
Abstract
Pancreatic cancer (PC) is one of the deadliest cancers worldwide with low survival rates and poor outcomes. The treatment landscape for PC is fraught with obstacles, including drug resistance, lack of effective targeted therapies and the immunosuppressive tumor microenvironment (TME). The resistance of PC to existing immunotherapies highlights the need for innovative approaches, with the TME emerging as a promising therapeutic target. The recent advancements in understanding the role of macrophages, this context highlight their significant impact on tumor development and progression. There are two important types of macrophages: M1 and M2, which play critical roles in the TME. Therapeutics strategies including, depletion of tumor-associated macrophages (TAMs), reprogramming TAMs to promote anti-tumor activity, and targeting macrophage recruitment can lead to promising outcomes. Targeting macrophage-related pathways may offer novel strategies for modulating immune responses, inhibiting angiogenesis, and overcoming resistance to chemotherapy in PC treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
With only 12% of patients living for 5 years following diagnosis, pancreatic ductal adenocarcinoma (PDAC), often commonly referred to as PC, has an extremely poor prognosis when compared to other cancers [1]. PDAC accounts for over 90% of PC occurrences [2]. In 2018, the global age-standardized incidence rate of PC was 4.8 per 100,000 [3]. While the occurrence of PC is increasing in most regions worldwide, high-income countries have the highest levels of incidence and mortality [4]. PC is associated with a significant degree of resistance to systemic therapy strategies [5]. The close proximity of numerous organs-at-risk and wide spatial variations make radiation therapy for PC very difficult [6]. Additionally, the PC is extremely resistant to current immunotherapies due to its immunosuppressive TME, which restricts the effectiveness of T cell infiltration [7]. Given its crucial involvement in both the cancer progression and treatment resistance, the TME has been recognized as a viable target for therapeutic intervention in a variety of malignancies [8]. TME consists of various cell types, including immune cells such as B and T lymphocytes, TAMs, myeloid-derived suppressor cells (MDSCs), as well as cancer-associated fibroblasts and pericytes [9].
PDAC is characterized by the desmoplastic stroma, an immunosuppressive microenvironment that forms a barrier to protect tumor cells from immunologic elimination and prevents chemotherapeutic penetration [10]. The stroma, mainly composed of proliferating fibroblasts, deposits collagen, fibronectin, and other extracellular matrix components. It also produces cytokines and chemokines to crosstalk with lesion cells and immune cells [11, 12].
PC suppresses cytotoxic T lymphocytes (CTLs) through immunosuppressive interaction with stromal cells in the TME. Combining immunotherapeutic approaches is suggested for infiltrating active T lymphocytes or rescuing exhausted CTLs. Maintaining activated CTLs in the TME is crucial for tumor immunotherapy [13]. The TME is characterized by hypoxia, acidity, increased permeability, and a high number of growth factors and hydrolases and numerous growth factors, leading to insensitivity to apoptosis, resistance to growth inhibition and immune escape [14]. The immunosuppressive microenvironment in PC is essential for malignant tumor development and is an obstacle for immunotherapy [15]. The cancer lacks the expression of immunogenic antigens and their major histocompatibility antigens (MHC) class I molecules, leading to decreased activity of effector T cells. In addition, the functions of effector T cells such as natural killer (NK) cells, dendritic cells (DCs), and macrophages are greatly reduced, while suppressive immune cells such regulatory T-cells, MDSCs, and M2-TAMs accumulate in the TME [16, 17]. One of the main agents in this regard that promotes carcinogenesis, tumor growth, and progression, as well as treatment resistance, are macrophages [18]. Pre-clinical PDAC mouse models have clearly shown the significance of these cells in promoting angiogenesis, matrix remodeling, immunosuppression, tumor cell invasion, and treatment resistance [19, 20].
Understanding the function of macrophages within the PC TME is essential for developing innovative approaches for treatment and management of this diseases. In this review, we aimed to outline the role of macrophages in PC TME and investigate their potential as therapeutic targets.
2 Macrophages: the double agents
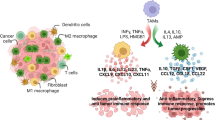
As crucial elements of the immune system, macrophages are essential for the modulation of immunological responses, regulation of inflammatory processes, tissue remodeling, and surveillance of immune activities [21]. These cells participate in both the innate and adaptive immune responses, among their many other roles in the immune system. Macrophages become activated in response to cytokines and other environmental stimuli, such as microbial compounds. M1 and M2 are the two different subtypes of activated macrophages (Fig. 1). The M1 type represents the phenotype that is traditionally active, whereas the M2 type represents the phenotype that is alternatively activated [22]. The M1 macrophages play a crucial role in immunity against intracellular infections and malignancies. Through the creation of tumoricidal substances like interleukin (IL)-12, IL-6, and tumor necrosis factor-α (TNF-α), they eliminate harmful tissue reactions [23]. Conversely, M2 macrophages suppress the immune system and are often activated by IL-4 and IL-13. They release cytokines including transforming growth factor-β (TGF-β) and IL-10 which trigger a T helper 2 (Th2) immune response [24]. Larger tumors, early hepatic recurrence, local recurrence, and a shorter survival time for PC patients are all associated with higher concentrations of M2 macrophages [25].

Subtypes of activated macrophages and their receptors, signaling pathways, related cytokines, and functions
Macrophages have notable plasticity, enabling them to effectively react to diverse stimuli and experience significant physiological alterations in reaction to different stimuli from the environment [26]. The plasticity that macrophages demonstrate enables them to transition between several activation states, among them are the anti-inflammatory M2 phenotype and the pro-inflammatory M1 phenotype, in reaction to distinct signals originating from their surrounding environment [27].
The signals encompass a range of factors including cytokines, microbial compounds, and various environmental stimuli [28]. Polarization is notably impacted by diverse signaling pathways.
Notch signaling system assumes crucial functions in the determination of macrophage polarization, namely in distinguishing between M1 and M2 phenotypes. These paths include PI3K-Akt, WNT, MAPK, NF-κB, STAT, IRF, JAK, and hedgehog signaling pathway [29, 30].
2.1 Angiogenesis
An angiogenic switch allows cancer cells to hijack blood supplies to enhance their progress and persist. In this key process which was recruited and reprogrammed by tumor cells, the role of monocyte-derived macrophages, as a major source of angiogenic factors, is undeniable [31]. In TME, TAMs reprogrammed by tumor cells mediated by exudation of some factors such as CC chemokine ligand 2, TGF-β, and VEGF-A which can facilitate tumor angiogenesis. It was proven that these factors play the main role in the switching of the macrophage phenotype from the M1 into the M2 phenotype, as the M2 phenotype has a high capacity to enhance tumor angiogenesis [32, 33]. Macrophages regulate the presence of VEGF-A in tumors by altering its availability through the action of matrix metalloproteinases (MMPs) [34]. Moreover, an increase in VEGF in contrast to a decrease in colony-stimulating factor 1 (CSF-1) activity results in the recruitment of macrophages and the continuance of vascularization and angiogenesis, all of which work toward an accelerated advancement of cancer [35]. Esposito et al. [36] reported the expression of proangiogenic and prolymphangiogenic molecules including VEGF-A, VEGF-C, and bFGF in patients with PC. They found significantly more mast cells and macrophages in these patients than in normal individuals, which had a direct correlation with intratumorous microvessel density. Lewis et al. [37] reported that TAMs upregulate VEGF expression in hypoxic and avascular regions of the beast tumor, as macrophages accumulation was found in higher numbers in poorly vascularized than highly vascularized areas. VEGF seems to exert a chemotactic action on macrophages to enhance their migration into avascular tumor regions. Furthermore, compared to VEGF-negative locations, TAMs are more common in VEGF-positive, weakly vascularized regions, indicating a potential function for VEGF in macrophage chemoattraction. In addition, in hypoxic conditions, macrophages upregulate MMPs and hypoxia-inducible factor (HIF)-regulated genes, which enable cancer cells to survive in hostile environments and result in the revascularization of tumor ischemia sites [38].
2.2 Immunosuppression
MDSCs are a kind of immune cell that has gained significant attention in recent years due to its immunosuppressive properties. They are closely linked to macrophages. MDSCs are a diverse collection of cells with a myeloid origin that gather in pathological circumstances, such as cancer. From a morphological perspective, these populations are composed of immature myeloid cells, granulocytes, and monocytes. They may be distinguished by their capacity to prevent responses from cytotoxic T cells. The granulocyte marker Gr1 and the myeloid cell marker CD11b are co-expressed by MDSCs in mice. Given the traditional definition of neutrophils, the presence of these markers suggests that MDSCs are not mononuclear phagocytes. The examination of cell surface markers has been further supported by gene expression analysis, which demonstrates significant differences between MDSCs and TAMs [39, 40].
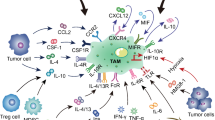
As mentioned, monocytes in tumors change into polarized or M2 macrophages in response to exposure to tumor-derived anti-inflammatory chemicals. The antigen presentation capacity of these macrophages is deficient, and they secrete substances that suppress the activity and proliferation of T-cells [33]. They possess superior adaptations for scavenging detritus, facilitating angiogenesis, and healing and rebuilding injured or damaged tissues. This is in stark contrast to the profile of traditionally activated type I, or M1 macrophages, are immune effector cells with a high degree of effectiveness capable of eliminating pathogens and tumor cells, presenting antigens, and producing significant amounts of immunostimulatory cytokines [41]. Comparable with regard to their molecular and functional profile, TAMs are distinguished by decreased TNF levels, increased constitutive expression of IL-6 and IL-1, and decreased expression of differentiation-associated macrophage markers such as carboxy peptidase M and CD51 [42, 43]. TAMs also release chemokines (e.g. C–C Motif chemokine ligand-7 (CCL7) and CCL2) and anti-inflammatory/immunosuppressive cytokines (e.g. TGF-β, PGE2), which encourage T-cell exhaustion and create a self-replicating tumor-permissive environment [29, 44]. Furthermore, NK cell proliferation is hindered by IL-10 produced from macrophages, and NK cell cytolytic activity is inhibited by TGF-β signaling in TAMs in a contact-dependent way [45, 46].
