
Abstract
Background
As the most common primary bone cancer, osteosarcoma (OS) still lacks satisfactory therapeutic outcomes. Therefore, it is crucial to further evaluate OS at different risk levels and identify new intervention targets. Many evidences suggest the important role of angiogenesis in OS, but further exploration is needed.
Methods
We utilized public databases TARGET and GEO and employed bioinformatics algorithms such as LASSO, univariate and multivariate Cox regression analyses, and unsupervised consensus clustering to explore the role of angiogenesis-related genes (AGRGs) in OS. By calculating AGRG scores, we further analyzed OS molecular subtypes based on AGRGs. The correlation between AGRG scores and immune infiltration was subsequently examined. In vitro experiments, including WB, PCR, siRNA, migration, and invasion assays, were used to determine the value of the selected targets for OS.
Results
Ultimately, we established an OS prognosis model based on five AGRGs (COL5A2, CXCL6, FSTL1, NRP1, and TNFRSF21) that can independently validate prognosis levels. In vitro experiments confirmed the aberrant expression of CXCL6 in OS and its potential role in migration and invasion.
Conclusion
Our study reveals the impact of angiogenesis on OS from a novel perspective and provides potential intervention targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Osteosarcoma (OS), the most common primary bone cancer, develops from mesenchymal cells [1]. Because of the local invasion and early metastasis of OS, the therapeutic outcomes for OS are still not ideal despite the continuous optimization of treatment regimens in recent years [2]. With the increasing understanding of the role of the tumor microenvironment (TME) in the pathogenesis of OS, targeted therapy and immunotherapy have become novel treatment options for sarcomas [3]. However, because of tumor heterogeneity and the complexity of the TME, some patients with OS do not respond well [4]. Therefore, it is clinically significant to further stratify OS risk and identify new biomarkers in order to find new treatment strategies and improve OS prognosis.
Angiogenesis is typical during tumor progression due to the need for tumor growth and proliferation [5]. Endothelial cells are activated by angiogenic factors. The proteolytic enzymes produced during this process can degrade the extracellular matrix and further enable endothelial cells to proliferate and migrate to surrounding tissues to form new blood vessels [6]. These new blood vessels provide oxygen and nutrients to cancer cells and play an important role in cancer cell survival and metastasis [7, 8]. During OS development, multiple features, including proliferation, migration, and metastasis, were associated with angiogenesis [9]. Typically, anti-angiogenic factors and angiogenic factors jointly regulate local vascular formation and play a crucial balancing role in skeletal and cartilage homeostasis [10, 11]. Imbalances in bone homeostasis caused by changes in angiogenesis and vascular secretion activity may lead to further development of osteosarcoma [12, 13]. Angiogenic factors activate endothelial cells, which further produce proteolytic enzymes and degrade the extracellular matrix [7]. The degradation of the underlying basal membrane facilitates endothelial cell proliferation and migration to surrounding tissues to form new blood vessels, which allows for the further development of osteosarcoma and potential distant metastasis [6].
The role of anti-angiogenic therapies in the treatment of OS has gained increasing recognition. As an important regulator of angiogenesis, the vascular endothelial growth factor (VEGF) pathway in OS can be activated by a variety of inducers [14, 15]. Abnormal activation of VEGF pathway during OS carcinogenesis suggests abnormal activation of angiogenesis process in OS patients [16]. Antiangiogenic drugs have shown great potential in preclinical trials and the clinical treatment of OS by inhibiting the VEGF related pathways [17, 18]. However, more evidence is needed to further confirm the involved targets in the process. Furthermore, the risk stratification of anti-angiogenesis targeted therapies for OS needs further evaluation with supporting evidence.
The cells surrounding the tumor and external factors constitute the tumor microenvironment (TME) and promote tumor growth by exerting immunosuppressive functions. In addition to immune-cytotoxic CD8+ T cells, CD4+ T cells and natural killer cells, tumor-associated macrophages (TAM), myeloid-derived suppressor cells (MDSC), regulatory T cells (Treg cells) are common tumor-associated participants in TME [19, 20]. There was abundant infiltration of inflammatory cells in OS tissues, forming a complex immune microenvironment [21]. This complex immune microenvironment plays a key role in the development and progression of OS [22, 23]. Factors such as hypoxia in TME can induce angiogenesis. Moreover, induced tumor-associated angiogenesis can accelerate tumor growth and further induce tumor-associated TME formation [24]. By regulating the angiogenesis process, and then changing this complex crosstalk can not only slow down tumor growth, but also activate the anti-tumor immunity [25]. Therefore, it is necessary to further study the correlation between angiogenesis and immune infiltration in OS.
In this study, we used bioinformatics algorithms to explore the role of angiogenesis related genes (AGRGs) in OS. We established OS molecular typing based on AGRGs. AGRGs scores were then calculated and their correlation with immune infiltration was investigated. We further established AGRGs-related OS prognostic model that could independently predict the prognosis of patients with OS. Finally, in vitro experiments further determined the abnormal expression of the screened target CXCL6 in OS cells and its effects on migration and invasion of OS cells, thus partially confirming the reliability of the bioinformatics results. Our study suggests the important role of angiogenesis in OS patients from a new perspective.
2 Materials and methods
2.1 Transcriptome data acquisition and preprocessing
Gene features of OS transcriptomes used in this study were obtained from the TARGET and GEO (GSE21257 and GSE39058) databases. To enhance clarity, consider splitting into two sentences: “OS samples without clinical baseline data were excluded. A total of 140 OS samples (TARGET: 87 samples, GEO: 53 samples) were extracted from the two independent databases for final analysis. The “sva” script was employed to standardize the transcriptome data from the two independent databases, eliminating batch effects between the data [26, 27]. Clinical baseline data of OS from both databases were extracted in a Perl language environment and incorporated into subsequent analyses. Angiogenesis-related genes (AGRGs) were obtained from the MSigDB database (Supplementary Table 1) [28].
