, including co-segregation analysis, population frequency data, in-trans observations with a deleterious mutation, and direct evidence of the functional impact, are often not available for these rare variants. As a result, in silico prediction tools are frequently used to evaluate the pathogenicity of missense variants (Pesaran et al. 2016; Thompson et al. 2014).
These computational tools are based on the premise that the evolutionary conservation of an amino acid in a protein reflects its absolute requirement for protein function. In hereditary cancer testing, several in silico tools are available to evaluate the pathogenicity of missense variants based on sequence conservation across multiple mammalian species (Ng and Henikoff 2001). Some algorithms also incorporate biochemical and/or biophysical parameters of the protein in order to improve their predictive accuracy (Adzhubei et al. 2010; Chao et al. 2008; Mathe et al. 2006; Tavtigian et al. 2006).
In silico tools were initially developed for academic research, where computational methods are often used to screen large amounts of data to identify candidate genes/variants for further investigation. With the increased use of clinical genetic testing and the challenges of classifying missense variants, in silico tools are now incorporated into professional society guidelines for variant classification (Easton et al. 2007; Plon et al. 2008; Richards et al. 2015) and are used for clinical variant classification. However, several studies have demonstrated that the specificity (correct identification of benign variants) of these tools can be as low as 13% (Chan et al. 2007; Doss and Sethumadhavan 2009; Flanagan et al. 2010; Gnad et al. 2013; Leong et al. 2015; Miosge et al. 2015; Ng and Henikoff 2002; Schiemann and Stowell 2016; Thusberg et al. 2011; Valdmanis et al. 2009) and with poor correlation between results from multiple programs (Thusberg et al. 2011). While these limitations may be acceptable in selecting variants for research, false positives and variability between methods may compromise patient care in a clinical setting.
Existing comprehensive reports on the clinical use of in silico tools have been limited to small cohorts (Hicks et al. 2011; Thompson et al. 2013) or variant datasets that are poorly curated (Thusberg and Vihinen 2009). In order to provide a comprehensive evaluation of their clinical use, we evaluated the predictive functionality of several in silico tools in a large cohort of individuals who underwent clinical genetic testing. Classifications for missense variants in BRCA1, BRCA2, MLH1, and MSH2 that were based on independent lines of evidence were compared to classifications from six commonly used in silico tools: Align-GVGD, CONDEL, Grantham Analysis, MAPP-MMR, PolyPhen-2, and SIFT.
Materials and methods
Variant selection
All missense variants identified in BRCA1, BRCA2, MLH1, and MSH2 by a commercial testing laboratory (Myriad Genetic Laboratories, Inc., Salt Lake City, UT) as of August 2013 were evaluated here. Variation in these high penetrance genes has the potential to significantly affect medical management. These variants were classified as pathogenic or benign based on multiple, independent lines of evidence that did not include sequence conservation (Eggington et al. 2013; Richards et al. 2015). This includes statistical methods (Pruss et al. 2014), biochemical/biophysical assays in the literature, population allele frequency thresholds, and co-occurrences with mutations in the same gene or pathway. Variants whose pathogenicity may arise from a mechanism not purely related to the amino acid substitution (variants that impact mRNA splicing, translation from the initiating 5′ AUG start codon, silent mutations) were excluded. This resulted in a final dataset of 1118 missense variants.
In silico classification tools
The final set of variants was classified using the following commonly used in silico tools: SIFT, PolyPhen-2 (two available models: HumDiv and HumVar), Grantham matrix score, Align GVGD, MAPP-MMR, and CONDEL server. For Align-GVGD, an additional 316 BRCA1 and 434 BRCA2 variants were excluded from analysis, as they were used to train the algorithm and represent a possible source of statistical bias.
A brief summary of these algorithms is provided in Table 1 and the full details can be found in Online Resource 1. Briefly, these tools base variant classifications on sequence homology (SIFT, PholyPhen-2, MAPP-MMR, CONDEL), structural features (PolyPhen-2, CONDEL), and/or physiochemical properties (Grantham, Align-GVGD, MAPP-MMR). All in silico tools were applied to variants in BRCA1, BRCA2, MLH1, and MSH2 with the exception of MAPP-MMR, which is only applicable for MLH1 and MSH2 variants. Each in silico classification tool utilizes different parameters for variant classification, the full details of which are in Online Resource 1.