Additionally, TAMs have the ability to increase the production of ligands that attach to inhibitory receptors on the surface of T cells to prevent T-cell activation, proliferation, and effector activities [47]. These consist of CD80/CD86 and PDL1/PDL2, which attach to the surface of CD8 T-cells in response to CTLA4 and PD1, respectively. Dectin1 is one of the other ligands; in mouse and human PDAC, macrophages express it at a high level. Dectin1 binds to Galectin-9 on the surface of cancerous and invading immune cells, suppressing adaptive immunological responses and training tolerogenic macrophages [48].
2.3 Metastasis
Macrophages have the capacity to create an autocrine loop that stimulates cancer cell migration, which in turn encourages invasion and eventual metastasis from the initial tumor location. CSF-1, which is produced by cancer cells, stimulates macrophages to create EGF. This autocrine signaling process leads to comigration of cancer cells and macrophages toward tumor blood vessels, where VEGF-A derived from macrophages facilitate the infiltration of cancer cells into the blood vessels [49, 50]. Tissue macrophage hemostasis is aided by a unique cytokine known as IL-34. The colony-stimulating factor 1 receptor (CSF-1R) can bind to it and initiate a signaling pathway that leads to macrophage proliferation, survival, and differentiation [51]. It has been demonstrated that this function can be performed by it as an alternative ligand for CSF-1R. One important regulator of macrophage populations is CSF-1 which also functions as a chemotactic factor for macrophages [52]. Myeloid progenitor cells can differentiate into monocytes and macrophages with the help of CSF-1, which also regulates the growth, survival, and migration of macrophages [53].
Macrophages facilitate the movement of stalled disseminated tumor cells (DTCs) from capillary networks to the secondary location. Dysplastic tumor cells secrete CCL2, which attracts bloodstream-derived inflammatory monocytes to the metastatic location [54]. In this context, inflammatory monocytes release VEGF-A to facilitate the extravasation of DTCs by enhancing vascular permeability. Furthermore, CCL2 stimulates the production of CCL3 by metastasis-associated macrophages (MAMs), which in turn enhances the ability of MAMs to remain at the site of metastasis. This enhances the direct interaction between macrophages and cancer cells by means of VCAM1-α4 integrin mediated signaling, hence facilitating the retention of cancer cells at the metastatic location [55]. Moreover, DTCs can draw platelets and start the coagulation cascade by the production of tissue factor, which activates thrombin and deposits fibrin, finally causing blood clots to form. Macrophages are attracted to the clots that form around cancer cells. They support the survival of these cancer cells by a process that does not involve NK cells [56]. This could be explained by the direct relationship that exists between MAMs and DTCs, since studies have shown that macrophages enhance the survival of DTCs by establishing physical contact between the cells. In this context, pro-survival signaling in cancer cells is activated via the PI3K/Akt signaling pathway by DTCs expressing higher levels of VCAM-1 interacting with α4-integrins on MAMs [57].
MAMs are essential in promoting the spread of PC cells by facilitating the development of a metastatic environment in the liver. Upon the entrance of DTCs, there is an accumulation of monocytes with inflammatory characteristics in the liver, which thereafter differentiate into monocyte-derived macrophages. Local hematopoietic stem and progenitor cells are stimulated to become αSMA+ myofibroblasts by MAMs through the release of granulin. These activated myofibroblasts secrete periostin, an extracellular matrix substance, which augments the colony forming capabilities of PC cells. In the bone marrow region, the absence of granulin eliminates the accumulation of periostin and extracellular matrix in the metastatic growths, leading to decreased proliferation of cancer cells that have already spread to other body regions [58].
2.4 Matrix remodeling
Recent evidence suggests that macrophages can influence the behavior of cancer stem cells (CSCs) and/or tumor starting cells. TAMs have been demonstrated to augment the properties of CSCs in lung and colon cancer, such as boosting their tumorigenicity and resistance to chemotherapy [48]. The milk-fat globule-epidermal growth factor-8 (MFG-E8) produced from TAMs was found to be an essential CSCs activity regulator. This factor triggers the activation of the sonic hedgehog and STAT3 pathways in CSCs. Suppression of the recruitment of macrophages to the PC tumor location, leads to a reduction in PC cells that express high amounts of the CSCs marker aldehyde dehydrogenase. It is noteworthy that in this paradigm, the tumor-initiating characteristics of murine PC cell lines were intensified by conditioned media from macrophages educated by tumors [59].
2.5 Resistance to chemotherapy
In PC cells, macrophages promote the synthesis of cytidine deaminase, the primary enzyme that is responsible for metabolizing the chemotherapeutic agent gemcitabine. Chemotherapy increases the tumor cells' chance of survival when orthotopically implanted pancreatic tumors are treated. This effect can be mitigated by reducing the number of macrophages or inhibiting CCR2+ inflammatory monocytes [60]. Insulin-like growth factor1 (IGF-1) and IGF-2 are another way that TAMs encourage chemoresistance in PC. In an orthotopic PC model, gemcitabine with antibody-mediated neutralization of IGF enhances the response to chemotherapy, reducing tumor growth and increasing cancer cell death [61]. Cathepsin expression in macrophages can also be directly induced by chemotherapeutic agents. The amount of cathepsins produced by macrophages could be adequate to prevent tumor cells from dying, and blocking cathepsins made cancer cells more susceptible to various chemotherapy agents [62].
3 Therapeutic strategies targeting macrophages
Recently, there has been a focus on developing various strategies to counteract detrimental impacts of TAMs, considering their role in promoting tumor growth.
3.1 Depletion of TAMs
Reducing the total number of TAMs within a tumor can be accomplished by either compromising their survival or eliminating preexisting TAMs. Due to the reliance of macrophages on CSF-1R signaling, the primary method for reducing TAMs involves specifically targeting the CSF-1R. CSF-1R is a type III protein kinase receptor that interacts with two ligands, CSF-1 and IL-34. When the receptor binds to its ligands, it triggers the formation of CSF-1R homodimers and activates signaling pathways that are essential for macrophage survival [63]. Blocking CSF-R1 leads to decreased number of TAMs through the inhibition of monocyte differentiation and reduced survival of existing TAMs [64]. Notably, there is ongoing focus on alternative strategies to reduce TAMs directly. These strategies include the use of Trabectedin, which activates caspase 8 through TNF-related apoptosis-inducing ligand (TRAIL), causing monocytes and macrophages within the tumor to undergo apoptosis [65, 66]. The engagement of CSF-1R was validated by a rise in CSF-1 levels and a decrease in CD14lowCD16high monocytes in peripheral blood mononuclear cells [67]. It is important to mention that while these strategies have been explored, their application in reducing TAMs survival has not been thoroughly studied in relation to PC. Ongoing investigations are assessing the effectiveness of different antibodies and receptor antagonists that specifically target CSF-1R to compromise the survival of TAMs. These studies are being conducted in diverse preclinical studies and clinical trials, examining the use of CSF-1R inhibitors as standalone treatments or in combination with other therapies including immunotherapies. Several small molecule inhibitors are employed in the treatment of PC. PLX3397, also known as Pexidartinib, is a receptor tyrosine kinase inhibitor (TKI) that targets multiple receptors including CSF-1R, KIT, and FLT3. However, it is more accurately described as a conformation-specific inhibitor rather than a traditional TKI for CSF-1R, which specifically binds to the autoinhibited state of CSF-1R and directly contacts with the juxtamembrane region [68, 69]. In 2019, the FDA approved its use for treating symptomatic tenosynovial giant cell tumors [70]. To evaluate the safety and efficacy of pexidartinib in combination with durvalumab (an anti-PD-L1 antibody) for patients with advanced colorectal cancer (CRC) and PDAC, a phase I clinical trial was carried out [71]. The objective of the study was to improve PD-L1 blockade responses by the removal of CSF-1-dependent suppressive TAMs. 15% of patients had stable disease, 2% of patients had a partial response, and there were no unexpected toxicities noted BLZ945 is a selective, orally administered investigational inhibitor of CSF-1R tyrosine kinase that can penetrate the brain. In various animal models, including those with intracranial glioblastoma multiforme (GBM), the blockade of CSF-1R by BLZ945 has shown potential in reducing the TAMs’ recruitment to the TME, inhibiting tumor growth, and overcoming resistance to PD-1 inhibitors [64, 69]. Patients with tumors linked to TAMs upregulation, including as GBM (n = 24), PC (n = 19), CRC (n = 10), as well as other types including breast cancer, mesothelioma, and soft tissue sarcoma., were included in a phase I trial to assess the efficacy of BLZ945, both as a monotherapy and in combination with spartalizumab (also called PDR001, an anti-PD-1 antibody). The administration of BLZ945 with or without spartalizumab showed an acceptable safety and efficacy profile [72, 73]. Anti-CSF-R1 antibodies for PC have also garnered attention from researchers. The anti-tumor effectiveness and safety of cyclophosphamide, pembrolizumab (an antibody that inhibits negative signals to T cells), GVAX (a PC vaccination), and IMC-CS4 (also known as LY3022855) in combination are being evaluated in an early phase I clinical trial [74]. The results of this investigation have not yet been published. AMG820 is a fully human monoclonal antibody (mAb) targeting CSF-1R. It is currently under investigation and functions by blocking the ligands CSF-R1 and IL-34 from attaching to the receptor, which prevents the ligand-induced activation of the receptor [69]. A Phase 1b/2 clinical trial is being conducted at multiple centers to evaluate the efficacy of combining AMG820 with pembrolizumab in individuals diagnosed with specific advanced solid tumors including PC. According to Razak et al., a satisfactory safety profile was shown by the combination of 1100 mg AMG820 with 200 mg pembrolizumab at the recommended dose. Nevertheless, in the particular patient populations examined, the demonstrated anticancer activity did not meet the requirements for additional assessment of this combination [75, 76]. BMS-986227 is another antibody that had been studied in a clinical trial and showed pharmacodynamic activity for anti-PD-1/PD-L1–resistant tumors including PC. Additionally, Clodronate has been investigated for its ability to decrease macrophage tumor infiltration [77, 78]. In a recent study, targeting proliferating F4/80+ macrophages enhanced the antitumor response mediated by cytotoxic CD8+ T-cell. Depletion of macrophages using clodronate liposomes decreased proliferating macrophages in PDAC mouse models and increased the proximity of CD8+ T cells to tumor cells. This in turn, led to an increase in IFN-γ+ CD8+ T cells infiltration, promoting an antitumor effect. These findings highlight the potential of targeting proliferating TAMs to enhance CD8+ T-cell infiltration and improve antitumor immune responses in PDAC [79] (Table 1).