2.2 Identification of AGRG prognostic signature and molecular subtype features
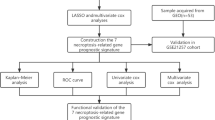
The univariate Cox algorithm was used to analyze the relationship between AGRG gene signature and OS clinical survival time. The LASSO regression analysis algorithm was utilized to screen AGRG prognostic variables related to OS prognosis, and the multivariate Cox analysis algorithm was applied to determine AGRG variables with independent prognostic value. Based on these AGRG prognostic variables with independent prognostic value, OS samples were subjected to unsupervised consensus clustering analysis using the “ConsensusClusterPlus” script, and subtypes were classified according to the optimal k value. The “ggplot2” script was used to explore the distribution patterns among different molecular subtypes. Survival curves of OS samples from different molecular subtypes were visualized using the “survival” script for analysis. The STRING database was used to predict protein-protein interactions among AGRG and to construct a correlation network diagram.
2.3 Construction and independent performance evaluation of AGRG scoring model
Using the hazard ratio of AGRG signature obtained from multivariate Cox analysis and AGRG expression levels, AGRG scores were calculated for each OS sample, and a scoring model based on AGRG scores was constructed. The “ggpubr” script was used to analyze the distribution of AGRG scores among different molecular subtype subgroups. The “ggalluvial” script analyzed associations among AGRG subtypes, score subgroups, and OS survival outcomes. Based on the median value of AGRG scores, OS samples were randomly categorized into high and low AGRG score subgroups. The “survival” R package was used to explore clinical survival outcomes between the two AGRG score subgroups. PCA plots of AGRG score subgroups were generated based on the “ggplot2” script to explore the distribution of the two subgroups. To better explain the reliability and accuracy of AGRG score prediction for OS clinical survival outcomes, the “caret” script was executed to randomly divide OS samples into training and validation sets at a ratio of 7:3. AGRG scores were calculated using the same method for OS samples in both independent cohorts, and different score subgroups were classified. The “survivalROC” script was used to calculate the AUC of ROC curves for 1-year, 3-year, and 5-year survival to assess the accuracy of AGRG scores.
2.4 Independent prognostic evaluation of AGRG scores
Based on the clinical pathological characteristics of OS samples and AGRG scores, hazard ratios (HR) for each clinical feature and AGRG score were calculated using univariate and multivariate Cox analysis algorithms, and the independent prognostic value of each variable was evaluated. Using AGRG scores and various clinical pathological features, a novel nomogram model was constructed using the “rms” script to predict survival probabilities of OS samples at different time points. The “pec” and “ggDCA” scripts were used to analyze the consistency and accuracy of AGRG score and clinical feature variables in predicting OS clinical survival probabilities.
2.5 Evaluation of immune microenvironment infiltration features and drug sensitivity analysis
Based on the gene reference set of KEGG pathways “c2.cp.kegg.v7.2.symbols,” the “GSVA” script was executed to evaluate potential molecular mechanisms between AGRG score subgroups. Using the ssGSEA algorithm, relative proportions of 23 immune cell types and immune function scores were calculated based on transcriptome data of OS samples, with reference to marker genes for immune cells reported in the “immune.gmt” gene sequences. Additionally, based on transcriptome features of OS samples, the “ESTIMATE” script was used to evaluate immune infiltration features of AGRG score subgroups and calculate four indices including stromal score, immune score, ESTIMATE score, and tumor purity. Pearson correlation algorithm was used to assess the correlation between AGRG prognostic signature and immune microenvironment features, exploring the potential associations. Using the GDSC database, the “pRRophetic” script was employed to predict potential small molecule compounds with effects on AGRG score subgroups based on transcriptome features of OS.
2.6 Cell culture
The MG63 osteosarcoma cell line was obtained from the China Center for Type Culture Collection (CCTCC). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 atmosphere. Cells were passaged every 2–3 days to ensure they were in the logarithmic growth phase for experiments.
2.7 Construction and transfection of CXCL6 interference plasmid
Gene silencing was performed using small interfering RNA (siRNA) specifically designed to target the CXCL6 gene. Specific siRNA sequences were designed and synthesized based on the CXCL6 gene sequence (GenePharma), with the sequence number: 5′-GCUCAGAGAUUGAGCUGUA-3′. The siRNA sequence was cloned into the pSilencer 2.1-U6 hygro plasmid vector to construct the CXCL6 interference plasmid. The correctness of the plasmid construction was verified by sequencing. The CXCL6 interference plasmid was transfected into MG63 cells using Lipofectamine 2000 transfection reagent (Invitrogen). One day prior to transfection, MG63 cells were seeded into 6-well plates at an appropriate density to ensure approximately 70–80% confluence at the time of transfection. Following the Lipofectamine 2000 reagent manual, the plasmid DNA was mixed with Lipofectamine 2000 reagent at a ratio of 1:2 and incubated at room temperature for 20 min before being added to the cell culture medium. After 24 h of transfection, the medium was replaced with fresh culture medium for further incubation.
2.8 Quantitative real-time RT-PCR (qRT- PCR) analysis
RNA extraction from both normal and Mg63 OS cell lines was conducted utilizing Trizol reagent (Catalog number: 15596018, Thermo). Subsequent cDNA synthesis was carried out using the RT kit with gDNA Eraser (Perfect Real Time). Real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Catalog number: RR047A, Takara) was employed for further analysis. The mRNA expression levels were determined using SYBR Pre-mix Ex Taq II (TliRNaseH Plus) (Catalog number: RR820B, Takara). The assessment of relative RNA expression levels was performed using the 2–ΔΔCT method. The GAPDH and CXCL6 PCR primers were as follows: GAPDH: Forward: 5′ TGAAGCAGGCATCTGAGGG 3′, Reverse: 5′ CGAAGGTGGAAGAGTGGGAG 3′; CXCL6 Forward: 5′ TTGGTAAACTGCAGGTGTTCCC 3′, Reverse: 5′ CAGACAAACTTGCTTCCCGTTC 3′.