Evaluation of in silico classification tool performance
For all genes and algorithms, we calculated the overall accuracy (ratio of overall correct predictions to the total number of predictions), specificity (correct identification of benign variants; true negative rate), and sensitivity (correct identification of pathogenic variants; true positive rate) relative to the testing laboratory classifications. Classifications were evaluated in three tiers (benign, uncertain, and pathogenic), and those of the same pathogenicity were considered concordant. For example, a laboratory classification of deleterious and an in silico classification of damaging would be considered concordant while a laboratory classification of benign and an in silico classification of unclassified would be considered discordant.
The positive predictive values (PPV, the proportion of positive results that were true positives) and negative predictive values (NPV, proportion of negative results that were true negatives) were also calculated.
As variant cohort size varied between algorithms, the Matthews correlation coefficient was used to provide a balanced comparison between in silico tools. This metric is not affected by sample size or the proportion of neutral to pathogenic variants in each dataset. A value of +1 indicates a perfect prediction while a value of −1 indicates a total disagreement between the in silico predicted classification and the laboratory classification. The full details of all statistical evaluations performed here can be found in Online Resource 1.
Results
Overall, a maximum of 1118 variants were available for analysis. Table 1 shows the distribution of variants evaluated by gene and by in silico method. Aside from MAPP-MMR, which was only available for MLH1 and MSH2, Align-GVGD had the lowest number of evaluable variants (n = 368). This was due to the large number of variants that were used to initially train the algorithm and that we eliminated from our analyses to mitigate possible statistical bias.
Overall in silico algorithm performance
Table 2 shows the results of the in silico classification evaluation for the overall analysis, with the highest accuracy being observed for Align-GVGD (90.8%). This algorithm also showed the least divergence between specificity (91.7%) and sensitivity (84.1%). Still, approximately 1 in 10 patients would receive a false negative with this tool and about 1 in 5 would receive a false positive. The number of true negatives, true positives, false positives, and false negatives are provided in Online Resource 2. While the NPV for Align-GVGD was 97.7%, the PPV was only 57.8%.
SIFT, PolyPhen-2, CONDEL, and Grantham had overall accuracies ranging from 58.7% (PolyPhen-2 HumDiv) to 71.1% (PolyPhen-2 HumVar) (Table 2). Although the sensitivity of these algorithms ranged from 78.2% (Grantham) to as high as 99.0% (SIFT), the specificities were much lower, ranging from 55.6% (PolyPhen-2 HumDiv) to 70.1% (PolyPhen-2 HumVar). The performance of the CONDEL algorithm (69.8%), which is a weighted average of SIFT, PolyPhen-2, and MutationAssessor, was not superior to SIFT and PolyPhen-2 in isolation. In addition, the NPVs for all algorithms were >95%; however, the PPVs were substantially lower at 16–20% (Table 2). Both the low specificities and PPVs reflect the high frequency of false positives for these in silico tools.
MAPP-MMR, which is specific to MLH1 and MSH2, had an overall accuracy of 74.6% (Table 2). There was a large discrepancy between the sensitivity (100.0%) and specificity (56.1%). As such, the PPV was 62.5%.
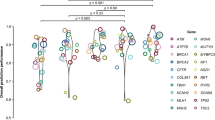
As the number of variants available for analysis for each in silico tool ranged from 71 (MAPP-MMR) to 1118 (SIFT, PolyPhen-2), we compared the performance of each algorithm in a manner that is independent of cohort size. Figure 1 (top) shows the overall Matthews correlation coefficients, where a value of 1 indicates complete concordance with the laboratory classifications. The highest degree of correlation was observed for Align-GVGD (0.65) then MAPP-MMR (0.59). The remaining in silico tools showed lower correlation with the laboratory classifications, which utilized multiple, independent lines of evidence, as detailed in the “Materials and methods,” with correlation coefficients ranging from 0.26 to 0.32.

Overall (top) and gene-specific (bottom) Matthews correlation coefficients for in silico tools relative to laboratory classification
Classification of BRCA1 and BRCA2 variants
All algorithms except MAPP-MMR were used to classify missense variants in BRCA1 and BRCA2. As observed for the overall algorithm performance, Align-GVGD had the highest overall accuracy for classification of BRCA1 variants (97.1%) (Table 2). Again, the sensitivity and specificity were consistent, at 91.7 and 97.8%, respectively. As such, both the PPV (84.6%) and NPV (98.9%) were high. However, this same degree of predictive accuracy was not observed for the classification of missense variants in BRCA2. Although the overall accuracy, sensitivity, and specificity are relatively high, the PPV is only 7.1%. This means that there will be a very high frequency of false positives for BRCA2 variants classified with Align-GVGD. This discrepant algorithm performance between BRCA1 and BRCA2 is also clear in Fig. 1, which shows the correlation between the algorithm classifications and the laboratory classifications. Despite a relatively high degree of correlation for BRCA1 (0.86), the correlation coefficient is only 0.26 for Align-GVGD classification of BRCA2 variants.
