
Abstract
The electrochemical oxidation and infrared spectral features for carbon monoxide adsorbed on Pt(100) and Pt(110) in 0.1 M NaOH were studied as a function of the annealing/cooling treatment with the objective to establish the influence of the platinum surface site geometry and the role of surface order in the electro-oxidation of CO on these two surfaces. Two common cooling methods (Ar and H2/Ar atmospheres) were employed for both surface electrodes. Additionally, CO cooling and I2 vapour cooling methods were used for Pt(100) and Pt(110), respectively. Multiple CO vibration bands were observed on both electrode surfaces, their distribution and potential dependence strongly depending on the surface treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The electrochemical adsorption/oxidation of CO has been one of the major subjects in fundamental and applied studies in the fields of electrocatalysis and electrochemical surface science because of the adverse effect of CO in low-temperature fuel cells. Carbon monoxide oxidation is also used as a simple model reaction because CO acts as a neutral probe to surface activity and structure sensitivity.
Adsorption and electrooxidation of CO on platinum surfaces have been studied by several groups. Previous studies paid special attention to the surface structure and the effect of pH [1–9]. Several studies have been carried out to elucidate the kinetics and mechanism of the CO oxidation on platinum electrodes in acidic media [1–4, 6] as well in alkaline media [7–9]. Employing stepped electrodes, it was established that the CO oxidation reaction in acidic media takes place preferentially at step sites and that CO diffusion from the (111) terrace to these sites is very fast [1–4]. Surprisingly, in a recent work employing platinum stepped surfaces in alkaline media, four different active oxidation sites on the surface, i.e. sites with (111), (110) and (100) orientation, as well as kink sites were observed during CO stripping voltammetry [8, 9]. The fact that, in alkaline media, the oxidation of CO can be observed on different sites was attributed to the blockage of the reactive step site by absorbed carbonate, which is the product of CO oxidation in alkaline media [8–10].
The electrooxidation of CO on Pt(100) and Pt(110) has received much less attention in comparison to Pt(111), especially in alkaline media. For these surfaces, it is well known that the conditions under which the crystal is cooled down to room temperature after the flame treatment have a strong effect on the crystalline order of the surface [11–17]. Previous studies showed that cooling down the surface in the absence of oxygen, i.e. in an H2/Ar atmosphere, is required for Pt(100) and stepped Pt surfaces [13–17] to obtain a well-ordered surface. It was also found by our group that different annealing procedures of the Pt(111) produce different surfaces with different CO vibrational signatures and CO oxidation behavior [18]. The effect of the annealing/cooling preparation on the surface structure of the Pt(110) surface has also been studied. The Pt(110) surface can exhibit two different superficial structures: Pt(110)–(1 × 2) and Pt(110)–(1 × 1). Bittner et al. showed by electrochemical scanning tunneling microscopy (EC-STM) that for a Pt(110) cooled in iodine, at very negative potentials (in H2SO4 solution), the iodine desorbs and gives rise to an unreconstructed (1 × 1) surface with a surface topography consisting of small rectangular and isotropic terraces [19]. However, as will be shown below, our results suggest the coexistence of both superficial structures Pt(110)−(1 × 2) and Pt(110)−(1 × 1), in better agreement with an older work from Vogel et al. [20].
In this paper, we present a study on the electrooxidation of CO on Pt(100) and Pt(110) single crystal electrodes in alkaline media. As in our previous study on the electro-oxidation of CO on Pt(111) [18], our experiments involved a combination of cyclic voltammetry and in situ Fourier transform infrared (FTIR) spectroscopy. The influence of different surface preparations of the Pt(100) and Pt(110) surface on the CO adsorption and oxidation will also be examined. This study was carried out in order to establish the influence of the platinum surface site geometry and the superficial order of these two surfaces on the electro-oxidation of CO.
Experimental
For the electrochemistry experiments, bead-type single crystals of Pt(111), Pt(100) and Pt(110) orientation prepared according to the method of Clavilier et al. [21] were used in this study. For the in situ infrared experiments, Pt disk electrodes (Mateck®) of 1 cm diameter with the corresponding orientation were employed. Before each experiment, the single-crystal electrode was flame annealed and cooled down to room temperature according to the following methods:
-
1.
Cooling down in an Ar atmosphere, after which the electrode was transferred to the cell under the protection of a droplet of de-oxygenated water
-
2.
Cooling down in air and quenching with water after which the electrode was transferred to the cell under the protection of a droplet of water
-
3.
Cooling down in an H2/Ar atmosphere, after which the electrode was transferred to the cell under the protection of a droplet of water saturated with the cooling gases.
-
4.
Cooling down in an CO/Ar atmosphere, after which the electrode was transferred to the cell under the protection of a droplet of water saturated with the cooling gases
-
5.
Cooling down in an I2 atmosphere, after which the electrode was transferred to a first cell. In order to remove the I2 from the surface, the electrode was immersed at controlled potential of −0.1 V vs reversible hydrogen electrode (RHE). In order to avoid any contribution of the dissolved I2, the electrode was subsequently transferred to a second electrochemical cell with the working electrolyte.
In order to control the quality of the system, the blank voltammetric profile of the Pt(hkl) surface in 0.1 M NaOH was recorded before each experiment and compared with the literature [8, 21, 22]. CO stripping experiments were performed after bubbling CO through the cell for 2–3 min while keeping the Pt electrode at 0.05 V, followed by argon purging for 20 min to remove the excess CO from solution. When the electrode was flame annealed and cooled down in a CO/Ar atmosphere, the adsorbed CO was removed from the surface by stripping voltammetry and next readsorbed again by the procedure described above. All the CV stripping voltammograms shown here correspond to the stripping profile of the CO adsorbed in the electrochemical glass cell.