2.9 Western blot analysis
Total protein was extracted from normal and Mg63 cell lines using RIPA lysis buffer (Catalog number: R0010, Solarbio). Protein quantification was performed using the BCA assay kit (Catalog number: PC0020, Solarbio). A 20 µL protein sample was mixed with 200 µL of BCA working solution and incubated at 37 ℃ for 30 min. After quantification, protein samples were mixed with loading buffer (4:1 v/v), boiled for 10 min, subjected to SDS-PAGE, and transferred to a PVDF membrane. The membrane was blocked with 5% skimmed milk powder for 1 h at room temperature. Following this, the membrane was incubated overnight at 4 °C with primary antibodies targeting β-actin (Catalog number: E-AB-40517, Elabscience), CXCL6 (Catalog number: PA5-67571, Invitrogen), MMP2 (catalog: MA5-14186, Invitrogen), MMP9 (catalog: MA5-13595, Invitrogen). After washing with TBST buffer solution three times, the transferred membrane was incubated with the secondary antibody (1:20000) at room temperature for 1 h, followed by additional washes with TBST. Ultimately, the protein bands were visualized using an Odyssey Clx system (Li-Cor, America).
2.10 Transient transfection
The Mg63 cells were seeded in 6-well plates. Once the cell density reached 40–60%, transfections were made following the Lipofectamine 2000 transfection reagent instructions. The transfected cells were collected for further experimentation after 48 to 72 h.
2.11 Cell Migration and Invasion Assay
For the cell invasion experiment, ABW Matrigel stored at −80 °C was thawed in a −4 °C refrigerator overnight until it became liquid. Serum-free medium was used to dilute Matrigel to 1 mg/mL, and 100µL of the solution was added to Transwell chambers (pore size 8 μm, TCS003006, BIOFIL). The chambers were placed in a 37 °C incubator for 4–5 h. After incubation, 100µL serum-free medium was added, and the plates were placed in a 37 °C incubator for an additional 30 min for hydration. The culture medium was then aspirated and set aside. OS cells and transfected cells were cultured to logarithmic growth phase, detached, washed once with PBS and serum-free medium, and then suspended in serum-free medium. Cell concentration was adjusted to 2 × 10^5/mL. In the lower chamber, 600µL of medium containing 10% serum was added, and in the upper chamber, 150µL of cell suspension was added. The cells were incubated for 24 h. Carefully remove the Transwell chambers, aspirate the liquid from the upper chamber, wash, and transfer to wells containing 4% PFA. After fixing at room temperature for 20 min, wash with PBS, stain with 10% crystal violet for 5–10 min, rinse with carbonate-free water until no purple color is observed. Gently wipe the upper side of the chambers with a cotton swab, use the tip of a syringe needle to carefully remove the PC membrane, place it upside down to air dry, and mount it on a glass slide using neutral gum. Count five random fields under a microscope and record the results.
2.12 Statistical analysis
In this study, data extraction and processing were carried out in both the Perl and R programming environments. The potential correlation between AGRG scores and immune cells was computed using the Pearson correlation algorithm. For comparisons between two groups, the Wilcoxon rank-sum test was employed to assess the significance of data differences. In the case of comparisons involving three groups, the one-way ANOVA test algorithm was utilized to determine the significance of data differences. A significance level of P < 0.05 was considered indicative of substantial data differences.
3 Results
3.1 Identification of prognosis-associated AGRGs in OS and molecular subtype recognition analysis
We collected 34 AGRGs to explore their potential roles in the occurrence and progression of OS. Figure 1A shows the protein-protein interaction (PPI) network of the 34 AGRGs. We analyzed the HR and p-values of these 34 AGRGs based on their expression profiles and clinical survival data in OS. Results from LASSO-univariate Cox regression analysis indicated that 8 AGRGs features were significantly associated with the clinical survival prognosis of OS, including 4 risk factors (APOH, COL5A2, CXCL6, and LPL) and 4 favorable factors (FSTL1, NRP1, SLCP2A1, and TNFRSF21) (Fig. 1B–D). Based on the univariate Cox analysis results, we further conducted multivariate Cox analysis on these 8 AGRGs associated with the clinical survival outcome of OS to evaluate their independent predictive value in predicting OS clinical prognosis. The results indicated that 5 AGRGs have independent predictive value for OS prognosis. According to the expression profiles of these 5 AGRGs, we performed unsupervised consensus clustering analysis and classified OS samples into 4 different AGRGs molecular subtypes based on the optimal K value. Unsupervised PCA showed significant distribution patterns among the 4 AGRG subtypes, indicating their independence (Fig. 1E). Analysis of clinical prognosis outcomes showed that among these 4 AGRGs molecular subtypes, AGRG subtype C had the worst clinical prognosis (Fig. 1F, p = 0.04).

Identification of prognosis-associated AGRG and recognition of AGRG-related molecular subtypes. A The PPI network diagram reveals potential interactions among 34 AGRG. B–D LASSO-univariate Cox analysis demonstrates AGRG associated with clinical survival outcomes of OS samples. E Unsupervised PCA analysis suggests distribution patterns among different AGRG molecular subtypes. F Clinical survival outcomes of OS samples in different AGRG subtypes are calculated using the Log-rank algorithm
3.2 Analysis of immune infiltration features of AGRG molecular subtypes
The pathogenesis of OS, a highly invasive malignant bone tumor, involves changes in immune cell infiltration. To better explore the key factors influencing clinical survival outcomes among different AGRG molecular subtypes, we evaluated the immune infiltration status and relative proportions of immune cells among AGRG molecular subtypes based on the ESTIMATE and ssGSEA algorithms. The ESTIMATE results show significant differences in immune infiltration statuses among the AGRG molecular subtypes. Notably, AGRG subtype C, which has the worst clinical outcome, shows significantly lower stromal, immune, and ESTIMATE scores, and higher tumor purity compared to subtypes A, B, and D (Fig. 2A–D).The proportion of immune cell infiltration results suggest that in AGRG molecular subtype C, most immune cell proportions among the 23 types of immune cells are significantly reduced in the subtype with the worst clinical survival outcome, such as activated B cells, CD4+ T cells, CD8+ T cells, and immature B cells (Fig. 2E). Meanwhile, immune function score results also indicate that in AGRG molecular subtype C, most immune function indicators such as CCR, checkpoint, parainflammation, and Type I IFN response were significantly reduced (Fig. 2F). These results suggest that poor prognosis in OS is closely associated with low immune infiltration.