All other algorithms were poor predictors of the clinical classification of both BRCA1 and BRCA2 variants (Table 2). For BRCA1 variant classification, overall accuracies ranged from 48.9% (SIFT) to 69.2% (PolyPhen-2 HumVar). Again, algorithm specificities (41.5–59.2%) and PPVs (19.9–27.4%) were low, which is representative of a large number of false positives (Online Resource 2). For example, BRCA1 c.5317A>T (p. Thr1773Ser) was classified as benign by the reference laboratory based on published literature showing that the variant does not affect gene function (Lee et al. 2010) and phenotypic evidence from a family history weighting algorithm (Pruss et al. 2014) (Table 3). Although the variant was classified as benign by PolyPhen-2, it was classified as pathogenic by Align-GVGD and SIFT.
For BRCA2 variant classification, SIFT, PolyPhen-2, CONDEL, and Grantham had overall accuracies ranging from 60.9 to 71.8%. Although nearly all algorithms had a sensitivity of 100%, the specificities ranged from 59.9% (SIFT) to 72.9% (PolyPhen-2 HumVar) (Table 2). Again, this was accompanied by extremely low PPVs (4.3–9.2%) due to a high frequency of false positives. For example, BRCA2 c.7994A>G (p. Asp2665Gly) was classified as benign by the reference laboratory based on an in-trans observation (occurring in the same gene but different allele) with a known pathogenic BRCA2 variant (Table 3). However, all four in silico tools that produced a classification for this variant called it either pathogenic or likely pathogenic.
The analysis in Fig. 1, which is independent of cohort size, shows that there was low correlation between the remaining algorithms and laboratory classifications. Most in silico tools had lower correlation coefficients for BRCA2 classifications than for BRCA1 classifications. This was most significant for Grantham, which dropped from 0.38 for BRCA1 to 0.06 for BRCA2.
Classification of MLH1 and MSH2 variants
The highest overall accuracy for MLH1 missense variants was observed for MAPP-MMR (86.1%) (Table 2). However, the sensitivity and specificity were not consistent, at 100.0 and 68.8%, respectively. As a result, the NPV was high (100.0%) while the PPV was only 80.0%. Align-GVGD also had a relatively high overall predicative accuracy for the classification of MLH1 variants at 79.6%. This algorithm also had similar sensitivity (76.2%) and specificity (82.1%). SIFT, PolyPhen-2, CONDEL, and Grantham had overall accuracies ranging from 59.2 to 71.8%. Algorithm specificities (32.1–73.9%) and PPVs (51.3–64.7%) were relatively low due to a high frequency of false positives (Online Resource 2). For example, MLH1 c.394G>C (p. Asp132His) was classified as benign by the reference laboratory based on an in-trans observation with a known pathogenic variant and phenotypic evidence from a family history weighting algorithm (Pruss et al. 2014) (Table 3). However, this variant was classified as pathogenic or likely pathogenic by Align-GVGD, PolyPhen-2, SIFT, and MAPP-MMR.
For MSH2 variant classification, the overall accuracy of MAPP-MMR dropped to 62.9%. Align-GVGD had the highest overall accuracy for MSH2 variant classification, at 84.3%. SIFT, PolyPhen-2, CONDEL, and Grantham had lower overall accuracies, which ranged from 52.9 to 66.0%. Again, the specificity for nearly all algorithms was much lower than the sensitivity. As such, the PPVs ranged from 28.1% (SIFT) to 56.3% (Align-GVGD) (Table 2). This again shows that the use of in silico tools for MSH2 missense variant classification results in a high incidence of false positives. Figure 1 shows that the correlation coefficients for MLH1 were generally higher than those for MSH2; however, all algorithms showed relatively low degrees of correlation with the reference laboratory classifications, with the exception of MAPP-MMR for MLH1 classification.