A Pt sheet was used as a counter electrode, and an RHE in the supporting electrolyte was employed as the reference electrode. Electrochemical measurements were performed with a computer-controlled Autolab PGSTAT12 potentiostat-galvanostat. The FTIR experiments were carried out with a Bruker Vertex 80V Infrared spectrophotometer. A spectroelectrochemical glass cell with a 60° CaF2 prism was used, designed for the external reflection mode in a thin layer configuration. FTIR spectra were collected from an average of 20 scans obtained with 6 cm−1 resolution at selected potentials, by applying single potential steps from a reference potential (E 0 = 0.05 V) in the positive-going direction up to 0.9 V. Spectra are represented as the ratio R/R 0, where R and R 0 are the reflectance measured at the sample and the reference potential, respectively. Consequently, positive and negative bands correspond to the loss and gain of species at the sample potential, respectively. S- and p-polarised light were used to distinguish between adsorbed and solution species: s-polarised light probes only species in solution, whereas p-polarised light probes species on the electrode surface as well as in solution [23].
All glasswares were cleaned by boiling in concentrated sulfuric acid, followed by washing and boiling with ultra-pure water. Solutions were prepared from high purity reagents (99,998%, Sigma-Aldrich) and ultra-pure water (Millipore MilliQ gradient A10 system, 18.2 MΩ cm, 2 ppb total organic carbon). Argon (N66) was used to deoxygenate all solutions and CO (N47) to dose CO.
Results and Discussion
Pt(100)
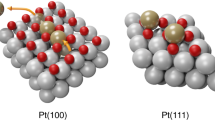
The blank voltammetric profiles of the Pt(100) electrode in alkaline media after different surface treatments are shown in Fig. 1. The cyclic voltammogram of the Pt(100) electrode recorded after cooling in a H2/Ar mixture (Fig. 1a) shows four well-defined quasi-reversible peaks that correspond to the adsorption/desorption process of H and OH. Unfortunately and opposite to the case of the Pt(111), where it is possible to distinguish between the Hads/des region and the OHads/des, the discrimination between the Hads/des and the OHads/des on the Pt(100) cannot be done in 0.1 M NaOH because of the overlapping peaks. It is important to note that the current densities of the four peaks are all similar. We will use the notation I, II, III and IV for these peaks, with peak I at the least positive potential (0.25 vs RHE) and peak IV at the most positive potential (0.57 V vs RHE). Feliu et al. [13, 14], using ex situ STM, and Kibler et al. [15], using in situ STM, showed that when a Pt(100) electrode is cooled in a H2 containing atmosphere after flame-annealing, the surface presents flat terraces separated by monoatomic high steps and covered by monoatomically high square-shaped islands (Fig. 2). This result indicates that the reconstructed Pt(100)–hex–R0.7° (see Fig. 2), which is the stable surface in UHV, was lifted to a Pt(100)–(1 × 1) surface during the cooling step.

Cyclic voltammogram of the same Pt(100) electrode pretreated by flame annealing and cooling in a H2/Ar (3:1) atmosphere, b Ar atmosphere and c CO/Ar (3:1) atmosphere. The supporting electrolyte is 0.1 M NaOH. Scan rate = 50 mV/s

Hard-sphere model of the Pt(100) with the different adsorption sites as a result of the different annealing treatments
The corresponding cyclic voltammogram of the Pt(100) cooled in Ar (Fig. 1b) is characterised by the same peaks I–IV. However, the intensities of the peaks have changed, peak II now being more pronounced. This peak is expected to correspond to defect sites caused by the cooling step. This is in agreement with previous ex and in situ STM images, where the surface is covered by many small monotatomic islands making the surface rich in step and defect sites [13–15]. Tentatively, it was proposed that the formation of monoatomic islands originates from lifting of the thermally induced hex reconstruction and further roughening of the surface by oxygen adsorption present on the cooling system [15].
On the other hand, if the electrode is flame annealed and cooled down in a CO/Ar saturated atmosphere (voltammogram shown in Fig. 1c), the changes with respect to the Fig. 1a are the opposite to those observed in Fig. 1b. Peaks I and II at lower potentials overlap and have low intensities in comparison with the peaks III and IV. The intensities of peaks III and IV are higher than in the case of the electrode cooled down in H2/Ar and much higher than for the electrode cooled in Ar. In this regard, it is important to mention that the EC-STM images presented by Kibler et al. show that when the surface was cooled in CO atmosphere, the CO adsorption increases the mobility of the surface atoms, and after the lifting of the reconstruction of the Pt(100)–hex surface into Pt(100)–(1 × 1), no monoatomic square islands were present on the surface [15]. Additionally, the STM images of the Pt(100) annealed under these conditions do not show the presence of kink sites [15].
Summarising the analysis of the blank voltammetric profiles of the Pt(100) electrode under different surface treatment conditions, we assign peaks I and II to low coordinated adsorption sites (i.e. steps and defects) and the peaks III and IV to adsorption sites at terraces [17].
Next, we will discuss the influence of the cooling method on the oxidative stripping of CO adsorbed on the Pt(100). Figure 3 shows the CO stripping voltammetry of the Pt(100) electrodes obtained by the different annealing treatments. In the potential region between 0.1 and 0.5 V, all the profiles are characterised by a complete blocking of hydrogen adsorption. At higher potential, the profiles exhibit marked differences. The surface cooled down in Ar (Fig. 3) presents a pre-wave presumably associated with the CO oxidation on low coordination sites and a main peak at higher potential (0.64 V). The pre-wave is absent in the other two voltammograms, in which the main peak appears at potentials higher than 0.69 V. The voltammogram of the CO stripping at the surface cooled down in Ar presents only a very small feature at 0.67 V that would correspond to a small amount of well-ordered terraces.