Analysis of immune microenvironment features in AGRG molecular subtypes. A–D Assessment of immune infiltration status in AGRG molecular subtypes based on the ESTIMATE algorithm. E Evaluation of immune cell infiltration characteristics in AGRG molecular subtypes. F Assessment of immune function scores in AGRG molecular subtypes
3.3 Construction of AGRG score and comprehensive analysis of prognostic prediction models
Using the AGRG gene signature and independently prognostic model parameters from multivariate Cox regression analysis, we constructed a novel AGRG scoring system to predict the clinical survival outcome of OS samples. In the AGRG molecular subtypes, AGRG subtype C, which has the worst clinical outcome, had significantly higher AGRG scores than other subtypes, suggesting that a high AGRG score may be associated with poor prognosis in OS (Fig. 3A). Furthermore, we performed a Sankey diagram to assess the potential associations among AGRG molecular subtypes, AGRG scores, and OS clinical survival outcomes (Fig. 3B). Based on the median value of AGRG scores, we divided OS samples into low and high AGRG score subgroups. Remarkably, based on the prognostic signature of AGRG, we found that the AGRG score subgroups could be significantly separated by a PCA model (Fig. 3C, D). The heatmap results suggest that in the high AGRG score subgroup, the expression of COL5A2 and CXCL6 was significantly increased, while FSTL1, NRP1, and TNFRSF21 were significantly decreased in the high AGRG score subgroup (Fig. 3E). The clinical prognosis of OS was significantly worse in the high AGRG score subgroup compared to the low AGRG score subgroup (Fig. 3F). These results indicate that the AGRG scoring system can accurately stratify OS risk, with a high AGRG score closely associated with poor prognosis.

Construction of AGRG score model based on AGRG prognostic signature. A Analysis of differences in AGRG scores among AGRG molecular subtypes. B Sankey diagram revealing potential relationships among AGRG molecular subtypes, AGRG scores, and clinical survival outcomes. C PCA analysis based on AGRG prognostic signature. D Stratification of AGRG score subgroups. E Expression profile analysis of AGRG prognostic signature in AGRG score subgroups. F Analysis of clinical survival outcomes in AGRG score subgroups of OS
3.4 Independent prognostic analysis of AGRG score
Time-dependent ROC curve analysis showed that the AGRG score had satisfactory predictive ability for 1-year, 3-year, and 5-year clinical survival prognosis of OS samples, with AUCs of 0.791, 0.769, and 0.751, respectively (Fig. 4A). We then evaluated the independent prognostic value of different clinical features and the AGRG score in predicting clinical survival outcomes. Univariate Cox analysis results suggested that metastases (HR = 5.839, p < 0.001) and AGRG score (HR = 1.152, p < 0.001) were significantly associated with poor prognosis (Fig. 4B). Multivariate Cox analysis results indicated that metastases (HR = 5.461, p < 0.001) and AGRG score (HR = 1.093, p = 0.049) as independent prognostic factors for OS could be used to evaluate clinical survival outcomes of OS (Fig. 4C). Based on clinical characteristics and AGRG score, we constructed a nomogram model to assess the survival probabilities of OS samples at 1 year, 3 years, and 5 years (Fig. 4D). Calibration curve results showed high consistency between the predicted and actual 1-year, 3-year, and 5-year survival probabilities of OS samples (Fig. 4E). Furthermore, the concordance index results indicated that the predictive ability of AGRG score for predicting clinical prognosis of OS was significantly better than other clinical indicators (Fig. 4F). These results indicate that predicting clinical survival outcomes of OS using AGRG score is reliable and can serve as an independent prognostic factor for predicting survival probabilities of OS.

Independent prognostic analysis based on AGRG score and clinical pathological features. A Time-dependent ROC curve analysis based on AGRG score. B, C Univariate and multivariate Cox analysis based on AGRG score and clinical features. D Construction of nomogram model. E Calibration curve. F Concordance index analysis of AGRG score and clinical features
3.5 Validation of independence of AGRG score
To validate the independence and stability of the AGRG score in predicting OS clinical survival outcomes, we used the “caret” script to randomly divide the OS samples into training and validation sets. Based on the prognostic signature of AGRG, we calculated AGRG scores of OS samples in two independent cohorts and divided OS samples into low and high AGRG score subgroups according to the median value. In the training set, clinical survival outcome results suggested that the prognosis status of OS in the low AGRG score subgroup was significantly better than that in the high AGRG score subgroup (p = 0.001). Similarly, in the validation set, we found that the prognosis status of OS samples in the AGRG score subgroup was significantly better than that in the high AGRG score subgroup, which was consistent with the results of the training set (Fig. 5A, B). These results indicate that the application of the AGRG scoring system to predict the clinical survival outcomes of OS is stable and reliable. In the training set, the AUCs for 1-, 3-, and 5-year ROC curves based on AGRG score were 0.774, 0.825, and 0.796, respectively. In the validation set, the AUCs were 0.824, 0.608, and 0.639, respectively (Fig. 5C, D). Importantly, in both the training and validation sets, PCA based on the prognostic signature of AGRG could significantly differentiate AGRG score subgroups (Fig. 5E, F). Based on these results, we can confirm the independence and stability of AGRG score system in predicting clinical survival outcomes of OS.