Discussion
We assessed the predictive functionality of six commonly used in silico tools for the assessment of missense variants in a large dataset of BRCA1, BRCA2, MLH1, and MSH2 variants identified during the course of clinical genetic testing. All algorithms in this study generated high error rates (high frequency of false positives, low correlation coefficients) when compared to the reference classifications. These results are consistent with previous studies that also reported a high rate of error from in silico-derived classifications (Flanagan et al. 2010; Gnad et al. 2013; Hicks et al. 2011; Martelotto et al. 2014; Miosge et al. 2015; Thusberg et al. 2011).
Sources of error for in silico tools
There are several sources of error that may contribute to the inconsistent and unreliable performance of in silico tools. First is the need for accurate sequence alignments in order for the algorithm to accurately assess conservation. As this process is essentially heuristic, the accuracy of the alignment and the predicted result falls off dramatically in poorly conserved regions of the protein. As demonstrated by Align-GVGD, sequence alignments may be corrected by incorporating information from available protein structures; however, it is likely that many homologous structures would be required to sufficiently improve the accuracy for clinical use. It would be more appropriate to perform a full structural analysis, which would also provide direct evidence regarding the effect of the variant on protein structure and function. While attempts may be made to manually curate sequence alignments, this can lead to misaligned regions of the proteins in the absence of additional information to help guide the process. For this reason, manual sequence conservation analysis should be avoided as it is likely to incorporate even more uncertainty.
The second source of error is the assumption that a conserved position in multiple sequence alignments is more important for protein function than one that is less well conserved. As recently alluded to by MacArthur et al., this hypothesis is limited by a lack of proper biological context (MacArthur et al. 2014). Pathogenic variants can be observed at sites of relatively low conservation while benign variants may be found in areas of the protein sequence that are highly conserved throughout mammalian evolution (Hicks et al. 2011).
Challenges with training in silico tools
The poor overall predictive accuracy of the in silico tools assessed here may be related to the challenges in “training” the algorithms. Given the rarity of individual missense variants and the resulting limited available evidence of pathogenicity, it is challenging to obtain enough robustly classified variants to both train and independently evaluate the algorithm. When these two datasets overlap, the data can be “overfit” and the reported predictive accuracy of the tool is often artificially inflated. A recent study by Grimm et al. highlighted that accurate predictions from in silico tools are less frequently observed for variants which overlap between the training dataset and the evaluation dataset (Grimm et al. 2015). This overfitting may also occur when predictions from tools are combined (i.e., CONDEL).
For Align-GVGD, we were able to selectively evaluate the algorithm on a curated set of variants, to maximize the overall predictive accuracy. As such, while this in silico tool had the best overall performance, it is unlikely that this accuracy could be replicated outside of the current study. Additionally, this approach is not feasible for genes in which few discrete missense variants have been identified and robustly, clinically classified.
The exclusion of a large number of BRCA1 and BRCA2 variants that were used to train Align-GVGD resulted in the smallest cohort size in this study. In the case of BRCA1, this may have affected the specificity, which is seemingly the highest across all genes studied using any of the six in silico tools employed. However, the PPV indicates that the ratio of true positives (TP) to the total number of positives (TP + FP) in the BRCA1 dataset was still only 84.6% for Align-GVGD.
For the remaining algorithms, we were unable to avoid any potential overfitting, instead relying on the built-in algorithm training. As such, it is not surprising that the performance of these in silico tools in this study was much lower than previously reported. The low PPVs observed for all algorithms across all genes investigated here indicate that there is a high frequency of false positive results when using in silico tools for the classification of variants in cancer-risk genes. This is exemplified by the three case examples given in Table 3, where independent evidence of pathogenicity including published literature and in-trans observations with a pathogenic variant resulted in a benign classification. In the absence of additional evidence, these false positives may result in inappropriate clinical management. This is especially troubling for BRCA1 and BRCA2, as pathogenic variants in these genes are potential indications for prophylactic surgeries (Daly et al. 2016).
Academic vs clinical utility
Establishing the relationship between genotype and phenotype is a fundamental principle of genetics. In an academic research laboratory, biological samples and corresponding phenotypic data are used to gain knowledge of this relationship. Given the size of the human genome, spectrum of variation, and ever decreasing costs of genotyping, researchers may be left with large volumes of candidate variants after association analysis. In such situations, in silico prediction tools are useful for contextualizing and prioritizing candidate variants for further follow-up, especially when functional experimental assays are not readily available or are cost prohibitive. One of the consequences of using these tools is that relevant data points may be discarded. However, the knowledge gained from these studies is often used to guide further research and not direct the medical management of a patient.