CO stripping voltammogram of the Pt(100) electrode prepared by flame annealing and cooling in (solid line) H2/Ar (3:1) atmosphere (dashed line) Ar atmosphere and (dotted line) CO/Ar (3:1) atmosphere. The supporting electrolyte is 0.1 M NaOH. Scan rate = 20 mV/s
One of the more interesting observations is the difference in the CO oxidation profile between the surface treated in H2/Ar (Fig. 3) and the surface treated in CO atmosphere. The presence of a pre-wave is clearly observed between 0.62 and 0.69 V in the case of the surface cooled down in presence of CO. One could assign the pre-wave to the oxidation of CO on the step edges of the square monoatomic island, as suggested by the potential at which it appears. However, as was described in [15], the surface cooled down in the presence of CO results in a flat surface free of square islands. The absence of these square islands was tentatively associated to the higher mobility of the atoms under these conditions leading to the formation of small islands. In such a case, the pre-wave in Fig. 3 (electrode cooled down in CO atmosphere) can be attributed to the oxidation of CO on the edges of this “nanoislands”. Unfortunately, the ECSTM results in [15, 33] does not show a clear evidence of the presence of this nanoislands [15, 33].
Another tentative explanation of the pre-wave on the voltametric profile of the surface cooled down in CO is the presence of (110) step sites on the surface. One important observation has to be done with respect the EC-STM images reported in [15]. The step edges of the square monoatomic islands observed when the electrode was cooled down in the presence of H2/Ar are parallel and perpendicular to the [0,−1,−1] direction that corresponds to (111) step site. In contrast, the step edge on the surface cooled down in presence of CO, in the same report show a large curvature, suggesting that the result is a combination of (111) and (110) sites. In this regard, one may suggest that the pre-wave observed in the CO stripping voltammogram of the surface cooled down in presence of CO could correspond to the CO oxidation on the (110) step sites (see Fig. 2), which could be more active than the (111) step sites present at the edges of the monoatomic islands. Although ECSTM images in [15] are just a local proof and those not reflect the behavior of the complete surface, the idea of the presence of (110) sites cannot a priori be discarded. Previous results of electrochemical oxidation of CO on platinum stepped surfaces Pt(S) − [n(100) × (110)] in acidic media show that the most active site corresponds to the (110) step sites [24]. In contrast with the report of Hoshi et al. [24], the Alicante group recently presented a report in which it was shown that the CO oxidation activity on the Pt(n,1,0) surface in acidic media decreases with increasing the step density [6]. In order to true assign the origin of the pre-wave and the reactive site, more detailed ECSTM studies are required.
In previous reports, the low potential oxidation peak on the Pt(111) in alkaline media was associated with the presence of kink sites [8]. Based on experimental data of voltammograms at low scan rates and chronoamperometries of the CO oxidation on Pt(111) and step surfaces in alkaline media [9, 10] and Pt(100) in acidic media [25], the authors suggest that the oxidation of CO follow a nucleation and grow behavior. The instantaneous nucleation of OH (responsible of the oxidation) takes place on the steps sites. The presence of large amount of defects on the surface when the electrode is cooled down in Ar atmosphere should be the responsible of the early oxidation of CO. Tentative and based on previous observation, we can suggest that the (110) step sites or small islands present on the surface cooled down in the presence of CO acts as nucleation centers of OH adsorption, and it is easier than on the (111) step edges at the monoatomic islands present on the Pt(100) cooled down in H2/Ar.
In relation to the charge associated with the CO oxidation, the surface cooled down in the presence of CO gives a charge of 520 ± 12 μC cm−2 (uncorrected by anion adsorption), which is around 10% smaller in comparison with the surface cooled down in Ar (560 ± 25 μC cm−2 uncorrected by the anion adsorption). On the other hand, it is 10% higher that the value obtained for the surface cooled down in the presence of H2/Ar (480 ± 18 μC cm−2 uncorrected by the anion adsorption). The difference in the charge may be partially associated to the excess of platinum atoms on the surface as a result of the adsorption of oxygen traces of during the cooling treatment (Ar atmosphere) and is smaller for a well-ordered surface (CO/Ar atmosphere). However, the different charges mentioned above can also be associated to different coverage of the CO layer on a fully blocked surface. It is well documented in the literature [26, 27] that a fully CO blocked Pt(111) surface in acidic media presents two different adlayer domains: (2 × 2) − 3CO (θ = 0.75) and (√19 × √19)R23.3° − 13CO (θ = 0.68). It is important to mention that the compressive forces required to create the (2 × 2) − 3CO adlayer characteristic of the Pt(111)/CO was only achieved with a substantial long-range substrate order. Such condensed CO adlayers have also been observed on Pt(100) in acidic media by Wakisaka et al. [28], having a coverage of ca. 0.75. One way to discriminate between the two possibilities, surface atoms excess or differences in the coverage, is by looking at charge of the hydrogen adsorption region in Fig. 1. The Pt(100) cooled down in Ar atmosphere presents a charge of 280 μC cm−2, which is 5% and 10% larger than the charge associated to the hydrogen adsorption region on the Pt(100) cooled down on H2/Ar and CO/Ar atmosphere respectively. The fact that the surface cooled down in Ar presents the larger charge of H2 adsorption and CO oxidation reflect a higher roughness of the surface. On the other hand, the Pt(100) cooled down in CO/Ar atmosphere shows the least hydrogen adsorption charge but larger CO oxidation charge compared to the surface annealed under H2/Ar atmosphere. This suggests the presence of a well-order structure and a more compact adlayer of CO with higher coverage. An alternative way to look in the different coverage is by following the potential dependence infrared spectra of the CO oxidation. As the compressed adlayers exhibit different adsorption geometries, the electrochemical oxidation of adsorbed CO on the Pt(100) after different annealing procedures was also followed by FTIR in order to determine the preferential site of CO adsorption in each case. Figures 4 and 5 show the different spectra and the integrals of the CO bands on the Pt(100), as a function of potential, for the three different annealing treatments, respectively.