3.2 Reprogramming TAMs to promote anti-tumor activity
Reprogramming strategies for TAMs can be divided into two categories. The first category comprises strategies that aim to convert TAMs from a pro-tumoral phenotype to an anti-tumoral one. These strategies focus on targeting either the signaling pathways responsible for macrophage polarization or specific targets/markers expressed by TAMs. The second group, referred to as function-based reprogramming of TAMs, involves therapeutic strategies that specifically target the functions of TAMs, such as immunosuppression and phagocytosis. Furthermore, there are strategies aimed at engineering macrophages to enhance their anti-tumoral properties [80]. While some specific strategies have not been utilized for PC, a brief explanation is given for each method.
3.3 Phenotype reprogramming of TAMs
3.3.1 Reprogramming macrophage phenotype through polarization signaling pathways
PI3K encompasses multiple subclasses, with class 1B PI3Kγ primarily found in hematopoietic cells. Inhibiting this target through genetic and pharmacological agents means results in an increase in the expression of major histocompatibility complex class II (MHC II) molecules, along with elevated IL-12 levels and reduced secretion of IL-10. Consequently, PI3Kγ inhibition in TAMs promotes the recruitment of cells linked to adaptive immunity against tumors and inhibits tumor growth [81, 82]. Histone deacetylase inhibitors (HDACs) are vital in the elimination of acetyl groups from histones in order to epigenetically regulate the gene expression. This may result in a pro-inflammatory phenotype by altering the expression of CCL1 and CCL2 in monocytes. The research conducted by Guerriero et al., demonstrated that inhibitors targeting class IIa HDACs have the ability to reprogram TAMs into anti-tumoral macrophages. In preclinical cancer models, this reprogramming has been demonstrated to improve the effectiveness of immunotherapy and chemotherapy [83, 84]. Toll-like receptors (TLRs) are pattern recognition receptors of the innate immune system and are essentials in activating the innate immune response. TLRs have diverse functions in macrophage activation. They control cytokine production, promote cell survival, participate in the identification of invading pathogens and self and non-self antigens [85]. Numerous anti-TLRs therapeutics have been developed with the aim of repolarizing macrophages from an M2-like to an M1-like activation state [86], thereby enhancing the response of immune system to cancerous cells. The elevation of SNAIL and vimentin in PC cells and the considerable decline of the E-cadherin levels were achieved with the use of TLR4 siRNA and neutralizing antibodies against TLR4 and IL-10, expressed on M2-polarized TAMs [87]. By affecting TAMs polarization, histidine-rich glycoprotein (HRG), an antiangiogenic and immunomodulatory substance generated by the host, has been demonstrated to augment anti-tumor immune responses and promote vascular normalization. Specifically, it shifts TAMs from the M2 phenotype associated with tumor promotion to the tumor-inhibiting M1 phenotype [88]. HRG interacts with various molecules involved in tumorigenesis, including Fcγ receptors (FcγR), thrombospondins (TSPs), heparin. FcγR, expressed in macrophages, has been implicated in promoting the M2 macrophages [89]. However, it is important to note that FcγR can also activate macrophage cytotoxicity in response to Trastuzumab [90, 91].
TMP195 is a specific inhibitor of class IIa HDACs. By targeting these HDACs, TMP195 can alter the epigenomic profile of monocytes and macrophages. Ivaltinostat, Romidepsin, and Vorinostat are inhibitors of class IIa HDACs that several clinical trials have been conducted to investigate their effectiveness on PC. TLR3 agonists such as Poly- ICLC, Rintatolimod, and BO-112, TLR7 agonists such as SHR2150, TransCon, BDC-1001, and TLR9 agonists such as CMP-001 have been evaluated in several clinical trials. Resiquimod (R848) is an immunostimulatory drug that targets TLR 7/8. Both soluble R848 and R848 in nanoparticle formulation have demonstrated effectiveness in various preclinical tumor models. These effects include enhancing survival in a murine pancreatic model [92], transforming M2 macrophages into M1 phenotype, boosting cellular phagocytosis dependent on Ab [93], reorganizing the TME’s myeloid compartment to promote tumor regression [94], and enhancing the efficacy of chemotherapy [95]. Zoledronic acid (ZA) is another therapeutic that aims to reduce the number of TAMs and alter their polarization status [78, 96]. In the context of mesothelioma, ZA was found to reduce the M2 macrophage markers’ expression, including Arg-1, CD206, and TGF-β. Additionally, ZA prevented monocytes from differentiation into macrophages [97, 98]. FGD5-AS1 is a long non-coding RNA (lncRNA), is linked to high IL-6 expression and poor prognosis in PC patients. Through the STAT3/NF-κB pathway, exosomal FGD5-AS1 promotes PC progression and M2 macrophage polarization. Increased transcriptional activity and STAT3 acetylation result from FGD5-AS1's interaction with p300. These findings suggest that FGD5-AS1-rich exosomes are released by PC cells which induces M2 phenotype polarization and promotes aggressive behavior in PC. Targeting exosomal FGD5-AS1 could have diagnostic and therapeutic implications for PC [99] (Table 1).
3.3.2 Reprogramming macrophage phenotype through TAMs-preferentially expressed targets
The Macrophage Receptor with Collagenous Structure (MARCO) is predominantly expressed on M2 phenotype in the TME, which is associated with an immunosuppressive gene profile [100]. The presence of anti-inflammatory cytokine IL-37 and a higher concentration of regulatory T cells are associated with TAMs expressing MARCO. It is also linked to a decline in the quantity of NK cells and a decrease in CD8+ T cell activity [101]. In preclinical studies using murine models of melanoma, breast cancer, and colon cancer, the use of mAbs targeting MARCO has shown promising results. It led to a reduction in tumor growth and impaired metastasis [102]. According to Shi et al., PC cells showed a significant increase in the presence of M2 macrophages. They demonstrated an elevated expression of both MARCO and CD163 in PC cells when compared to adjacent para-cancerous tissues. Moreover, there was considerable variation in the levels of CD163 and MARCO expression among different cases of PC, indicating a heterogeneous expression pattern of these two markers among patients. Additionally, the elevated expression of MARCO and CD163 had a negative impact on both overall survival rates and disease-free survival in patients with PC. More analyses confirmed that expression of MARCO and CD163, independently served as indicators of PC prognosis [103]. Moreover, Sarhan et al., showed that using antibodies directed against MARCO repolarizes myeloid cells toward a pro-inflammatory phenotype, preventing them from suppressing T cells and NK cells through processes that include the release of extracellular ATP, the activation of inflammasomes, and the production of IL-18 [104]. Researchers have identified overexpression of endocannabinoid receptor-2 (CB-2) on TAMs and monoacylglycerol lipase (MGLL) in PC cells. A reduction-responsive nanoplatform has been created by them to co-deliver CB-2 siRNA and MGLL siRNA. This nanoplatform repolarized TAMs into a tumor-inhibiting phenotype and also efficiently inhibited the formation of free fatty acid in PC cells. In xenograft and orthotopic PC tumor models, this approach achieved an anticancer effect by inhibiting nutrient supply and inducing TAMs to secrete tumoricidal cytokines [105]. A recent study uncovered the mechanism by which exosomal miR-193b-3p generated from M2 macrophages promotes the glutamine uptake, migration, proliferation, and invasion of PC cells. This impact was achieved by targeting TRIM62, leading to a decrease in c-Myc ubiquitination. These results shed light on the interaction between PC cells and M2 macrophages and suggested a potential therapeutic target for PC [106].
3.4 Function-based reprogramming
3.4.1 Targeting phagocytic activity of macrophages
Tumor cells have the ability to evade phagocytosis by exploiting MHC I molecules through their interaction with members of the leukocyte immunoglobulin-like receptor subfamily B (LILRB) family. LILRB1, a protein that binds to MHC molecules, is extensively expressed on immune cells and is particularly abundant on TAMs. An inhibitory signal is transmitted by the immunoreceptor tyrosine-based inhibition motif (ITIM) contained in LILRB1. The expression of LILRB1 has been associated with the inhibition of cancer cell phagocytosis. Research on cancer cell lines resistant to anti-CD47 antibodies revealed its function as a myeloid checkpoint. It was found that tumor cells' production of MHC I molecules correlated with their resistance to anti-CD47 therapy. Additionally, inhibiting LILRB1 with particular antibodies could reverse the phagocytic activity that the anti-CD47 antibody had induced [107]. Additionally, macrophages express LILRB2, and blocking it resulted in enhanced phagocytic activity increased and pro-inflammatory activation [108]. The goal of ongoing research is to identify binding partners for LILRB molecules in order to create novel therapies that target macrophages. These binding partners are expected to provide a means of controlling the tumor's ability to evade phagocytosis. In this context, a human monoclonal antibody called MK-4830, which specifically targets LILRB2, has been evaluated in a phase I dose-escalating study for the treatment of advanced-stage solid tumors [109]. The study assessed the antibody both as a monotherapy and in combination with pembrolizumab. Initial findings from the study have shown promising outcomes, with durable responses observed. These results lend credence to MK-4830’s continued exploration as a potential therapeutic agent. Furthermore, it has been reported that the LILRB4 gene plays a critical role in influencing the clinical outcomes of early-stage PDAC and is strongly correlated with the prognosis of these patients [110] (Fig. 2). CD47 is a commonly found protein in various cells and plays a regulatory role in several biological processes, including cytokine production, cell migration, T cell activation, and axon extension [111]. Also, High expression of CD47 has been observed in a variety of tumor types [112]. CD47 has an interaction with signal regulatory protein-α (SIRPα), also referred to as SHPS1, and thrombospondin 1. Through its interaction with SIRPα, CD47 is involved in metastasis, tumor invasion, and, most significantly, the innate immune system's regulation of phagocytosis [112]. These interactions primarily occur with myeloid cells, among them macrophages and DCs. The interaction between SIRPα, which is expressed on macrophages, and CD47, which is expressed on cancer cells, prevents phagosome formation and, consequently, prevents macrophages from engulfing cancer cells. It has been shown that CD47 antagonists enhance the phagocytic ability of macrophages to uptake cancer cells. They additionally promote the presentation of antigens, which increases T cell cross-priming [113]. CD24 is a molecule anchored to the cell membrane by a glycophosphatidylinositol linkage, and it plays a significant role in cancer development [114]. CD24 exhibits remarkably high expression levels at the transcript level across various tumor types, particularly in triple-negative breast cancer (TNBC) and ovarian cancer, and this overexpression is associated with an unfavorable prognosis [115]. Additionally, CD24 participate in regulating important cellular processes including cell proliferation, migration, and invasion [116]. Furthermore, CD24 is considered a potential marker for CSCs [117], indicating its involvement in tumor progression and resistance to therapy. A recent discovery has provided insight into the role of CD24 in inhibiting macrophages [115]. It has been demonstrated that CD24 acts as an immune checkpoint by interacting with Siglec-10, which is TAMs, leading to the suppression of phagocytosis. This interaction establishes CD24 as a novel “don’t eat me” signal in the context of cancer. The expression of PD1 by TAMs has been found to suppress phagocytosis and hinder tumor immunity [118]. This challenges the conventional understanding of the PD1-PDL1 axis as a T cell-specific checkpoint. Simultaneously, the expression of PDL1 on cancer cells enables them to evade both T cell cytotoxicity and macrophage-mediated phagocytosis. Therefore, blocking this axis may potentially unleash anticancer immunity through both innate and adaptive mechanisms. The precise mechanism through which PD1 engagement on macrophages inhibits phagocytosis and the signals that induce PD1 upregulation are yet to be fully understood [119].