Assessment of independence and stability of AGRG score in predicting clinical survival prognosis of OS. A, B Clinical survival prognosis analysis of AGRG score subgroups in the training and validation sets. C, D Analysis of 1-, 3-, and 5-year ROC curves in two independent cohorts. E, F PCA analysis based on the prognostic signature of AGRG
3.6 Analysis of immune infiltration features and correlation in AGRG score subgroups
Using the ‘GSVA’ algorithm, we analyzed molecular regulatory mechanisms among AGRG score subgroups. Immune-related pathways, including toll-like receptor signaling, natural killer cell mediated cytotoxicity, B cell receptor signaling, and chemokine signaling, were significantly downregulated in the high AGRG score subgroup (Fig. 6A). We used correlation analysis to explore the relationship between AGRG scores and immune infiltration features. The results indicated a significant negative correlation between AGRG score and most immune cells, suggesting that low immune status may be an important factor contributing to worse prognosis in the high AGRG score subgroup (Fig. 6B). Quantitative results of immune infiltration features showed that in the high AGRG score subgroup, the proportions of most immune infiltrating cells were significantly decreased, such as activated B cell, CD8+ T cell, activated dendritic cell, and CD56bright natural killer cell (Fig. 6C). Immune-related functional results demonstrated a significant decrease in immune regulation functions in the high AGRG score subgroup, involving cytolytic_activity, inflammation − promoting, MHC_class_I, and parainflammation (Fig. 6D). Quantitative results of immune status showed that compared to the low AGRG score subgroup, the stromal score, immune score, ESTIMATE score was significantly decreased, while tumor purity was significantly increased in the high AGRG score subgroup (Fig. 6E). Correlation analysis revealed that NRP1 and TNFRSF21 were positively correlated with most immune cells, whereas COL5A2 was negatively correlated (Fig. 6F). In summary, significant differences in immune infiltration were observed among AGRG score subgroups. The decreased immune infiltration in the high AGRG score subgroup may contribute to its poor clinical prognosis.

Analysis of immune infiltration features in AGRG score subgroups. A Analysis of potential regulatory mechanisms among AGRG score subgroups. B Correlation analysis between AGRG score and immune infiltration features. C Quantitative analysis of immune infiltrating cells in AGRG scores subgroups. D Prediction of immune functions in AGRG subgroups. E Assessment of immune infiltration status based on ESTIMATE. F Correlation analysis between AGRG prognostic signature and immune infiltrating cells
3.7 Analysis of drug sensitivity in AGRG score subgroups
To improve precision treatment for OS, we used the GDSC database and gene expression profiles from AGRG score subgroups to predict potential therapeutic compounds with anticancer effects. As shown in Fig. 7, the drug sensitivity analysis results indicated that in the low AGRG score subgroup A, the IC50 values of Pyrimethamine, KIN001-135, Imatinib, GW843682X, BMS-509,744, BI-2536, and AKT inhibitor VIII were significantly higher than those in the high AGRG score subgroup, while the IC50 values of TGX221 and Shikonin were significantly lower than those in the high AGRG score subgroup. These results suggest that AGRG score-based subgroups reflect drug sensitivity, offering a new approach for personalized treatment of OS.

Prediction of drug response in AGRG score subgroups. Prediction of IC50 value for A TGX221, B Shikonin, C Pyrimethamine, D KIN001-135, E Imatinib, F GW843682X, G BMS-509,744, H BI-2536 and I AKT inhibitor VIII
3.7.1 CXCL6 promotes migration and invasion of OS cells
Due to the highest correlation coefficient, we selected CXCL6 among the prognostic ARAGs to verify the results of the bioinformatics analysis. The qRT-PCR and WB results indicated that the mRNA and protein expression levels of CXCL6 were significantly elevated in OS cell lines compared to normal cell lines, which is consistent with our previous bioinformatics data (Fig. 8A–C). To further clarify the role of CXCL6 in OS, we transiently transfected Mg63 cells to interfere with the expression of CXCL6. As shown in Fig. 8C–F, the interference of CXCL6 expression significantly reduced the levels of metastasis-related proteins MMP9 and MMP2 in Mg63 cells compared to cells transfected with the empty vector. Migration and invasion assays demonstrated that interfering with CXCL6 expression significantly inhibited the migration and invasion abilities of Mg63 cells compared to cells transfected with the empty vector (Fig. 8G). Based on these results, we highlight the potential role of CXCL6 in OS and partially validate the reliability of the bioinformatics analysis results, providing a new target for future OS treatment.

CXCL6 promotes migration and invasion of OS cells. A mRNA expression level of CXCL6 in control cell line and Mg63 OS cell line. B Western blot analysis of CXCL6 in Mg63 OS cells and control cells. The statistical results of western blot (n = 3). C Verification of CXCL6 transfection efficiency and expression levels of metastasis-related proteins MMP9 and MMP2. The statistical results of western blot (n = 3). G Cell migration and invasion assay of Mg63 cells by Transwell after transient transfection. Control (left), siCXCL6 (right) (x 200). Significant differences are indicated as: *p < 0.05, **p < 0.01, ***p < 0.001
4 Discussion
In this study, our research on AGRGs and OS suggests the important role of angiogenesis in OS from a new perspective. Many effective clinical treatments for OS target angiogenesis through factors such as VEGF, PDGF, fibroblast growth factor (FGF), integrins, cyclooxygenase (COX-2), metalloproteinases (MMP-2, MMP-9), and HIF-1 [29,30,31]. However, the prediction and evaluation of the therapeutic effect of antiangiogenic drugs on OS depends on further exploration of the participants and mechanisms of action [7].
Among the screened angiogenesis related genes, our results showed that COL5A2 and CXCL6 were significantly increased in the high AGRG score subgroup and were associated with poor prognosis. It has been reported that COL5A2 is highly expressed in OS cells [32]. In addition, COL5A2 expression was higher in patients with metastatic osteosarcoma, suggesting that high COL5A2 expression was associated with a poorer prognosis [33]. TGF-β and Wnt/β-catenin signaling promote OS progression and influence prognosis, with COL5A2 potentially affecting these pathways [34, 35]. CXCL6 overexpression promotes tumor growth and metastasis, and targeting CXCL6 shows potential clinical value, consistent with our findings [36].