FTIR spectra for the oxidation of CO adsorbed on Pt(100) in 0.1 M NaOH for the electrode annealed in a H2/Ar (3:1) atmosphere, b Ar atmosphere and c CO/Ar (3:1) atmosphere. The potential was stepped from 0.05 V in 0.05 V steps up to 0.8 V. Adsorption potential E ad = 0.05 V vs RHE and reference potential is E ref = 0.8 V vs RHE. The sample potentials corresponding to each spectrum are indicated in the figure

Potential dependence of the integrated band intensities for COL (upper panel), COB (middle panel) and carbonate (bottom panel). Pt(100) electrode in 0.1 M NaOH, annealed in H2/Ar (3:1) atmosphere (●), Ar (■) and CO/Ar (3:1) atmosphere (∆). The integral values were obtained from the spectra of Fig. 4 using 0.8 V as reference spectra
Figure 4 shows the potential dependence of the FTIR spectra of adsorbed CO on Pt(100) in 0.1 M NaOH. The rate at which these spectra were taken corresponds to an effective scan rate of 10 mV/s. As a general characteristic in the spectra, two negative bands that diminish in intensity are observed. These two bands are associated to the stretching mode of CO adsorbed on top (COL) and bridge sites (COB). From Fig. 4, it can be also observed that at more positive potentials, these features are accompanied by the appearance of (initially) weak positive bands at about 1,650 and 1,400 cm−1. The first band is described in the literature as the O–H bending mode of water and is related to the instability of the thin layer [29], while the band at 1,400 cm−1 can be associated to the formation of a carbonaceous species [30, 31]. Table 1 summarises the peak positions of the COL and COB for the three annealing procedures. For the sake of comparison, the values corresponding to the CO adsorption in acidic media are included in parentheses.
The values corresponding to the frequencies of the CO adsorbed on the Pt(100) as presented in Table 1 are in agreement with the values reported in the literature based on experimental and DFT methods [32, 33]. However, in comparison with previous results in acidic media, the CO adsorbed on bridge site (about 1,865 cm−1) presents the major contribution in the spectra independently of the atmosphere used for cooling down the surface. As can be seen in Fig. 4 and most clearly in the values of the integrals in Fig. 5, the COL stretching band is almost negligible in comparison to the COB feature when the electrode was cooled down in a CO/Ar atmosphere. In comparison with the other two cooling treatments, the band of the COL is most significant when the electrode was cooled down in Ar atmosphere. In contrast with our results, in acidic media, Lopez-Cudero et al. obtained a very different contribution from the COL and COB adsorbed on the Pt(100) surface, the major contribution being the band at around 2,056 cm−1 assigned to the COL and a smaller band at about 1,820 cm−1 [34]. As for the latter band, based on the irreproducibility and also on the non-linear dependence of these bands with the potential, the authors suggest that it is related to the topography of the surface and the coverage of the CO, the band being more intense at lower coverage of CO. However, for higher coverages (maximum coverage of 0.79) or in the presence of CO in solution, the band related to COB becomes larger as is reported [34, 35].
The difference in the behavior of CO adsorption on Pt(100) between acidic and alkaline media may be addressed mainly to two factors. First, the higher COB abundance observed in alkaline media has to be related to an electronic effect. The “real” electrode potential is more negative in alkaline media (or the effective Fermi level more positive), with respect to the pH-independent levels in solution (i.e., the NHE scale), and consequently, an enhanced effect of back-donation into the Pt-CO bond is expected [36]. DFT calculations have shown that the latter effect may produce a stronger bonding interaction between Pt and CO and that multifold coordination is favored at these negative electrochemical potentials [37, 38]. Second, the introduction of a small amount of defects dramatically changes the CO adsorption state, while defect density increases the COB decreases (or which is the same the COL increases). Apparently, a long-range order of the (100) terrace is necessary to detect only COB. Additionally, a red shift of the COL and COB band centers is observed, compared to the experiment carried out with the Pt(100) surface annealed in CO/Ar. Again, this band center shift may be attributed to the higher amount of defects on these surfaces [18]. These latter observations emphasise the importance of long-range order to obtain a compressed CO adlayer and confirm the previous observation obtained by analysis of the charges, that is, on the CO/Ar annealed surfaces, the CO coverage is larger due to the large order surface structure and the absence of islands.
In Fig. 5, it can be also seen how the signals related to the COL decrease at more negative potentials than the corresponding one of the COB for all the different flame annealed treatments. Moreover, from the same figure is observable that CO adsorbed in bridge configuration is the most strongly adsorbed CO (i.e. the least reactive) and is completely removed from the Pt surface at potentials higher than 0.65 V. The latter was observed during the CO oxidation on slightly disordered Pt(111) electrode in alkaline media [18], in which it was associated to CO adsorbed in a bridge site on the top of the step site. Therefore, this COb is oxidised by OH adsorbed on the Pt(100) terrace at very positive potentials. It should be noted that the last affirmation is in perfect agreement with the anodic currents observed during the CO stripping voltammograms (Fig. 3).