Different stages of pancreatic cancer
Several studies have employed Anti-CD47/SIRPα antibodies, including Hu5F9-G4 and BI 765063. Researchers have also shown interest in CD47-Fc fusion proteins, such as TTI-622, TTI-621, IMM-01, SL-172154, and ALX148. Several antibodies [120] and antibody fusions have been developed to target CD24, such as those conjugated with CD30 or CD20 [121]. For patients with metastatic melanoma, a clinical trial is presently being conducted to evaluate the safety and efficacy of combining ipilimumab, nivolumab, and the recombinant fusion protein CD24Fc [122]. The evaluation of combining anti-PD1 or anti-PDL1 therapies with other strategies, such as PI3Kγ inhibitors [82] and TLRs [123, 124] agonists, has been conducted.
3.4.2 Targeting the immunosuppressive and immunostimulating activity of macrophages
A subset of macrophages express SIGLEC1, sometimes referred to as sialoadhesin or CD169, which has been reported to be elevated in some cancers. Its expression levels are correlated with a poorer prognosis [80, 125]. Depletion of CD169+ TAMs was found to be beneficial in lowering tumor burden and metastasis in animal models of breast cancer. Furthermore, the depletion of CD169+ macrophages has been shown to reduce lung metastasis and tumor growth in mouse models of TNBC [126]. The underlying mechanism involves a notable proliferation of CD8+ T cells in the spleen and circulation, in addition to a heightened concentration of these T cells inside the tumors. When CD169+ macrophages were cultivated with tumor cells in vitro, PDL1 was found to be upregulated in the macrophages. This implies that TAMs in the TME might be involved in evading T cell-mediated immune surveillance [80]. A class of small non-coding RNAs known as miRNAs is involved in sequence-specific regulation of transcription and translation. The maturation process of miRNAs is controlled by the enzyme DICER, which belongs to the RNase-III family [127]. A recent study demonstrated that blocking the activity of DICER in macrophages has an impact on the programming of TAMs. This inhibition resulted in tumor regression and modified immune cell infiltration. The inhibition of DICER led to the reprogramming of TAMs, causing them to express an IFNγ-STAT1 signature and exhibit an antitumoral phenotype. Additionally, the inhibition of DICER in TAMs was found to enhance the response to immunostimulatory antibodies, suggesting a potential therapeutic target (benefit) [128]. Several antigen-presenting cells (APCs), including as macrophages, monocytes, B cells and DCs, express CD40, a member of the TNF receptor superfamily. The primary cells that express CD40 Ligand (CD40L) are mast cells, basophils, and CD4+ T cells [129]. MHC molecules are upregulated as a result of the interaction between CD40 and CD40L, and pro-inflammatory cytokines, such as IL-12, are produced. This interaction is essential in order to prime naïve CD4+ and CD8+ T cells and direct their differentiation into Th cells and cytotoxic cells, respectively. Targeting CD40, a number of therapeutic approaches have been developed, including agonistic CD40 antibodies, recombinant human CD40L and its fusion proteins, and CD40L gene therapy using adenoviral vectors expressing CD40L. Notably, the administration of CD40 agonists to TAMs in conjunction with anti-CSF-1R antibodies leads to a significant reprogramming of TAMs prior to their depletion. These reprogrammed TAMs help foster an environment that is pro-inflammatory and stimulates potent T cell responses. Importantly, this approach has shown promise in eliciting T cell responses in tumors that were previously unresponsive to immune checkpoint inhibitors [130, 131].
There are ongoing clinical trials investigating at least ten therapeutics that target CD40. Some of these therapeutics have progressed to phase 2 trials, including ChiLob7/4, which is being evaluated for the management of head and neck cancer and PC [132]. Also, another clinical trial has been conducted to evaluate the combination of CP870,893 and Gemcitabine for patients diagnosed with newly resectable PC [133]. In addition, CP-870,893 in combination with gemcitabine demonstrated favorable tolerability and showed indication of anti-tumor efficacy in PDAC patients [134]. APX005M has recently obtained FDA approval as an orphan drug for the treatment of PDAC, esophageal and gastroesophageal junction cancer [135, 136]. RO7009789 is another anti-CD40 antibody that has been evaluated on PC [137].
3.4.3 CAR macrophage
There have been advancements in utilizing macrophages that express chimeric antigen receptors (CARs) to target particular molecules including CD47 and CSF-1R. These approaches aim to enhance immune responses by directing to SIRPα-expressing macrophages. This targeted amplification of immune responses has shown promise in limiting tumor growth [138, 139]. CAR-macrophages (CAR Ms) have been found to express proinflammatory cytokines and chemokines, enabling them to recruit immune cells and present antigens to T cells. To summarize the process of developing and using CAR Ms, monocytes are stimulated through subcutaneous administration of G-CSF before being isolated through leukapheresis and CD14+ monocyte selection. These monocytes are then cultured to differentiate into macrophages and genetically modified with Adf535, which encodes the CAR transgene. Subsequently, the CAR M cells are cryopreserved in a suitable medium for infusion and undergo testing to ensure their quality before being used in therapy [140, 141] (Fig. 3). Remarkably, a single infusion of human CAR Ms improved overall survival and reduced tumor burden in two tumor xenograft mouse models [140]. The efficacy of adenovirus-transduced macrophages expressing an anti-HER2 CAR is currently under evaluation in a clinical investigation [142].

Development of CAR Macrophages from induced pluripotent stem cells (iPSCs) and their mechanism of action
3.5 Targeting macrophage recruitment
Recruitment of circulating monocytes replenishes the TAMs population on a continual basis. Preventing the recruitment of new TAMs is another method for lowering the total number of TAMs within a tumor. Due to the involvement of the chemokine CCL2 in the recruitment of monocytes to tumors, significant research has focused on targeting the CCL2/CCR2 axis [78]. Several investigations have demonstrated that in a variety of tumor models, reducing CCL2 signaling through the use of neutralizing antibodies or small-molecule inhibitors can result in a decrease in tumor size and metastases [143,144,145]. However, despite promising results in preclinical models, clinical trials investigating CCL2/CCR2 blockade have not shown comparable efficacy [146]. Studies indicate that CCL2 may serve as a significant negative regulator of PC progression. While there was an inverse relationship between blood CCL2 levels and tumor proliferative activity, a positive association was established in PC patients between serum CCL2 levels and TAM infiltration [147]. Patients with tumors displaying high CCL2 expression and low CD8 T-cell infiltration experience notably reduced survival rates. In mouse models, inflammatory monocytes and macrophages from the main tumor and premetastatic liver are efficiently reduced by inhibiting CCR2. This depletion subsequently enhances antitumor immunity, reduces tumor growth, and diminishes metastasis [148]. TAMs generate TNF-α, which inhibits PDAC cells’ production of IL-33. In mouse models, TNF-α was decreased and IL-33 expression was up when monocyte recruitment was inhibited by CCR2 deletion, which enhanced survival and decreased metastases. High CCL5 levels in the TME aid in the accumulation of macrophages and lymphocytes that have high CCR5 expression levels. Apart from its part in immune cell recruitment, CCL5 is involved in various processes, including tumor growth, drug resistance, expansion of CSCs, invasion of cancer cells, promotion of neoangiogenesis, and polarization of macrophages towards an immunosuppressive phenotype [149]. CXCL12 is a chemokine that is involves in regulating the migration of monocytes and angiogenesis [150, 151]. Targeting the angiogenic and chemotactic factor CXCL12, which signals through CXCR4, has been explored as a strategy to decrease TAMs infiltration [151].