We found that FSTL1 levels were lower in the high AGRG score subgroup. Evidence suggests FSTL1 weakens NK cell immunity, which is linked to poorer prognosis in OS patients [37]. In addition, the potential interaction of FSTL1 with TGF-β and NF-κB pathways in OS may also influence the progression and therapeutic efficacy of OS [38, 39]. NRP1 is a co-receptor of vascular endothelial growth factor (VEGF). Its overexpression in OS can lead to tumor progression and poor prognosis [40]. The regulation of TGF-β and wnt/β-catenin signaling pathways may also be one of the specific mechanisms by which NRP1 affects the prognosis of OS [41]. TNFRSF21 expression was down-regulated in the OS cell line [42]. Its overexpression can lead to apoptosis of the OS cell line [43]. However, it has not been reported how TNFRSF21 affects OS cells. The observed gene functions align with reported evidence, supporting the reliability of our results.
Our results indicate that macrophages are the immune cells most associated with AGRG scores. Lower levels of macrophages were linked to better prognosis in OS patients. In a tumor microenvironment (TME) characterized by inflammation, local hypoxia, and high lactic acid levels, macrophages differentiate into tumor-associated macrophages (TAMs) under the influence of cytokines such as IL-34, CSF1, VEGF, and chemokines like CCL5 and CCL2 [44]. TAMs secrete various cytokines, chemokines, and enzymes, and further induce immunosuppression through activation of regulatory T cells (Tregs), resulting in loss of T cell function [45]. The role of TAMs in OS has been emphasized, with high TAM infiltration generally correlating with poorer overall survival and increased metastasis [46, 47]. This correlates with our observation that lower macrophage levels were associated with better outcomes in patients with OS. There are several ongoing experimental protocols to reduce macrophage recruitment and target repolarization of activated TAMs to anti-tumor phenotypes with some success [48, 49]. Although multiple evidence, including our results, supports the role of macrophages in OS, further exploration is needed regarding the role of macrophages in the pathogenesis of OS and the development of effective targeted therapies.
The correlation between AGRG risk stratification and drug sensitivity highlights the importance of personalized therapy based on gene-related risk models. Multiple genes in OS have been shown to be associated with chemotherapy resistance signal transduction pathways [50, 51]. Specific genes like ERBB4 and STAT3 can partially resist apoptosis induced by chemotherapeutic drugs [52, 53]. Multidrug resistance (MDR) is also influenced by genes regulating the p-glycoprotein (p-GP) pump mechanism [54]. The prognostic factors we identified, such as NRP1 and COL5A2, have been associated with MDR in other diseases [55, 56]. In OS, although there is preliminary evidence that these genes are associated with chemotherapy sensitivities, the specific mechanism that causes sensitivity differences needs to be further explored.
While the TNM staging system is a gold standard for predicting tumor prognosis, it does not account for individualization and tumor location [57, 58]. Therefore, the prediction of OS risk stratification still has room for improvement. The current view is that personalized, smart medical services can provide a new perspective on prognosis and survival of malignant diseases, including OS [59]. The risk stratification system developed in this study with a p < 0.001 result has clinical potential to further improve OS risk prediction to support prospective treatment decisions. At the same time, it also provides the basis and potential intervention target for further comprehensive analysis of multi-risk models.
In summary, our study performed molecular typing and prognostic risk stratification of OS based on AGRGs, identifying five key risk markers. However, public database research may be biased by race and region, and treatment data in databases were limited. Additionally, the molecular pathway of screened targets affecting OS biology was not further elucidated. Future research should focus on validating these findings and exploring the role of angiogenesis in OS.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Esperanca-Martins M, Fernandes I, Soares do Brito J, Macedo D, Vasques H, Serafim T, Costa L, Dias S. Sarcoma metabolomics: current Horizons and Future perspectives. Cells 2021, 10(6).
Saerens M, Brusselaers N, Rottey S, Decruyenaere A, Creytens D, Lapeire L. Immune checkpoint inhibitors in treatment of soft-tissue sarcoma: a systematic review and meta-analysis. Eur J Cancer. 2021;152:165–82.
Grunewald TG, Alonso M, Avnet S, Banito A, Burdach S, Cidre-Aranaz F, Di Pompo G, Distel M, Dorado-Garcia H, Garcia-Castro J, et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol Med. 2020;12(11):e11131.
Anderson TS, Wooster AL, Piersall SL, Okpalanwaka IF, Lowe DB. Disrupting cancer angiogenesis and immune checkpoint networks for improved tumor immunity. Semin Cancer Biol. 2022;86(Pt 3):981–96.
Li YS, Liu Q, Tian J, He HB, Luo W. Angiogenesis process in Osteosarcoma: an updated perspective of pathophysiology and therapeutics. Am J Med Sci. 2019;357(4):280–8.
Liu Y, Huang N, Liao S, Rothzerg E, Yao F, Li Y, Wood D, Xu J. Current research progress in targeted anti-angiogenesis therapy for osteosarcoma. Cell Prolif. 2021;54(9):e13102.
Sheng G, Gao Y, Yang Y, Wu H. Osteosarcoma and metastasis. Front Oncol. 2021;11:780264.
Giner F, Lopez-Guerrero JA, Machado I, Garcia-Casado Z, Peydro-Olaya A, Llombart-Bosch A. The early stages of tumor angiogenesis in human osteosarcoma: a nude mice xenotransplant model. Virchows Arch. 2015;467(2):193–201.
Broadhead ML, Akiyama T, Choong PF, Dass CR. The pathophysiological role of PEDF in bone diseases. Curr Mol Med. 2010;10(3):296–301.
Zhu S, Qiu H, Bennett S, Kuek V, Rosen V, Xu H, Xu J. Chondromodulin-1 in health, osteoarthritis, cancer, and heart disease. Cell Mol Life Sci. 2019;76(22):4493–502.