It is also clearly seen in Fig. 5 that the formation of carbonate starts slightly earlier in the case of the electrode cooled down in Ar. It is also important to note that, when the electrodes are cooled down in H2/Ar or CO/Ar, the CO and carbonate bands disappear/appear respectively in a very similar way and at similar potentials. The oxidation on the surface cooled down in CO/Ar starts slightly earlier respect to the annealed with H2/Ar, as was also observed in the voltammogram of Fig. 1. The low-potential oxidation of CO on this surface could be associated with the oxidation of CO at the (110) step sites as was mentioned before. However, it can be also attributed to the oxidation of the metastable compressed adlayer.
Pt(110)
Figure 6a and b shows the cyclic voltammograms of the Pt(110) electrode in 0.1 M NaOH recorded after cooling the electrode in a H2/Ar atmosphere and air, respectively. Additionally, the voltametric profile of the Pt(110) cooled down in I2 vapor (see “Experimental”) is shown in the Fig. 6c. As can be seen in the figure, the profiles present a very similar behavior below 0.3 V vs RHE, although some differences can be observed for each treatment. In the case of the electrodes annealed and subsequently cooled down in Air and H2/Ar, the main peak seems to present two separate current contributions at 0.28 and 0.30 V. The more negative contribution is significantly higher in the surface cooled down in presence of H2 and shifted slightly to more negative potential (0.26 V), and the contribution at 0.3 V is more important when the electrode was cooled down in absence of H2. On the other hand, if the electrode is cooled down in presence of I2 vapor, the voltammetric profile presents an apparently single peak at 0.28 V where the two contributions cannot be distinguished. However, at potentials higher than 0.3 V, remarkable differences can be observed between the profiles of the different surfaces. These differences can be ascribed to the different adsorption sites present on the surface. The peak at 0.39 V(reversible) and at 0.73 V in the anodic scan (0.69 V in the cathodic scan) present in the profiles of the surfaces cooled down in air and I2 vapor could be ascribed to the presence of defects on the surface [8]. Similar features are observed when the well-ordered and freshly annealed electrode is cycled up to oxidation potentials [39]. It has been also reported that the surface contains a non-negligible number of defects even after hydrogen cooling [40]. Regarding the surface structure, previous studies by ex situ low-energy electron diffraction [41–43] have shown that the (1 × 2) Pt(l10) surface is stable in perchloric and sulfuric acid solution if the potential is restricted to potentials below 0.7 V. There is no previous report based on in situ techniques that shows the presence of the (1 × 2) reconstruction of the Pt(110) in an electrochemical environment. If there were a fast potential-induced reconstruction, the conclusions about the stability range for Pt(110) could be wrong (like for Au(110) [43]) because the (1 × 2) structure could be formed back from (1 × 1) at the immersion potential. However, Pt atoms have a much lower surface mobility than Au atoms, and hence, a potential-induced surface reconstruction should be kinetically strongly hindered.

Cyclic voltammogram of the same Pt(110) electrode pretreated by flame annealing and cooling in a H2/Ar (3:1) atmosphere, b air atmosphere and c I2 vapor. The supporting electrolyte is 0.1 M NaOH. Scan rate = 50 mV/s
Since the surface structure of the Pt(110) is strongly affected by the annealing treatment, it is expected that significant changes in the CO oxidation stripping curves and also in the adsorption sites of the CO will be observed. In order to confirm this expectation, stripping voltammetry and FTIR experiments were performed.
Figure 7 shows the potential sweeps in 0.1 M NaOH at 20 mV s−1 for the anodic stripping of CO adsorbed on the Pt(110) treated in the previously described atmospheres. As in the case of Pt(100), the complete blockage of the surfaces was confirmed by the absence of current density in the region between 0.1 and 0.3 V associated to the adsorption/desorption of hydrogen. Significant differences were found as a function of the annealing treatment. In the case of the electrode quenched with water (i.e. fast cooling down in air), the voltametric profile of the Fig. 7b presents a “pre-wave” (0.3–0.5 V) that was found previously for the Pt(111) surface, and it could be ascribed to oxidation of CO at defect sites, more specifically kink sites [8, 18]. Increasing the electrode potential from 0.5 to 0.7 V, two overlapping contributions in the CO oxidation can be observed, and those might be associated to the oxidation of CO on terrace sites. The broadness of these voltammetric features should be associated to a large distribution of terraces sites, with peak potentials suggesting relatively short terraces with (1 × 1)–Pt(110) structure. Additionally, a small contribution in current is observed at higher potentials. As we reported for the oxidation of CO on Pt(111) in alkaline media [8–10, 18], the late oxidation of CO may be attributed to the CO adsorbed on the step site oxidised by OH adsorbed on terrace site. The broad potential window for CO oxidation likely corresponds to the contribution of terraces with different width.

CO stripping voltammogram of the Pt(110) electrode prepared by flame annealing and cooling in a H2/Ar (3:1) atmosphere, b air and c I2 vapor. The supporting electrolyte is 0.1 M NaOH. Scan rate = 20 mV/s
When the electrode was flame annealed and cooled down in a H2/Ar saturated atmosphere, the voltammogram (shown in Fig. 7a) shows two well-defined potential regions: a small pre-peak between 0.4 and 0.6 V, as in Fig. 7b but now with lower current density, and a main peak at 0.6 V. As was mentioned before the peak at 0.6 V can be associated to the oxidation of CO at the terrace sites with (1 × 1)–Pt(110) structure. Analysing the voltammogram more in detail, the peak at 0.6 V presents a small shoulder at 0.68 V that may be associated to a site with lower reactivity towards the CO oxidation, i.e. the produced surface has principal domains (terraces) and a low defect density (steps and kinks). Moreover, these defects seem to be more of long range (step) than short range (kink). The last assumption is supported by previous studies [8–10], in which the step site has two different catalytic activities towards the CO oxidation: CO adsorbed below the step is more reactive than the CO adsorbed further away on the terrace (pre-peak at about 0.5 V), while CO adsorbed at the step is the least active. The curves of the CO stripping experiments confirm the observations that flame annealing in H2/Ar leads to a surface with homogeneous and larger well-ordered terraces in comparison with the surface cooled down in absence of H2.