Phase I clinical studies have been initiated for a number of therapeutic agents that target the CCL2/CCR2 pathway, such as small-molecule inhibitors and mAbs. These include the CCL2-neutralizing antibody CNTO888 (Carlumab) [152], and the anti-CCR2 antibody MLN1202 (plozalizumab) [153]. PF-4136309 which is a CCR2 antagonist have been evaluated on PDAC in combination with gemcitabine and Nab-paclitaxel [154]. At the suggested phase 2 dosage, PF-04136309 plus FOLFIRINOX did not result in any additional toxicity. Blockade of CCR2 resulted in a reduced infiltration of TAMs and demonstrated signs of an innate anti-tumor immune response. These findings indicate that CCR2-targeted therapy has an impact on the biology of PDAC [155]. A phase 1-b open-label study assessing the safety and efficacy of CCX872-B (a CCR2 antagonist) in patients with PDAC who were receiving FOLFIRINOX chemotherapy has been successfully completed [156]. They reported that combination of CCX872-B and FOLFIRINOX produced an 18-month overall survival rate of 29% without any safety concerns. Additionally, the treatment resulted in a decrease in circulating monocytes, inflammatory monocytes, and monocytic MDSCs [157]. By using CCX598, an orthosteric inhibitor, to target CCR2, PDAC became susceptible to checkpoint blocking, which decreased metastasis and improved survival. These findings suggest that disrupting immunosuppressive macrophage infiltration or their signaling could enhance immunotherapeutic efficacy for PDAC by promoting antitumor immunity mediated by IL-33, cytotoxic T cells, and DCs [158]. In preclinical studies conducted on mouse models of PC and prostate cancer [159, 160], the use of CCR5 antagonists demonstrated a reduction in tumor growth, as well as a decrease in tumor adhesion and invasion. The effectiveness of BMS-813160, an CCR2/CCR5 antagonist, either as a standalone treatment or in combination with chemotherapy, nivolumab, or chemotherapy plus nivolumab, has been evaluated in participants with metastatic CRC and PC [161]. In a clinical trial, the efficacy of treatment involving a CCR5 antagonist, Maraviroc, aCCR5 antagonist, in combination with nivolumab and ipilimumab, was investigated in patients with CRC and PC [162, 163]. The interaction between CCR5 and CCL5 has been identified as a potential indicator for metastatic PC, as it contributes to heightened invasion of PC cells [150]. Plerixafor (AMD3100) is one of the CXCR4 antagonists, which was approved in 2008 for autologous transplantation for the mobilization of hematopoietic stem cells. Currently, it is under investigation in clinical trials for its potential use in various types of cancers [164]. BL-8040 (motixafortide), a CXCR4 antagonist, demonstrated effectiveness when used in combination with pembrolizumab and chemotherapy in PDAC [165]. Moreover, the cooperation between CXCL8/IL-8 and CXCL12/stromal cell-derived factor-1α can enhance migration, invasion, and angiogenesis. This suggests that targeting CXCR2 (the receptor for CXCL8) and CXCR4 (the receptor for CXCL12) could serve as potential therapeutic targets for anti-angiogenic and anti-metastatic strategies in PC [151]. Plerixafor (AMD3100) is also a CXCR4 antagonist which had promising results in clinical trials (Table 1).
4 Challenges and limitations of targeting macrophages in PC
PC exhibits a higher level of aggressiveness compared to other gastrointestinal malignancies, primarily due to its early-stage dissemination through hematogenous or lymphatic metastasis. This aggressive behavior is linked to poor prognosis and challenging therapeutic options. Firstly, while the field of cancer research and therapy has witnessed rapid progress in targeting macrophages as a potential treatment option, determining the optimal therapeutic approach remains a challenge due to the heterogeneous nature of TAMs and their diverse roles influenced by environmental conditions. It was reported that pancreatic TME is highly heterogeneous, as this heterogeneity could influence the efficacy of future PC treatment [166,167,168]. Generally, TAMs numerates as the most abundant tumor-infiltrating immune cells in PC, which are divided into two subgroups including immune-stimulatory macrophages (M1) and immunoregulatory macrophages (M2) [166]. The ability of macrophages to switch between two pro-inflammatory and anti-inflammatory states has a dynamic characteristic and plays a significant role in the progress and development of PC [169]. Pan et al. [170] reported that the increase in the abundance of CD68+ macrophages and CD163+ M2 in tumors was significantly associated with worse survival in patients with PC. Chen et al. [171] reported that SPP1, as an immune-related marker, is mainly expressed in type 2 ductal cells and M2 macrophage in PC. Moffitt et al. [172] reported two tumor cell subtypes (“basal-like” and “classical” and two stromal subtypes (“normal” and “activated”) for PDAC. It was found that tumors with “basal-like” subtype presented a worse prognosis compared to either corresponding subgroup but additionally it seems that this subtype shows better responses towards adjuvant therapy. Bailey et al. [173] performed a genomic analysis and identified 4 subtypes including squamous; pancreatic progenitor; immunogenic; and aberrantly differentiated endocrine exocrine (ADEX). Therefore, it seems that the identification of these differences provides opportunities for therapeutic development [174].
Secondly, the dynamic TME, comprising infiltrating immune cells and external factors, further complicates the matter. Thirdly, recognizing tumor heterogeneity and its evolution during the transition to malignancy and post-therapy becomes crucial in comprehending the TAMs and other immune cells' dynamic interactions [80]. The effects of these interactions vary based on the stage of progression and cancer type.
Furthermore, as mentioned earlier, numerous studies have explored the combination of macrophage therapy with conventional treatments for PC and other cancers. It is worth noting that the success of combination therapy requires a comprehensive comprehension of the TME, including the heterogeneity of macrophage populations and their interactions with other immune cells. Compared to mouse PC tumors, human PC tumors show a higher degree of interpatient, intertumor, and intratumor heterogeneity altogether between individuals [172, 173, 175,176,177,178,179]. Therefore, designing treatment options with the approach of targeting macrophages will not be successful without understanding the complex system related to the PC TME and will have deleterious or negligible clinical outcomes. Finally, careful consideration should be given to the timing, dosing, and sequencing of treatments to maximize their synergistic effects and minimize potential adverse effects. For instance, depletion strategies, like CSF-1R inhibitors, have shown potential efficacy but pose the risk of non-specific depletion of monocytes and macrophages, leading to long-term toxicity [63, 78]. Plus, the complexity of TAM populations highlights the essential functionality of DCs and macrophages in the effectiveness of specific therapies, such as anti-PD1 and anti-CTLA4. Another example is careful timing and a comprehensive understanding of immune interactions throughout different tumor formation stages, which are crucial for successful implementation of Fcγ receptor inhibitors.
5 Emerging research and future directions
PC, the twelfth most common cancer worldwide, is associated with an extremely poor prognosis and survival rate. While resection offers a potential cure for patients with PC, 20% of patients are candidates for resection at the time of diagnosis, and despite the surgery, most of the patients die due to local recurrence or metastasis. It was estimated that the 1-year disease-related mortality is 30% [180,181,182]. Therefore, it is meaningful to reduce the tumor stage and improve the resectable rate through neoadjuvant therapy [183,184,185]. Given the various ways in which macrophages promote the development and progression of PC, they are considered as potential targets for anti-cancer treatment. Macrophages play a crucial role in cancer progression and have emerged as potential therapeutic targets. Some potential new targets and strategies for modulating macrophage function include inhibition of monocyte/macrophage recruitment, killing or depletion of macrophages, reprogramming of macrophage phenotypes at disease sites, inhibition of cytokines and chemokines involved in the recruitment and polarization of tumor-promoting macrophages, targeting macrophage receptor-ligand interactions, modulating macrophage polarization, chimeric antigen receptor macrophage therapy, and macrophage-mediated drug delivery systems. Some of the macrophage-targeting therapies are currently undergoing clinical evaluation, and there is growing interest in understanding how targeting different macrophage populations can affect immunosuppression, fibrosis, and responses to chemotherapy [186]. Macrophage-derived CCL18 has been shown to enhance the invasive ability of PC cells, and targeting this pathway could potentially impact tumor progression [187]. The use of chimeric antigen receptor (CAR) macrophages showed that in situ CAR engineering of macrophages with MUC1-specific CARs stimulated tumoricidal immunity against pancreatic adenocarcinoma [188]. CAR-engineered macrophages induced a shift in the TME from an immunosuppressive to a tumoricidal state, supporting the potential of applying CAR macrophages in the TME in pancreatic adenocarcinoma. Investigating the role of M2 macrophage-derived exosomes in PC progression demonstrates that exosomal miR-193b-3p, derived from M2 macrophages, enhances the proliferation, migration, invasion, and glutamine uptake of PC cells by targeting TRIM62, leading to a decrease in c-Myc ubiquitination. These findings provide insights into the mechanism involved in the crosstalk between M2 macrophages and PC, and shed light on the potential impact of targeting this pathway for the treatment of PC [106]. In another miRNA-based strategy, the role of miR-222-3p-containing extracellular vehicles (EVs) derived from M2 macrophages in conferring gemcitabine resistance in PC was evaluated by co-culture of PC cells with M2 macrophage-derived EVs enriched with miR-222-3p, which led to decreased sensitivity to gemcitabine, lower percentage of apoptosis and enhanced proliferation. Furthermore, the delivery of a miR-222-3p inhibitor by M2 macrophage-derived EVs suppressed tumor growth and elevated the sensitivity of cancer cells to gemcitabine. miR-222-3p also targets and suppresses TSC1 expression, while activating the PI3K/AKT/mTOR pathway, thereby contributing to gemcitabine resistance in PC, suggesting the potential of targeting miR-222-3p and the associated pathways as a therapeutic strategy to overcome gemcitabine resistance in PC [189]. Studying the impact of blocking the CCR4 chemokine receptor on intratumoral macrophage recruitment and survival in PC bearing mice demonstrated that defeat in the CCR4 chemokine receptor or blockade of CCR4 in mice exerted a significant survival benefit in PC bearing mice. Diminished intratumoral macrophage recruitment led to a decrease in tumor growth and an increase in survival rates, suggesting that the CCR4 chemokine receptor blockade may be a potential therapeutic approach for PC, as it reduces the recruitment of macrophages to the TME and enhances the survival of cancer-bearing mice [190]. Evaluation of the effect of the ALOX5 enzyme inhibitor (Zileuton) in regulating TAM polarization and its impact on PC invasion and metastasis showed regulated TAM M2 polarization by targeting the JAK/STAT pathway. The findings demonstrated that Zileuton inhibited PC invasion and metastasis by suppressing M2 TAM polarization [191]. Applying the combination of an mTOR inhibitor, gemcitabine, and a PD-L1 antibody on PC progression in Trp53flox/+LSL-KrasG12D/+Pdx-1-Cre murine models showed suppressed PC progression by inducing metabolic reprogramming and remodeling the immune microenvironment. mTOR inhibition could change the immune microenvironment of PC via metabolic reprogramming, and promoting the efficacy of PD-L1 blockade when combined with gemcitabine [192]. In another study, the role of arginase 1 (Arg1) in driving immune suppression in PC was evaluated and showed that Arg1 drives immune suppression in PC by depleting arginine and inhibiting T cell activation. Also, genetically inactivation of Arg1 in macrophages in a dual recombinase genetically engineered mouse model of PC led to delayed invasion and higher rate of CD8+ T cell infiltration. An arginase inhibitor designated CB-1158 was applied to treat established tumors. The results exhibited that there was an additional increase in CD8+ T cell infiltration beyond what was observed with the macrophage-specific knockout, and the tumors acquired more sensitivity to anti-PD1 immune checkpoint blockade. The research highlights the potential of targeting Arg1 as a promising immunotherapeutic target to enhance the response to immune therapy in PC [193]. Moreover, high CXCR4+ macrophage infiltration is associated with poor overall survival and disease-free survival in PDAC patients. Targeting CXCR4+ macrophages is a potential therapeutic strategy to improve the prognosis of PDAC patients since CXCR4+ macrophages promote PDAC progression by inducing immune suppression and remodeling the TME [194]. While macrophage-targeting therapies hold promise, there are also clinical challenges associated with these treatment modalities in human PDAC patients. The desmoplastic stroma which provides an immunosuppressive microenvironment with a dense barrier to protects tumor cells from immunological elimination and prevent penetration of chemotherapeutics, poses a significant obstacle to the development of therapies. Overcoming these challenges will require a deep understanding of the complex interactions between macrophages, the TME, and the immune system [195].