In a clinical laboratory, samples are tested due to clinical suspicion of hereditary cancer risk. Accurate variant classification using multiple lines of evidence is vital for appropriate clinical management based on NCCN guidelines. In this setting, incorrect variant classification based on in silico tools comes with more immediate consequences to the patient.
Additional considerations for clinical use
We have outlined many challenges associated with the use of several specific in silico tools in the clinical setting. As discussed previously, this includes the requirement of large training sets, unclear variant classification, and inaccurate biological assumptions. Caution must also be used when sequence conservation information is included in multifactorial models to establish posterior probability. Such methods calculate a prior probability of causality (calculated from in silico sequence conservation), which is then combined with various likelihoods of causality including co-segregation analysis, in-trans observations, family history, and histopathology data to give a final probability of causality for a variant (Lindor et al. 2013; Lindor et al. 2012).
The inherent problem with these methods is that the separate probabilities and likelihoods are combined (i.e., the various lines of evidence are never truly considered “independent” of one another). The analysis is therefore weighted by the prior sequence conservation analysis, which, as we have shown here, is a poor predictor of the clinical significance of a variant. In addition, any thresholds for pathogenicity used in such a multifactorial analysis would have to be thoroughly clinically validated.
Conclusions
Classification of missense variants can be a challenging process, particularly for clinical use. Prediction of their possible cancer association is especially difficult in pleiotropic genes such as BRCA1, where disruption of a specific function or domain may not necessarily affect downstream tumor suppression activity (Shakya et al. 2011). One must therefore be careful to ensure that a loss of function associated with a missense variant correlates with increased predisposition to a given cancer syndrome. In general, the shortcomings of in silico sequence conservation analysis may be described as applying a one- or two-dimensional solution to a much more complex, three-dimensional problem. While some algorithms may incorporate biochemical features or limited structural analysis, these additions were insufficient to overcome the inherent low accuracy in these methods for variant classification in the cancer-risk genes evaluated here. Although a full structural analysis is far more appropriate, this must be carefully undertaken by a trained structural biologist.
The low specificity of these methods is of particular concern given the potential for more aggressive medical management strategies including prophylactic surgeries following a false-positive result. Since the primary aim of clinical testing is to provide results to inform medical management, variant classification techniques must reach an exceptionally high level of confidence compared to academic research use. For the purposes of purely academic research, the rates of false-positives and false-negatives are less problematic as long as the results are not applied to patient care. However, to confidently inform a clinical decision, the thresholds for accuracy need to be exceedingly high, well above the average sensitivity/specificity seen in this study (Akobeng 2008). We therefore conclude from this analysis that the error rates of commonly used in silico tools are too high to warrant use as primary evidence to further assess variants of unknown significance in a clinical setting.
References
Adzhubei IA et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. doi:10.1038/nmeth0410-248
Akobeng AK (2008) Confidence intervals and p-values in clinical decision making. Acta paediatrica (Oslo, Norway: 1992) 97:1004–1007. doi:10.1111/j.1651-2227.2008.00836.x
Chan PA et al (2007) Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR). Hum Mutat 28:683–693. doi:10.1002/humu.20492
Chao EC et al (2008) Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR). Hum Mutat 29:852–860. doi:10.1002/humu.20735
Daly M et al (2016) Genetic/familial high-risk assessment: breast and ovarian. Version 2.2016. NCCN Clinical Practice Guidelines in Oncology
Doss CG, Sethumadhavan R (2009) Investigation on the role of nsSNPs in HNPCC genes--a bioinformatics approach. J Biomed Sci 16:42. doi:10.1186/1423-0127-16-42
Easton DF et al (2007) A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet 81:873–883. doi:10.1086/521032
Eggington JM et al (2013) A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. doi:10.1111/cge.12315
Flanagan SE, Patch AM, Ellard S (2010) Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genetic testing and molecular biomarkers 14:533–537. doi:10.1089/gtmb.2010.0036
Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z (2013) Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics 14(Suppl 3):S7. doi:10.1186/1471-2164-14-s3-s7
Grimm DG et al (2015) The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat 36:513–523. doi:10.1002/humu.22768
Hicks S, Wheeler DA, Plon SE, Kimmel M (2011) Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat 32:661–668. doi:10.1002/humu.21490