The most significant results were obtained when the electrode was cooled down in I2. The voltammogram looks similar to the profile of the electrode cooled down in H2/Ar, but the ratio of the peaks at 0.6 and 0.68 V is quite different; in this case, the peak developed at more positive potentials is well resolved and has a higher charge. As was mentioned before and similar to the behavior of the CO oxidation on Pt(111) [8–10], the feature at 0.68 V (late CO oxidation) may correspond to the CO strongly adsorbed on the step site oxidised by OH adsorbed on terrace site. CO adsorbed on such a low coordinated site should present significant difference in adsorption energy as well as in vibrational frequency with respect to the CO adsorbed on other platinum sites [36].
Figures 8 and 9 show the difference spectra and the integrals of the bands corresponding to CO oxidation on the Pt(110) after the three different annealing treatments. Table 2 summarises the peak positions corresponding to the observed CO frequencies. In general, Fig. 9 shows some very important differences with respect the spectrum of CO on Pt(110) in UHV and acidic media. In UHV and acidic media, CO adsorption on Pt(110) shows a single band located at around 2,150 and 2,050 cm−1, respectively, for a similar coverage of 0.5 [44]. By contrast, in alkaline media, Fig. 9 shows the coexistence of two or even three different adsorption sites of CO for the same maximum coverage of 0.5. The band corresponding to the linearly bonded CO (COL) appears at lower wavenumbers (about 2,030 cm−1) than in acidic media, and the band position depends sensitively on the cooling treatment. Density Functional Theory (DFT) calculations have shown that the effect of the more negative potential, or the effectively higher Fermi level, may produce a stronger bonding interaction between Pt and CO [37, 38, 45]. It will typically also lead to lower CO stretching frequencies and to an enhanced propensity to multifold coordination. A second small vibration band related to bridge CO (COB) appears at around 1,847 cm−1, but it does not depend on the employed cooling treatment. In addition to these two bands, the spectra recorded during the CO oxidation on the Pt(110) cooled down in I2 vapor exhibit a band not reported previously at 1,955 cm−1 (\( {\text{CO}}_{\text{L}}^{{*}} \)). As can be observed in the spectra of Fig. 8c, this negative band is present in the potential region between the initial potential and starts to decrease at higher potentials than 0.3 V. This band at 1,955 cm−1 appears close to a band at 1,920 cm−1 observed by Ito et al. on stepped Pt(2n−1,1,1) electrodes with (100) terraces and (111) step sites in acidic media [46]. In UHV environment, Bare et al. reported the formation of an intermediate state of CO adsorbed on a “short bridge state” at 1,915 cm−1, in the coverage range 0.5 < θ < 0.9 after partial desorption of the CO using high resolution electron energy loss spectroscopy [47]. In addition to these experimental values observed in UHV, Yamagishi et al. [32] proposed the presence of a CO linearly bonded on the (1 × 2)–Pt(110) with a tilt angle with respect to the (110) normal with a stretching frequency of 1,922 cm−1 on the basis of DFT calculations, which is close to the value obtained in this work. Unfortunately, the fact that the band overlaps with the other COL band at 2,025 cm−1, irrespective of which reference potential is taken (E ref = 0.05 V, E ref = 0.9 V or E ref = 0.4 V), complicates the accurate integration and the estimation of the Stark tuning shift of the bands. The lower frequency observed for the \( {\text{CO}}_{\text{L}}^{{*}} \) may be explained by an increase in the back donation produced by atoms with lower coordination (i.e. steps). Moreover, it is observed that the band intensity related to \( {\text{CO}}_{\text{L}}^{{*}} \) decreases at potentials higher than 0.3 V, which is about 0.15 V more positive than the observed for COB. Consequently, it is possible to suggest a picture of the system at the initial potential (0.05 V), with linear CO adsorbed at two different sites, i.e. COL adsorbed on terraces with (1 × 2)–Pt(110) and \( {\text{CO}}_{\text{L}}^{{*}} \) adsorbed on the missing row step. Next, as potential increases, the COB oxidises at E > 0.15 V, while the \( {\text{CO}}_{\text{L}}^{{*}} \) is oxidised at E > 0.3 V in a clear correlation with the CO stripping voltammogram depicted in Fig. 9a (the difference in the potentials has to be related to the different techniques used in the experiments carried out by FTIRS and cyclic voltammetry: stepping and sweeping potential respectively, even though the effective potential sweep rates are similar).

FTIR spectra for the oxidation of CO adsorbed on Pt(110) in 0.1 M NaOH for an electrode annealed in a H2/Ar (3:1) atmosphere, b air and c I2 vapors. The potential was stepped from 0.05 V in 0.05 V steps up to 0.8 V. Adsorption potential E ad = 0.05 V vs RHE and reference potential is E ref = 0.8 V vs RHE. The sample potentials corresponding to each spectrum are indicated in the figure

Potential dependence of the integrated band intensities for COL (upper panel), COB (middle panel) and carbonate (bottom panel). Pt(110) electrode in 0.1 M NaOH, annealed in H2/Ar (3:1) atmosphere (★), air (■) and I2 vapor (∆). The integral values were obtained from the spectra of Fig. 9 using 0.8 V as reference spectra
A linear Stark tuning shift of the CO stretching frequency with applied potential is observed for COL and COB with slopes of 37 and 36 cm−1 V−1, respectively. Similar Stark tuning slopes for COL and COB were found in a previous study of CO adsorption and oxidation on Pt(110) in acidic solution [45].