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- CAR Ms:
-
CAR-macrophages
- CARs:
-
Chimeric antigen receptors
- CB-2:
-
Endocannabinoid receptor-2
- CCML:
-
C–C Motif chemokine ligand
- CD:
-
Cluster of differentiation
- CRC:
-
Colorectal cancer
- CSCs:
-
Cancer stem cells
- CSF:
-
Colony-stimulating factor
- CSF-1R:
-
Colony-stimulating factor 1 receptor
- CTLs:
-
Cytotoxic T lymphocytes
- DCs:
-
Dendritic cells
- DTCs:
-
Disseminated tumor cells
- EGF:
-
Epidermal growth factor
- FGF:
-
Fibroblast growth factor
- GBM:
-
Glioblastoma multiforme
- HDACs:
-
Histone deacetylase inhibitors
- HIF:
-
Hypoxia-inducible factor
- HRG:
-
Histidine-rich glycoprotein
- IGF:
-
Insulin-like growth factor
- IL:
-
Interleukin
- LILRB:
-
Leukocyte immunoglobulin-like receptor subfamily B
- MAMs:
-
Metastasis-associated macrophages
- MARCO:
-
Macrophage Receptor with Collagenous Structure
- MDSCs:
-
Myeloid-derived suppressor cells
- MGLL:
-
Monoacylglycerol lipase
- MHC II:
-
Major histocompatibility complex class II
- MHC:
-
Major histocompatibility antigens
- MMP:
-
Matrix metalloproteinases
- NK:
-
Natural killer
- PC:
-
Pancreatic cancer
- PD:
-
Programmed cell death
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDL:
-
Programmed death ligand
- PlGF:
-
Placental growth factor
- SIRPα:
-
Signal regulatory protein-α
- TAMs:
-
Tumor-associated macrophage
- TGF-β:
-
Transforming growth factor-β
- Th:
-
T helper
- TKI:
-
Tyrosine kinase inhibitor
- TLRs:
-
Toll-like receptors
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- TNF-α:
-
Tumor necrosis factor-α
- VEGF:
-
Vascular-endothelial growth factor
- ZA:
-
Zoledronic acid
References
Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12–49.
Hu J-X, Zhao C-F, Chen W-B, Liu Q-C, Li Q-W, Lin Y-Y, et al. Pancreatic cancer: a review of epidemiology, trend, and risk factors. World J Gastroenterol. 2021;27(27):4298.
Pourshams A, Sepanlou SG, Ikuta KS, Bisignano C, Safiri S, Roshandel G, et al. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2019;4(12):934–47.
Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol. 2021;18(7):493–502.
Krug S, Michl P. New developments in pancreatic cancer treatment. Minerva Gastroenterol Dietol. 2012;58(4):427–43.
Momin S, Lei Y, Wang T, Zhang J, Roper J, Bradley JD, et al. Learning-based dose prediction for pancreatic stereotactic body radiation therapy using dual pyramid adversarial network. Phys Med Biol. 2021;66(12): 125019.
Luo W, Zheng L, Zhang T. Do novel treatment strategies enhance T cell-mediated immunity: opportunities and challenges in pancreatic cancer immunotherapy. Int Immunopharmacol. 2021;90: 107199.
Rihawi K, Ricci AD, Rizzo A, Brocchi S, Marasco G, Pastore LV, et al. Tumor-associated macrophages and inflammatory microenvironment in gastric cancer: novel translational implications. Int J Mol Sci. 2021;22(8):3805.
Belli C, Antonarelli G, Repetto M, Boscolo Bielo L, Crimini E, Curigliano G. Targeting cellular components of the tumor microenvironment in solid malignancies. Cancers (Basel). 2022;14(17):4278.
Evans A, Costello E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front Physiol. 2012;3:30097.
Wang W, Yan L, Guan X, Dong B, Zhao M, Wu J, et al. Identification of an immune-related signature for predicting prognosis in patients with pancreatic ductal adenocarcinoma. Front Oncol. 2021;10: 618215.
Xiao Z, Li J, Yu Q, Zhou T, Duan J, Yang Z, et al. An inflammatory response related gene signature associated with survival outcome and gemcitabine response in patients with pancreatic ductal adenocarcinoma. Front Pharmacol. 2021;12: 778294.
Borst J, Ahrends T, Bąbała N, Melief CJ, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–47.
Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17(9):527–40.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12:1–15.
Osipov A, Saung MT, Zheng L, Murphy AG. Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer. 2019;7:1–12.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:1–30.
Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. 2014;5:127.
Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47(2):323–38.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Can Res. 2013;73(3):1128–41.
Miao X, Leng X, Zhang Q. The current state of nanoparticle-induced macrophage polarization and reprogramming research. Int J Mol Sci. 2017;18(2):336.
Molawi K, Sieweke MH. Transcriptional control of macrophage identity, self-renewal, and function. Adv Immunol. 2013;120:269–300.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–43.
Sugimoto M, Mitsunaga S, Yoshikawa K, Kato Y, Gotohda N, Takahashi S, et al. Prognostic impact of M2 macrophages at neural invasion in patients with invasive ductal carcinoma of the pancreas. Eur J Cancer. 2014;50(11):1900–8.
Gonçalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2015;111(1):14.1.1.1.6.
He X, Chen X, Li B, Ji J, Chen S. FAK inhibitors induce cell multinucleation and dramatically increase pro-tumoral cytokine expression in RAW 264.7 macrophages. FEBS Lett. 2017;591(23):3861–71.
Simmons DP, Nguyen HN, Gomez-Rivas E, Jeong Y, Jonsson AH, Chen AF, et al. SLAMF7 engagement superactivates macrophages in acute and chronic inflammation. Sci Immunol. 2022;7(68):eabf2846.
Valilou SF, Keshavarz-Fathi M, Silvestris N, Argentiero A, Rezaei N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018;39:46–61.
Javadrashid D, Baghbanzadeh A, Derakhshani A, Leone P, Silvestris N, Racanelli V, et al. Pancreatic cancer signaling pathways, genetic alterations, and tumor microenvironment: The barriers affecting the method of treatment. Biomedicines. 2021;9(4):373.
Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014;5:75.
Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Ahmadpour S, Habibi MA, Ghazi FS, Molazadeh M, Pashaie MR, Mohammadpour Y. The effects of tumor-derived supernatants (TDS) on cancer cell progression: a review and update on carcinogenesis and immunotherapy. Cancer Treat Res Commun. 2024;40: 100823.
Tugues S, Honjo S, König C, Noguer O, Hedlund M, Botling J, et al. Genetic deficiency in plasma protein HRG enhances tumor growth and metastasis by exacerbating immune escape and vessel abnormalization. Cancer Res. 2012;72(8):1953–63.
Nielsen SR, Schmid MC. Macrophages as key drivers of cancer progression and metastasis. Mediators Inflamm. 2017;2017:9624760.
Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, et al. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57(6):630–6.
Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000;192(2):150–8.
Tazzyman S, Murdoch C, Yeomans J, Harrison J, Muthana M. Macrophage-mediated response to hypoxia in disease. Hypoxia (Auckl). 2014;2:185–96.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8.
Venneri MA, Palma MD, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood J Am Soc Hematol. 2007;109(12):5276–85.
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci Landmark. 2008;13(2):453–61.
Schmieder A, Michel J, Schönhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Seminars in cancer biology. Elsevier; 2012.
Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood. 2006;107(5):2112–22.
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–37.
Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol. 2000;165(5):2665–70.
Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol. 2020;10:3038.
Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, et al. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128(4):887–96.
Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife. 2014;3: e04177.
Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 2015;5(9):932–43.
Kadioglu E, De Palma M. Cancer metastasis: perivascular macrophages under watch. Cancer Discov. 2015;5(9):906–8.
Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity. 2012;37(6):1050–60.
Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, et al. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol. 2012;367(2):100–13.
Andersen LB, Nørgaard M, Rasmussen M, Fredsøe J, Borre M, Ulhøi BP, et al. Immune cell analyses of the tumor microenvironment in prostate cancer highlight infiltrating regulatory T cells and macrophages as adverse prognostic factors. J Pathol. 2021;255(2):155–65.
Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–3.
Gabrilovich D. Fatal attraction: how macrophages participate in tumor metastases. J Exp Med. 2015;212(7):976.
Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9.
Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L, et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9(5):453.
Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18(5):549–60.
Hao N-B, Lü M-H, Fan Y-H, Cao Y-L, Zhang Z-R, Yang S-M. Macrophages in tumor microenvironments and the progression of tumors. J Immunol Res. 2012;2012: 948098.
Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat. 2015;23:55–68.
Mutgan AC, Besikcioglu HE, Wang S, Friess H, Ceyhan GO, Demir IE. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol Cancer. 2018;17(1):66.
Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241–55.
Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53.
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62.
Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The role of macrophages in cancer development and therapy. Cancers. 2021;13(8):1946.
Voissiere A, Gomez-Roca C, Chabaud S, Rodriguez C, N'Kodia A, Berthet J, et al. The CSF-1R inhibitor Pexidartinib impacts dendritic cell differentiation through inhibition of FLT3 signaling and may antagonize the effect of durvalumab in patients with advanced cancer–results from a phase 1 study. medRxiv. 2023. 2002–23.
Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428–37.
Lin C-C. Clinical development of colony-stimulating factor 1 receptor (CSF1R) inhibitors. J Immunother Precis Oncol. 2021;4(2):105–14.
Lamb YN. Pexidartinib: first approval. Drugs. 2019;79(16):1805–12.