Alfranca A, Martinez-Cruzado L, Tornin J, Abarrategi A, Amaral T, de Alava E, Menendez P, Garcia-Castro J, Rodriguez R. Bone microenvironment signals in osteosarcoma development. Cell Mol Life Sci. 2015;72(16):3097–113.
Chim SM, Tickner J, Chow ST, Kuek V, Guo B, Zhang G, Rosen V, Erber W, Xu J. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 2013;24(3):297–310.
Tsai HC, Tzeng HE, Huang CY, Huang YL, Tsai CH, Wang SW, Wang PC, Chang AC, Fong YC, Tang CH. WISP-1 positively regulates angiogenesis by controlling VEGF-A expression in human osteosarcoma. Cell Death Dis. 2017;8(4):e2750.
Wang SW, Liu SC, Sun HL, Huang TY, Chan CH, Yang CY, Yeh HI, Huang YL, Chou WY, Lin YM, et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis. 2015;36(1):104–14.
Liao Z, Li M, Wen G, Wang K, Yao D, Chen E, Liang Y, Xing T, Su K, Liang C, et al. Comprehensive analysis of angiogenesis pattern and related immune landscape for individual treatment in osteosarcoma. NPJ Precis Oncol. 2023;7(1):62.
Kaya M, Wada T, Nagoya S, Yamashita T. Prevention of postoperative progression of pulmonary metastases in osteosarcoma by antiangiogenic therapy using endostatin. J Orthop Sci. 2007;12(6):562–7.
Xing P, Zhang J, Yan Z, Zhao G, Li X, Wang G, Yang Y, Zhao J, Xing R, Teng S, et al. Recombined humanized endostatin (Endostar) combined with chemotherapy for advanced bone and soft tissue sarcomas in stage IV. Oncotarget. 2017;8(22):36716–27.
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27(45):5904–12.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Ying H, Li ZQ, Li MP, Liu WC. Metabolism and senescence in the immune microenvironment of osteosarcoma: focus on new therapeutic strategies. Front Endocrinol (Lausanne). 2023;14:1217669.
Corre I, Verrecchia F, Crenn V, Redini F, Trichet V. The osteosarcoma microenvironment: a complex but targetable ecosystem. Cells. 2020;9(4):976.
Locy H, de Mey S, de Mey W, De Ridder M, Thielemans K, Maenhout SK. Immunomodulation of the tumor microenvironment: turn foe into friend. Front Immunol. 2018;9:2909.
Kim HJ, Ji YR, Lee YM. Crosstalk between angiogenesis and immune regulation in the tumor microenvironment. Arch Pharm Res. 2022;45(6):401–16.
Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–22.
Zhang Q, Liu J, Yao D, Shi JX, Liu YJ, Wei YG, Guo S. Comprehensive analysis to identify Rh Family C glycoprotein (RHCG) as the causative gene for psoriasis and search for alternative treatment modalities. Drug Des Devel Ther. 2023;17:2593–611.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40.
Egners A, Rezaei M, Kuzmanov A, Poitz DM, Streichert D, Muller-Reichert T, Wielockx B, Breier G. PHD3 acts as tumor suppressor in mouse osteosarcoma and influences tumor vascularization via PDGF-C signaling. Cancers. 2018;10(12):496.
Kuzmanov A, Wielockx B, Rezaei M, Kettelhake A, Breier G. Overexpression of factor inhibiting HIF-1 enhances vessel maturation and tumor growth via platelet-derived growth factor-C. Int J Cancer. 2012;131(5):E603–613.
Su Y, Gao L, Teng L, Wang Y, Cui J, Peng S, Fu S. Id1 enhances human ovarian cancer endothelial progenitor cell angiogenesis via PI3K/Akt and NF-kappaB/MMP-2 signaling pathways. J Transl Med. 2013;11:132.
Han YL, Luo D, Habaxi K, Tayierjiang J, Zhao W, Wang W, Aikebaier W, Wang L. COL5A2 inhibits the TGF-beta and Wnt/beta-catenin signaling pathways to inhibit the Invasion and metastasis of osteosarcoma. Front Oncol. 2022;12:813809.
Chen H, Liu W, Zhong L, Liao D, Zhang R, Kang T, Wu Y. NKX2-2 suppresses osteosarcoma metastasis and proliferation by downregulating multiple target genes. J Cancer. 2018;9(17):3067–77.
Zhao H, Peng C, Lu X, Guo M, Yang T, Zhou J, Hai Y. PDCD5 inhibits osteosarcoma cell metastasis via targeting TGF-beta1/Smad signaling pathway and is associated with good prognosis. Am J Transl Res. 2019;11(2):1116–28.
Matsuoka K, Bakiri L, Wolff LI, Linder M, Mikels-Vigdal A, Patino-Garcia A, Lecanda F, Hartmann C, Sibilia M, Wagner EF. Wnt signaling and Loxl2 promote aggressive osteosarcoma. Cell Res. 2020;30(10):885–901.
Liu G, An L, Zhang H, Du P, Sheng Y. Activation of CXCL6/CXCR1/2 Axis promotes the growth and Metastasis of Osteosarcoma Cells in vitro and in vivo. Front Pharmacol. 2019;10:307.
Ogiwara Y, Nakagawa M, Nakatani F, Uemura Y, Zhang R, Kudo-Saito C. Blocking FSTL1 boosts NK immunity in treatment of osteosarcoma. Cancer Lett. 2022;537:215690.
Zhang G, Jin C, Zhu Y, Fu F, Wang G, Li S. Sulforaphene inhibits the progression of osteosarcoma via regulating FSTL1/NF-kappaB pathway. Life Sci. 2020;263:118485.
Tanaka M, Murakami K, Ozaki S, Imura Y, Tong XP, Watanabe T, Sawaki T, Kawanami T, Kawabata D, Fujii T, et al. DIP2 disco-interacting protein 2 homolog A (Drosophila) is a candidate receptor for follistatin-related protein/follistatin-like 1–analysis of their binding with TGF-beta superfamily proteins. FEBS J. 2010;277(20):4278–89.