As can be observed in the spectra, the oxidation of COB occurs at lower potentials than the oxidation of COL, and the band intensity as well as the band position does not depend on the annealing method, suggesting that this oxidation peak is not very sensitive to the surface order. However, the band related to the CO adsorbed on top sites is more significantly affected by the employed annealing treatment. Two important characteristics are the high potential oxidation of the COL on the Pt(110) annealed in H2/Ar, in agreement with the CO stripping voltammogram of Fig. 7, and the increase in the intensity of the band at 1,955 cm−1 when the annealing is carried out in I2. The latter suggests shorter terraces or higher step density (i.e. conversion from COL → \( {\text{CO}}_{\text{L}}^{{*}} \)) when the electrode is cooled down in I2 atmosphere than annealed in H2/Ar gases. This fact could be related to the high mobility of the low-coordinated Pt atoms present on the missing row, a mobility induced by the COads oxidation that results in the faceting of the surface as was described earlier in UHV [48].
Conclusion
In this work, we presented a study on the electro-oxidation of CO on Pt(100) and Pt(110) single crystal electrodes in alkaline media and the effect of different surface preparations of the Pt(100) and Pt(110) surface on the CO adsorption and oxidation.
As in the case of acidic media [13–15], we found that the Pt(100) and Pt(110) present significant differences in the blank voltammogram in alkaline media as a function of the annealing treatment.
For the oxidation of CO adsorbed on the Pt(100), it was found that the CO oxidation takes place at low potentials on a surface with lower superficial order. Using FTIR, two different adsorption sites were identified on the Pt(100) regardless of the treatment employed: linearly bonded CO (COL) and bridged bonded CO (COB). In contrast with the observations in acidic media [34], the predominant adsorbate in alkaline media was the COB for all surfaces. The contribution of COL is more significant for the surface with lower superficial order (i.e. the one cooled down in Ar).
The CO oxidation depends strongly on the surface preparation of the Pt(110). The broad feature in the oxidation of CO when the surface was cooled down in air suggests a large distribution of terraces sites. When the electrode was flame annealed and cooled down in a H2/Ar saturated atmosphere, the stripping voltammogram shows two well-defined peaks that can be associated to the oxidation of CO at the terrace sites with (1 × 1)–Pt(110) and steps sites, respectively. When the electrode was cooled down in I2, the peak associated to the CO oxidation on defect sites is well resolved, is slightly shifted to positive potentials and has a higher charge. Voltametric and FTIR results suggest that CO adsorbed on the low coordinated platinum atoms is the least reactive.
Using FTIR, three different adsorptions sites were found on the Pt(110), i.e. COL, \( {\text{CO}}_{\text{L}}^{{*}} \) and COB. The CO linearly bonded (COL) and CO bridged bonded (COB) appear in the spectra regardless the treatment employed; however, the intensity of the bands depends strongly on the cooling treatment. The predominant adsorbate in alkaline media was the COL for all three cooling methods employed. The contribution of COB is more significant for the surfaces with lower superficial order (i.e. the ones cooled down in air and I2 vapor). Additionally, the identification of \( {\text{CO}}_{\text{L}}^{{*}} \) strongly adsorbed on low coordinated atoms was supported by FTIR, by a band at 1,955 cm−1. It was observed that the band intensity related to \( {\text{CO}}_{\text{L}}^{{*}} \) decreases at potentials higher than 0.3 V, which is about 0.15 V more positive than the observed for COL, suggesting a lower reactivity of \( {\text{CO}}_{\text{L}}^{{*}} \). Finally, it is possible to suggest a mechanism in which at the initial potential (0.05 V), COL is adsorbed at two different sites, i.e. COL adsorbed on terraces with (1 × 2)–Pt(110) and \( {\text{CO}}_{\text{L}}^{{*}} \) adsorbed on the missing row step, with the COL being more reactive than the \( {\text{CO}}_{\text{L}}^{{*}} \).