Kohonen-Corish M et al (1996) RNA-based mutation screening in hereditary nonpolyposis colorectal cancer. Am J Hum Genet 59:818–824
Lee MS et al (2010) Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res 70:4880–4890. doi:10.1158/0008-5472.CAN-09-4563
Leong IU, Stuckey A, Lai D, Skinner JR, Love DR (2015) Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC medical genetics 16:34. doi:10.1186/s12881-015-0176-z
Lindor NM, Goldgar DE, Tavtigian SV, Plon SE, Couch FJ (2013) BRCA1/2 sequence variants of uncertain significance: a primer for providers to assist in discussions and in medical management. Oncologist 18:518–524. doi:10.1634/theoncologist.2012-0452
Lindor NM et al (2012) A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat 33:8–21. doi:10.1002/humu.21627
MacArthur DG et al (2014) Guidelines for investigating causality of sequence variants in human disease. Nature 508:469–476
Martelotto LG et al (2014) Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol 15:484. doi:10.1186/s13059-014-0484-1
Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV (2006) Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res 34:1317–1325. doi:10.1093/nar/gkj518
Miosge LA et al (2015) Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci U S A 112:E5189–E5198. doi:10.1073/pnas.1511585112
Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874. doi:10.1101/gr.176601
Ng PC, Henikoff S (2002) Accounting for human polymorphisms predicted to affect protein function. Genome Res 12:436–446. doi:10.1101/gr.212802
Pagenstecher C et al (2006) Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet 119:9–22. doi:10.1007/s00439-005-0107-8
Pesaran T et al (2016) Beyond DNA: an integrated and functional approach for classifying Germline variants in breast cancer genes. Int J Breast Cancer 2016:2469523
Plon SE et al (2008) Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29:1282–1291. doi:10.1002/humu.20880
Provenzale D et al. (2016) Colorectal cancer screening V 1.2016. NCCN Clinical Practice Guidelines in Oncology 2015
Pruss D et al (2014) Development and validation of a new algorithm for the reclassification of genetic variants identified in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat 147:119–132. doi:10.1007/s10549-10014-13065-10549
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics 17:405–424. doi:10.1038/gim.2015.30
Schiemann AH, Stowell KM (2016) Comparison of pathogenicity prediction tools on missense variants in RYR1 and CACNA1S associated with malignant hyperthermia. Br J Anaesth 117:124–128. doi:10.1093/bja/aew065
Shakya R et al (2011) BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334:525–528. doi:10.1126/science.1209909
Tavtigian SV et al (2006) Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet 43:295–305. doi:10.1136/jmg.2005.033878
Thompson BA et al (2013) Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat 34:255–265. doi:10.1002/humu.22214
Thompson BA et al (2014) Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 46:107–115. doi:10.1038/ng.2854
Thusberg J, Olatubosun A, Vihinen M (2011) Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat 32:358–368. doi:10.1002/humu.21445
Thusberg J, Vihinen M (2009) Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum Mutat 30:703–714. doi:10.1002/humu.20938
Valdmanis PN, Verlaan DJ, Rouleau GA (2009) The proportion of mutations predicted to have a deleterious effect differs between gain and loss of function genes in neurodegenerative disease. Hum Mutat 30:E481–E489. doi:10.1002/humu.20939
Wu Y et al (1997) MSH2 and MLH1 mutations in sporadic replication error-positive colorectal carcinoma as assessed by two-dimensional DNA electrophoresis. Genes, chromosomes & cancer 18:269–278
Acknowledgements
This study was supported by Myriad Genetic Laboratories, Inc. We acknowledge the efforts of the clinicians and patients who have participated in Myriad Genetics Laboratories’ Variant Classification Program. We thank Dr. Sean V. Tavtigian, University of Utah and Huntsman Cancer Institute, for providing us with a list of variants used to train Align-GVGD. We would like to acknowledge the genetic counselors who attended the 2013 Annual NSGC meeting who shared their experiences on sequence conservation methods in the interpretation of genetic testing results. We would also like to thank Kirstin Roundy for her assistance with manuscript editing and submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All authors were employees for Myriad Genetic Laboratories, Inc. at the time of this study and received salary and stock as compensation.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Electronic supplementary material
Online Resource 1
Supplemental Methods detailing the in silico tools evaluated here, variant classification analysis, and statistical methods. (DOCX 36 kb)
Online Resource 2
Supplemental Table 1 showing the number of variants evaluated, true positive (TP), false positive (FP), false negatives (FN), and true negatives (TN) for each in silico tool overall and by gene. (DOCX 21 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kerr, I.D., Cox, H.C., Moyes, K. et al. Assessment of in silico protein sequence analysis in the clinical classification of variants in cancer risk genes. J Community Genet 8, 87–95 (2017). https://doi.org/10.1007/s12687-016-0289-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12687-016-0289-x