Zhu H, Cai H, Tang M, Tang J. Neuropilin-1 is overexpressed in osteosarcoma and contributes to tumor progression and poor prognosis. Clin Transl Oncol. 2014;16(8):732–8.
Wei QF, Yao JS, Yang YT. MicroRNA-1247 inhibits the viability and metastasis of osteosarcoma cells via targeting NRP1 and mediating Wnt/beta-catenin pathway. Eur Rev Med Pharmacol Sci. 2019;23(17):7266–74.
Li X, Sun Z, Ma J, Yang M, Cao H, Jiao G. Identification of TNFRSF21 as an inhibitory factor of osteosarcoma based on a necroptosis-related prognostic gene signature and molecular experiments. Cancer Cell Int. 2024;24(1):14.
Choi YJ, Lee YS, Lee HW, Shim DM, Seo SW. Silencing of translation initiation factor eIF3b promotes apoptosis in osteosarcoma cells. Bone Joint Res. 2017;6(3):186–93.
Fujiwara T, Healey J, Ogura K, Yoshida A, Kondo H, Hata T, Kure M, Tazawa H, Nakata E, Kunisada T, et al. Role of tumor-associated macrophages in sarcomas. Cancers. 2021;13(5):1086.
Sawa-Wejksza K, Kandefer-Szerszen M. Tumor-Associated macrophages as Target for Antitumor Therapy. Arch Immunol Ther Exp (Warsz). 2018;66(2):97–111.
Iseulys R, Anne GB, Corinne B, Gonzague DBP, Marie K, Jean-Yves B, Aurelie D. The immune landscape of chondrosarcoma reveals an immunosuppressive environment in the dedifferentiated subtypes and exposes CSFR1 + macrophages as a promising therapeutic target. J Bone Oncol. 2020;20:100271.
Cersosimo F, Lonardi S, Bernardini G, Telfer B, Mandelli GE, Santucci A, Vermi W, Giurisato E. Tumor-associated macrophages in osteosarcoma: from mechanisms to therapy. Int J Mol Sci. 2020;21(15):5207.
Koo J, Hayashi M, Verneris MR, Lee-Sherick AB. Targeting Tumor-Associated macrophages in the Pediatric Sarcoma Tumor Microenvironment. Front Oncol. 2020;10:581107.
Zajac AE, Czarnecka AM, Rutkowski P. The role of macrophages in sarcoma tumor microenvironment and treatment. Cancers. 2023;15(21):5294.
Mintz MB, Sowers R, Brown KM, Hilmer SC, Mazza B, Huvos AG, Meyers PA, Lafleur B, McDonough WS, Henry MM, et al. An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Res. 2005;65(5):1748–54.
Chiappetta C, Mancini M, Lessi F, Aretini P, De Gregorio V, Puggioni C, Carletti R, Petrozza V, Civita P, Franceschi S, et al. Whole-exome analysis in osteosarcoma to identify a personalized therapy. Oncotarget. 2017;8(46):80416–28.
Salas S, Jiguet-Jiglaire C, Campion L, Bartoli C, Frassineti F, Deville JL, De Maues A, Forest F, Jezequel P, Gentet JC, et al. Correlation between ERK1 and STAT3 expression and chemoresistance in patients with conventional osteosarcoma. BMC Cancer. 2014;14:606.
Huang Z, Wang SL, Chen H, Shen RK, Li XD, Huang QS, Wu CY, Weng DF, Lin JH. Clinicopathological and prognostic values of ErbB receptor family amplification in primary osteosarcoma. Scand J Clin Lab Invest. 2019;79(8):601–12.
Mthethwa PG, Marais LC, Ramsuran V, Aldous CM. A systematic review of the heterogenous gene expression patterns associated with multidrug chemoresistance in conventional osteosarcoma. Genes. 2023;14(4):832.
Yang Y, Wu J. Significance of the differential peptidome in multidrug-resistant tuberculosis. Biomed Res Int. 2019;2019:5653424.
Januchowski R, Zawierucha P, Rucinski M, Zabel M. Microarray-based detection and expression analysis of extracellular matrix proteins in drug–resistant ovarian cancer cell lines. Oncol Rep. 2014;32(5):1981–90.
Balachandran VP, Gonen M, Smith JJ, DeMatteo RP. Nomograms in oncology: more than meets the eye. Lancet Oncol. 2015;16(4):e173–180.
Kim MS, Lee SY, Lee TR, Cho WH, Song WS, Koh JS, Lee JA, Yoo JY, Jeon DG. Prognostic nomogram for predicting the 5-year probability of developing metastasis after neo-adjuvant chemotherapy and definitive surgery for AJCC stage II extremity osteosarcoma. Ann Oncol. 2009;20(5):955–60.
Ingley KM, Maleddu A, Grange FL, Gerrand C, Bleyer A, Yasmin E, Whelan J, Strauss SJ. Current approaches to management of bone sarcoma in adolescent and young adult patients. Pediatr Blood Cancer. 2022;69(2):e29442.
Acknowledgements
The bioinformatics analysis and basic experiment of this study were thanks to Dr Xiaolin Wu’s guidance and assistance.
Funding
This study was supported by the Natural Science Foundation of Shandong Province (ZR2022MH218); Youth Research Fund of Affiliated Hospital of Qingdao University in 2021(QDFYQN202101012); The Affiliated Hospital of Qingdao University Clinical Medicine + X Research Project (QDFY + X2023141).
Author information
Authors and Affiliations
Contributions
S.R and Z.W conceived and designed the study. R.P performed the expression verification in cell lines. All authors participated in the acquisition, analysis, interpretation of data, drafting of the manuscript, and approval of the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The relevant data are public and freely available; there are no human or animal experiments in this study, and therefore, ethical approval is not required.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, S., Pan, R. & Wang, Z. Development and experimental verification of novel angiogenesis related prognostic model and immune infiltration characterization in osteosarcoma. Discov Onc 15, 411 (2024). https://doi.org/10.1007/s12672-024-01292-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01292-7