References
N.P. Lebedeva, M.T.M. Koper, E. Herrero, J.M. Feliu, R.A. van Santen, J. Electroanal. Chem. 487, 37–44 (2000)
N. Hoshi, M. Tanizaki, O. Koga, Y. Hori, Chem. Phys. Lett. 336, 13–18 (2001)
N.P. Lebedeva, M.T.M. Koper, J.M. Feliu, R.A. van Santen, J. Electroanal. Chem. 524–525, 242–251 (2002)
N.P. Lebedeva, M.T.M. Koper, J.M. Feliu, R.A. van Santen, J. Phys.Chem. B 106, 12938–12947 (2002)
D.S. Strmcnik, D.V. Tripkovic, D. van der Vliet, K. Chang, V. Komanicky, H. You, G. Karapetrov, J.P. Greeley, V.R. Stamenkovic, N.M. Markovic, J. Am. Chem. Soc. 130, 15332–15339 (2008)
F.J. Vidal-Iglesias, J. Solla-Gullón, J.M. Campiña, E. Herrero, A. Aldaz, J.M. Feliu, Electrochimica Acta 54, 4459–4466 (2009)
J. Spendelow, G.Q. Lu, P.J.A. Kenis, A. Wieckowski, J. Phys. Chem. B 110, 9545–9555 (2006)
G. García, M.T.M. Koper, Phys. Chem. Chem. Phys. 10, 3802–3811 (2008)
G. García, M.T.M. Koper, J. Am. Chem. Soc. 131(15), 5384–5385 (2009)
G. García, M.T.M. Koper, Phys. Chem. Chem. Phys. 11, 11437–11446 (2009)
J. Clavilier, R. Faure, G. Guinet, R. Durand, J. Electroanal. Chem. 107, 205 (1980)
J. Clavilier, J. Electroanal. Chem. 107, 211 (1980)
J. Clavilier, J.M. Orts, J.M. Feliu, J. Phys. IV 4 (1994)
J.M. Feliu, A. Rodes, J.M. Orts, J. Clavilier, Pol. J. Chem. 68, 1575–1595 (1994)
L.A. Kibler, A. Cuesta, M. Kleinert, D.M. Kolb, J. Electroanal. Chem. 484, 73–82 (2000)
A. Al-Akl, G.A. Attard, R. Price, B. Timothy, J. Electroanal. Chem. 467, 60–66 (1997)
D. van der Vliet, M.T.M. Koper, Surf. Sci. 604, 1912–1918 (2010)
G. García, P. Rodriguez, V. Rosca, M.T.M. Koper, Langmuir 25(23), 13661–13666 (2009)
A.M. Bittner, J. Wintterlin, G. Ertl, J. Electroanal. Chem. 388, 225–231 (1995)
R. Vogel, I. Kamphausen, H. Baltruschat, Ber. Bunsenges. Phys. Chem. 96, 525 (1992)
J. Clavilier, D. Armand, S.G. Sun, M. Petit, J. Electroanal. Chem. 205, 267 (1986)
F.J. Vidal-Iglesias, N. García-Aráez, V. Montiel, J.M. Feliu, A. Aldaz, Electrochem Comm 5(1), 22–26 (2003)
J. Kollár, L. Vitos, B. Johansson, H.L. Skriver, Phys Stat. Sol. 217, 405–418 (2000)
K. Mikita, M. Nakamura, N. Hoshi, Langmuir 23(17), 9092–9097 (2007)
C.F. McFadden, P.S. Cremer, A.J. Gellman, Lagmuir 12(10), 2483–2487 (1996)
I. Villegas, M. Weaver, J. Chem. Phys. 101(2), 1648–1660 (1994)
A. Rodes, R. Gómez, J.M. Feliu, M.J. Weaver, Langmuir 16, 811–816 (2000)
M. Wakisaka, T. Ohkanda, T. Yoneyama, H. Uchida and M. Watanabe (2005) Chem. Commun. 2710–2712
B. Rasch, T. Iwasita, Electrochimica Acta 35, 989–993 (1990)
T. Iwasita, A. Rodes, E. Pastor, J. Electroanal. Chem. 383, 181–189 (1995)
K. Arihara, F. Kitamura, Takeo, K. Tokuda, J. Electroanal. Chem. 510, 128–135 (2001)
S. Yamagi shi, T. Fujimoto, Y. Inada, H. Orita, J. Phys. Chem B 109, 8899–8908 (2005)
R. Martin, P. Gardner, A.M. Bradshaw, Surf. Sci. 342, 69–84 (1995)
A. López-Cudero, A. Cuesta, C. Gutiérrez, J. Electroanal. Chem. 586, 204–216 (2006)
C.K. Rhee, J.M. Feliu, E. Herrero, P. Mrozek, A. Wieckowski, J. Phys. Chem. 97, 9730–9735 (1993)
B. Hammer, O.H. Nielsen, J.K. Norskov, Catalysis Lett. 46, 31–35 (1997)
G. Blyholder, J. Phys. Chem. 68, 2772 (1964)
M.T.M. Koper, R.A. van Santen, J. Electroanal. Chem. 476, 64 (1999)
C. Morallon, J. Electroanal. Chem. Interfacial Electrochem. 288, 217 (1990)
J. Souza-Garcia, V. Climent, J.M. Feliu, Electrochem. Commun. 11, 1515–1518 (2009)
E. Yeager, A. Homa, B.D. Cahan, D. Scherson, J. Vac. Sci. Technol. 20, 628–633 (1982)
R. Michaelis, D.M. Kolb, J. Electroanal. Chem. 328, 341–348 (1992)
R. Michaelis, D.M. Kolb, Surf. Sci. 234, L281–L284 (1990)
S.-C. Chang, M. Weaver, Surf. Sci. 238, 142–162 (1990)
M.T.M. Koper, R.A. van Santen, S.A. Wasileski, M.J. Weaver, J. Chem. Phys. 113, 4392 (2000)
S. Watanabe, J. Inukai, M. Ito, Surf. Sci. 293, 1 (1993)
S.R. Bare, P. Hofmann, D. King, Surf. Sci. 144(2–3), 347–369 (1984)
R. Ladas, Imbihl, G. Ertl, Surf. Sci. 198, 42–68 (1988)
Acknowledgements
This work was funded by The Netherlands Organization for Scientific Research (NWO) through a “VICI” grant awarded to M.T.M.K. The authors also acknowledge the support of the European Commission through the FP7 Initial Training Network “ELCAT” (grant agreement no. 214936-2).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Rodríguez, P., García, G., Herrero, E. et al. Effect of the Surface Structure of Pt(100) and Pt(110) on the Oxidation of Carbon Monoxide in Alkaline Solution: an FTIR and Electrochemical Study. Electrocatal 2, 242–253 (2011). https://doi.org/10.1007/s12678-011-0059-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-011-0059-9