Evaluation of safety and activity of an anti-PDL1 antibody (DURVALUMAB) combined with CSF-1R TKI (PEXIDARTINIB) in patients with metastatic/advanced pancreatic or colorectal cancers (MEDIPLEX): US National Library of Medicine. https://clinicaltrials.gov/study/NCT02777710.
A study of BLZ945 single agent or BLZ945 in combination with PDR001 in advanced solid tumors: US National Library of Medicine. https://clinicaltrials.gov/study/NCT02829723.
Lin C-C, Gil-Martin M, Bauer TM, Naing A, Lim DWT, Sarantopoulos J, et al. Abstract CT171: phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 2020;80(16):CT171-CT.
Pilot study with CY, pembrolizumab, GVAX, and IMC-CS4 (LY3022855) in patients with borderline resectable adenocarcinoma of the pancreas: US national library of medicine. https://clinicaltrials.gov/study/NCT03153410.
Safety and efficacy study of AMG 820 and pembrolizumab combination in select advanced solid tumor cancer: US National Library of Medicine. https://clinicaltrials.gov/study/NCT02713529.
Razak ARA, Cleary JM, Moreno V, Boyer M, Aller EC, Edenfield W, et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cncer. 2020;8(2): e001006.
Hiraoka K, Zenmyo M, Watari K, Iguchi H, Fotovati A, Kimura YN, et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 2008;99(8):1595–602.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904.
Yang X, Lin J, Wang G, Xu D. Targeting proliferating tumor-infiltrating macrophages facilitates spatial redistribution of CD8+ T cells in pancreatic cancer. Cancers. 2022;14(6):1474.
Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann N Y Acad Sci. 2021;1499(1):18–41.
Sotsios Y, Ward SG. Phosphoinositide 3-kinase: a key biochemical signal for cell migration in response to chemokines. Immunol Rev. 2000;177:217–35.
Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–42.
Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9(5):319–25.
Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543(7645):428–32.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80.
Van Dalen FJ, Van Stevendaal MHME, Fennemann FL, Verdoes M, Ilina O. Molecular repolarisation of tumour-associated macrophages. Molecules. 2018;24(1):9.
Liu C-Y, Xu J-Y, Shi X-Y, Huang W, Ruan T-Y, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. 2013;93(7):844–54.
Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44.
Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, et al. FcRγ activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17(2):121–34.
Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan XL. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget. 2016;7(31):50735–54.
Adair GM, Nairn RS, Wilson JH, Seidman MM, Brotherman KA, ClinicalTrials.gov, et al. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Proc Natl Acad Sci USA. 1989;86:4574–8.
Michaelis KA, Norgard MA, Zhu X, Levasseur PR, Sivagnanam S, Liudahl SM, et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat Commun. 2019;10(1):4682.
Li H, Somiya M, Kuroda S. Enhancing antibody-dependent cellular phagocytosis by Re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials. 2021;268: 120601.
Pilch Z, Tonecka K, Braniewska A, Sas Z, Skorzynski M, Boon L, et al. Antitumor activity of TLR7 is potentiated by CD200R antibody leading to changes in the tumor microenvironment. Cancer Immunol Res. 2018;6(8):930–40.
Figueiredo P, Lepland A, Scodeller P, Fontana F, Torrieri G, Tiboni M, et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomater. 2021;133:231–43.
Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. 2010;14(12):2803–15.
Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Hegmans J, et al. Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br J Cancer. 2010;103(5):629–41.
Lv J, Chen F-K, Liu C, Liu P-J, Feng Z-P, Jia L, et al. Zoledronic acid inhibits thyroid cancer stemness and metastasis by repressing M2-like tumor-associated macrophages induced Wnt/β-catenin pathway. Life Sci. 2020;256: 117925.
He Z, Wang J, Zhu C, Xu J, Chen P, Jiang X, et al. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. Cancer Lett. 2022;548: 215751.
La Fleur L, Boura VF, Alexeyenko A, Berglund A, Pontén V, Mattsson JSM, et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int J Cancer. 2018;143(7):1741–52.
La Fleur L, Botling J, He F, Pelicano C, Zhou C, He C, et al. Targeting MARCO and IL37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Can Res. 2021;81(4):956–67.
Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15(9):2000–11.
Shi B, Chu J, Huang T, Wang X, Li Q, Gao Q, et al. The scavenger receptor MARCO expressed by tumor-associated macrophages are highly associated with poor pancreatic cancer prognosis. Front Oncol. 2021;11: 771488.
Sarhan D, Eisinger S, He F, Bergsland M, Pelicano C, Driescher C, et al. Targeting myeloid suppressive cells revives cytotoxic anti-tumor responses in pancreatic cancer. iScience. 2022;25(11): 105317.
Cao S, Saw PE, Shen Q, Li R, Liu Y, Xu X. Reduction-responsive RNAi nanoplatform to reprogram tumor lipid metabolism and repolarize macrophage for combination pancreatic cancer therapy. Biomaterials. 2022;280: 121264.
Zhang K, Li Y-J, Peng L-J, Gao H-F, Liu L-M, Chen H. M2 macrophage-derived exosomal miR-193b-3p promotes progression and glutamine uptake of pancreatic cancer by targeting TRIM62. Biol Direct. 2023;18(1):1–14.
Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19(1):76–84.
Chen H-M, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Investig. 2018;128(12):5647–62.
Study of MK-4830 as monotherapy and in combination with pembrolizumab (MK-3475) in participants with advanced solid tumors (MK-4830-001): US National Library of Medicine. https://clinicaltrials.gov/study/NCT03564691.
Gao Q, Mo S, Han C, Liao X, Yang C, Wang X, et al. Comprehensive analysis of LILR family genes expression and tumour-infiltrating immune cells in early-stage pancreatic ductal adenocarcinoma. IET Syst Biol. 2023;17:39–57.
Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11(3):130–5.
Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci. 2015;112(45):E6215–23.
Dheilly E, Majocchi S, Moine V, Didelot G, Broyer L, Calloud S, et al. Tumor-directed blockade of CD47 with bispecific antibodies induces adaptive antitumor immunity. Antibodies. 2018;7(1):3.
Tarhriz V, Bandehpour M, Dastmalchi S, Ouladsahebmadarek E, Zarredar H, Eyvazi S. Overview of CD24 as a new molecular marker in ovarian cancer. J Cell Physiol. 2019;234(3):2134–42.
Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572(7769):392–6.
Zhou Z, Li Y, Kuang M, Wang X, Jia Q, Cao J, et al. The CD24+ cell subset promotes invasion and metastasis in human osteosarcoma. EBioMedicine. 2020;51: 102598.
Mirhashemi M, Ghazi N, Saghravanian N, Taghipour A, Mohajertehran F. Evaluation of CD24 and CD44 as cancer stem cell markers in squamous cell carcinoma and epithelial dysplasia of the oral cavity by q-RT-PCR. Dent Res J. 2020;17(3):208.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9.
Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21(11):799–820.
He H, Tu X, Zhang J, Acheampong DO, Ding L, Ma Z, et al. A novel antibody targeting CD24 and hepatocellular carcinoma in vivo by near-infrared fluorescence imaging. Immunobiology. 2015;220(12):1328–36.
Evers M, Ten Broeke T, Jansen JHM, Nederend M, Hamdan F, Reiding KR, et al. Novel chimerized IgA CD20 antibodies: improving neutrophil activation against CD20-positive malignancies. Taylor & Francis; 2020.
CD24Fc with ipilimumab and nivolumab to decrease irAE (CINDI) (CINDI): US National Library of Medicine. https://clinicaltrials.gov/study/NCT04060407.
Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578–88.
Study of SHR2150 (TLR7 Agonist) in combination with chemotherapy plus PD-1 or CD47 antibody in subjects with unresectable/metastatic solid tumors: US National Library of Medicine. https://clinicaltrials.gov/study/NCT04588324.
Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):588–602.
Jing W, Guo X, Wang G, Bi Y, Han L, Zhu Q, et al. Breast cancer cells promote CD169+ macrophage-associated immunosuppression through JAK2-mediated PD-L1 upregulation on macrophages. Int Immunopharmacol. 2020;78: 106012.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–24.
Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, et al. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol. 2016;18(7):790–802.
van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol. 2000;67(1):2–17.
Hoves S, Ooi C-H, Wolter C, Sade H, Bissinger S, Schmittnaegel M, et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med. 2018;215(3):859–76.
Perry CJ, Muñoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J Exp Med. 2018;215(3):877–93.
A Phase I study of the chimeric anti-CD40 monoclonal antibody ChiLob 7/4 to treat advanced malignancies refractory to conventional anti-cancer treatment: US National Library of Medicine. https://clinicaltrials.gov/study/NCT01561911.
Phase 1 study of preoperative gemcitabine plus CP-870, 893 followed by addition of CP-870,893 to standard -of-care adjuvant chemoradiation for patients with newly diagnosed resectable pancreatic carcinoma: US National Library of Medicine. https://clinicaltrials.gov/study/NCT01456585.
Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–95.
Safety and efficacy of APX005M with gemcitabine and nab-paclitaxel with or without nivolumab in patients with previously untreated metastatic pancreatic adenocarcinoma: US National Library of Medicine. https://clinicaltrials.gov/study/NCT03214250.
O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021;22(1):118–31.
Study of neo-adjuvant RO7009789 alone or neo-adjuvant RO7009789 plus nab-paclitaxel and gemcitabine followed by adjuvant RO7009789 plus nab-paclitaxel and gemcitabine for patients with newly diagnosed resectable pancreatic carcinoma: US National Library of Medicine. https://clinicaltrials.gov/study/NCT02588443.
Kulkarni A, Chandrasekar V, Natarajan SK, Ramesh A, Pandey P, Nirgud J, et al. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat Biomed Eng. 2018;2(8):589–99.
Harrer DC, Doerrie J, Schaft N. Chimeric antigen receptors in different cell types: new vehicles join the race. Hum Gene Ther. 2018;29(5):547–58.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53.
Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Can Res. 2021;81(5):1201–8.
CAR-macrophages for the treatment of HER2 overexpressing solid tumors: US National Library of Medicine. https://clinicaltrials.gov/study/NCT04660929.
Macanas-Pirard P, Quezada T, Navarrete L, Broekhuizen R, Leisewitz A, Nervi B, et al. The CCL2/CCR2 axis affects transmigration and proliferation but not resistance to chemotherapy of acute myeloid leukemia cells. PLoS ONE. 2017;12(1): e0168888.
Teng K-Y, Han J, Zhang X, Hsu S-H, He S, Wani NA, et al. Blocking the CCL2–CCR2 axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther. 2017;16(2):312–22.
Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112–23.
Pienta KJ, Machiels J-P, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31:760–8.
Monti P, Leone BE, Marchesi F, Balzano G, Zerbi A, Scaltrini F, et al. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: regulation of expression and potential mechanisms of antimalignant activity. Can Res. 2003;63(21):7451–61.
Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19(13):3404–15.
Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 axis in cancer progression. Cancers. 2020;12(7):1765.
Singh SK, Mishra MK, Eltoum IEA, Bae S, Lillard JW Jr, Singh R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep. 2018;8(1):1323.
Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4-dependent immunosuppressive Ly6Clow monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci. 2017;114(39):10455–60.
A study of the safety and efficacy of single-agent carlumab (an anti-chemokine ligand 2 [CCL2]) in participants with metastatic castrate-resistant prostate cancer: US National Library of Medicine. https://clinicaltrials.gov/study/NCT00992186.
S0916, MLN1202 in treating patients with bone metastases: US National Library of Medicine. https://clinicaltrials.gov/study/NCT01015560.
FOLFIRINOX Plus PF-04136309 in patients with borderline resectable and locally advanced pancreatic adenocarcinoma: US National Library of Medicine. https://clinicaltrials.gov/study/NCT01413022.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62.
Phase 1b study of CCX872-B in patients with pancreatic adenocarcinoma: US National Library of Medicine. https://clinicaltrials.gov/study/NCT02345408.
Linehan D, Noel MS, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: correlation with blood monocyte counts. Am Soc Clin Oncol. 2018. https://doi.org/10.1200/JCO.2018.36.5_suppl.92.
Dixit A, Sarver A, Zettervall J, Huang H, Zheng K, Brekken RA, et al. Targeting TNF-α–producing macrophages activates antitumor immunity in pancreatic cancer via IL-33 signaling. JCI insight. 2022;7(22): e153242.
Tan MCB, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–55.
Zhang X, Haney KM, Richardson AC, Wilson E, Gewirtz DA, Ware JL, et al. Anibamine, a natural product CCR5 antagonist, as a novel lead for the development of anti-prostate cancer agents. Bioorg Med Chem Lett. 2010;20(15):4627–30.
A Study of BMS-813160 in combination with chemotherapy or nivolumab in participants with advanced solid tumors: US National Library of Medicine. https://clinicaltrials.gov/study/NCT03184870.
Ipilimumab, maraviroc and nivolumab in advanced metastatic colorectal and pancreatic cancer the LUMINESCENCE Trial: US National Library of Medicine. https://clinicaltrials.gov/study/NCT04721301.
Haag GM, Halama N, Springfeld C, Grün B, Apostolidis L, Zschaebitz S, et al. Combined PD-1 inhibition (Pembrolizumab) and CCR5 inhibition (Maraviroc) for the treatment of refractory microsatellite stable (MSS) metastatic colorectal cancer (mCRC): First results of the PICCASSO phase I trial. Am Soc Clin Oncol 2020.
Whole brain radiation therapy with standard temozolomide chemo-radiotherapy and plerixafor in treating patients with glioblastoma: US National Library of Medicine. https://clinicaltrials.gov/study/NCT03746080.
Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26(6):878–85.
Byeon S, du Toit-Thompson T, Gillson J, Gill AJ, Samra JS, Mittal A, et al. Heterogeneous tumor microenvironment in pancreatic ductal adenocarcinoma: an emerging role of single-cell analysis. Cancer Med. 2023;12(17):18020–31.
Krieger TG, Le Blanc S, Jabs J, Ten FW, Ishaque N, Jechow K, et al. Single-cell analysis of patient-derived PDAC organoids reveals cell state heterogeneity and a conserved developmental hierarchy. Nat Commun. 2021;12(1):5826.
Juiz N, Elkaoutari A, Bigonnet M, Gayet O, Roques J, Nicolle R, et al. Basal-like and Classical cells coexistence in pancreatic cancer revealed by single cell analysis. bioRxiv. 2020:2020.01. 07.897454.
Ryu S, Lee EK. The pivotal role of macrophages in the pathogenesis of pancreatic diseases. Int J Mol Sci. 2024;25(11):5765.
Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol. 2019;12:1–18.
Chen K, Wang Q, Liu X, Wang F, Ma Y, Zhang S, et al. Single cell RNA-seq identifies immune-related prognostic model and key signature-SPP1 in pancreatic ductal adenocarcinoma. Genes. 2022;13(10):1760.
Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47(10):1168–78.
Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Carcinogenesis GYRI, Wandmacher AM, Mehdorn A-S, Sebens S. The heterogeneity of the tumor microenvironment as essential determinant of development, progression and therapy response of pancreatic cancer. Cancers. 2021;13(19):4932.
Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17(4):500–3.
Zhao L, Zhao H, Yan H. Gene expression profiling of 1200 pancreatic ductal adenocarcinoma reveals novel subtypes. BMC Cancer. 2018;18:1–13.
Lomberk G, Blum Y, Nicolle R, Nair A, Gaonkar KS, Marisa L, et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat Commun. 2018;9(1):1978.
Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet. 2020;52(2):231–40.
Evan T, Wang VMY, Behrens A. The roles of intratumour heterogeneity in the biology and treatment of pancreatic ductal adenocarcinoma. Oncogene. 2022;41(42):4686–95.
Hidalgo M, Cascinu S, Kleeff J, Labianca R, Löhr J-M, Neoptolemos J, et al. Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology. 2015;15(1):8–18.
Barugola G, Partelli S, Marcucci S, Sartori N, Capelli P, Bassi C, et al. Resectable pancreatic cancer: who really benefits from resection? Ann Surg Oncol. 2009;16:3316–22.
Hishinuma S, Ogata Y, Tomikawa M, Ozawa I, Hirabayashi K, Igarashi S. Patterns of recurrence after curative resection of pancreatic cancer, based on autopsy findings. J Gastrointest Surg. 2006;10:511–8.
Taboada AGM, Lominchar PL, Roman LM, García-Alfonso P, Martin AJM, Rodriguez JAB, et al. Advances in neoadjuvant therapy for resectable pancreatic cancer over the past two decades. Ann Hepato-biliary-pancreatic Surg. 2021;25(2):179.
Banks S, Hong W, Degeling K, Shapiro J, Thomson B, Ko H, et al. Impact of neoadjuvant FOLFIRINOX over upfront resection in borderline resectable pancreatic cancer—an international, multicentre, real-world analysis. ESMO Real World Data Digital Oncol. 2024;3: 100022.
Heinrich S, Besselink M, Moehler M, Van Laethem J-L, Ducreux M, Grimminger P, et al. Opinions and use of neoadjuvant therapy for resectable, borderline resectable, and locally advanced pancreatic cancer: international survey and case-vignette study. BMC Cancer. 2019;19:1–9.
Wang S, Yang Y, Ma P, Huang H, Tang Q, Miao H, et al. Landscape and perspectives of macrophage -targeted cancer therapy in clinical trials. Mol Ther Oncolytics. 2022;24:799–813.
Meng F, Li W, Li C, Gao Z, Guo K, Song S. CCL18 promotes epithelial-mesenchymal transition, invasion and migration of pancreatic cancer cells in pancreatic ductal adenocarcinoma. Int J Oncol. 2015;46(3):1109–20.
Liu Y, Jing W, Zhang J, Chen C, Gao L, Shi C, et al. In situ MUC1-specific CAR engineering of tumor-supportive macrophages stimulates tumoricidal immunity against pancreatic adenocarcinoma. Nano Today. 2023;49: 101805.
Guo Y, Wu H, Xiong J, Gou S, Cui J, Peng T. miR-222-3p-containing macrophage-derived extracellular vesicles confer gemcitabine resistance via TSC1-mediated mTOR/AKT/PI3K pathway in pancreatic cancer. Cell Biol Toxicol. 2023;39(4):1203–14.
Khabipov A, Trung DN, van der Linde J, Miebach L, Lenz M, Erne F, et al. CCR4 blockade diminishes intratumoral macrophage recruitment and augments survival of syngeneic pancreatic cancer-bearing mice. Biomedicines. 2023;11(6):1517.
Hu W-M, Liu S-Q, Zhu K-F, Li W, Yang Z-J, Yang Q, et al. The ALOX5 inhibitor Zileuton regulates tumor-associated macrophage M2 polarization by JAK/STAT and inhibits pancreatic cancer invasion and metastasis. Int Immunopharmacol. 2023;121: 110505.
Qiu J, Feng M, Yang G, Cao Z, Liu Y, You L, et al. mTOR inhibitor, gemcitabine and PD-L1 antibody blockade combination therapy suppresses pancreatic cancer progression via metabolic reprogramming and immune microenvironment remodeling in Trp53flox/+ LSL-KrasG12D/+ Pdx-1-Cre murine models. Cancer Lett. 2023;554: 216020.
Menjivar RE, Nwosu ZC, Du W, Donahue KL, Hong HS, Espinoza C, et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. Elife. 2023;12: e80721.
Liao Z, Ye L, Li T, Jin X, Lin X, Fei Q, et al. Tissue-resident CXCR4+ macrophage as a poor prognosis signature promotes pancreatic ductal adenocarcinoma progression. Int J Cancer. 2023;152(11):2396–409.
Zhao Y, Shen M, Wu L, Yang H, Yao Y, Yang Q, et al. Stromal cells in the tumor microenvironment: accomplices of tumor progression? Cell Death Dis. 2023;14(9):587.
Acknowledgements
The authors would like to thank Urmia University of Medical Sciences for the support.
Funding
The authors have not received any funding from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
PL, MD, NN, and MAH wrote the manuscript MA, BRJ, and SA reviewed and revised the manuscript. SA and ST, are corresponding authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lorestani, P., Dashti, M., Nejati, N. et al. The complex role of macrophages in pancreatic cancer tumor microenvironment: a review on cancer progression and potential therapeutic targets. Discov Onc 15, 369 (2024). https://doi.org/10.1007/s12672-024-01256-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01256-x