
Abstract
Objective
Gastric cancer (GC) is one of the most common malignancies worldwide and it is considered the fourth most common cause of cancer death. This study aimed to find critical genes/pathways in GC pathogenesis to be used as biomarkers or therapeutic targets.
Methods
Differentially expressed genes were explored between human gastric cancerous and noncancerous tissues, and Gene Ontology and Kyoto Encyclopedia of Genes and Genomes signaling pathway enrichment analyses were done. Hub genes were identified based on the protein–protein interaction network constructed in the STRING database with Cytoscape software. The hub genes were selected for further investigation using GEPIA2 and DrugBank databases.
Results
Ten overexpressed hub genes in GC were identified in the current study, including FN1, TP53, IL-6, CXCL5, ELN, ADAMTS2, WISP1, MMP2, CTGF, and THBS1. The study demonstrated the PI3K-Akt pathway's central involvement in GC, with pronounced alterations in essential components. Survival analysis revealed significant correlations between CTGF, FN1, IL-6, THBS1, and WISP1 overexpression and reduced overall survival times in GC patients.
Conclusion
A mutual interplay emerged, where PI3K-Akt signaling could upregulate certain genes, forming feedback loops and intensifying cancer phenotypes. The interconnected overexpression of genes and the PI3K-Akt pathway fosters gastric tumorigenesis, suggesting therapeutic potential. DrugBank analysis identified limited FDA-approved drugs, advocating for further exploration while targeting these hub genes could reshape GC treatment. The identified genes could be novel diagnostic/prognostic biomarkers or potential therapeutic targets for GC, but further clinical validation is required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
With a poor prognosis, gastric cancer (GC) is the fourth most frequent malignancy worldwide and the fifth most common cause of cancer-related mortality [1]. There have been multiple studies investigating the connection between different risk factors and the development of GC, but the exact molecular network processes are yet to be fully understood [2]. Understanding the molecular mechanisms involved in the development of GC can help in finding potential targets for early detection and classification, which may improve patient survival rates.
Carcinogenesis is a complex process that entails numerous genetic and epigenetic alterations [3]. The most common type of GC is adenocarcinoma, accounting for about 95% of cases [4]. Numerous studies have been conducted on the epigenetic and genetic changes in various types of GC, with a particular focus on changes in oncogenes, tumor suppressor genes, DNA repair genes, and cell cycle regulators [5]. Investigating dysregulated genes in various pathways can help in understanding the molecular pathophysiological mechanisms of carcinogenesis and developing new therapeutic approaches. High-throughput techniques for gene and network analysis can help in detecting and classifying cancer, predicting patient response to treatment, and determining disease prognosis. These tools have become increasingly important in cancer research and could significantly contribute to the development of personalized cancer treatments [6, 7]. A more comprehensive understanding of diseases and personalized treatment approaches can be achieved through the use of systems biology techniques, which can help in the identification of gene signatures and the clarification of underlying mechanisms [8]. By utilizing these kinds of systems-level observations, it becomes feasible to generate computational models of biological pathways that govern the development and progression of tumors with greater accuracy [9, 10].
Several investigations have been carried out on the altered genes in GC [11,12,13,14]. However, these studies are still inadequate in providing a complete understanding of the molecular causes of GC [15], and further studies are needed to achieve this objective. The GEO database (Gene Expression Omnibus) is a highly valuable open-source databank in the case of gene expression data. This database contains both array-based and sequence-based data. The experiments can be downloaded, and gene expression profiles can be curated [16]. With a specific focus on probable key genes that can be considered as contributing factors to gastric carcinogenesis, the current study examines the gene expression alterations in GC using bioinformatics tools to determine the crucial genes as well as their different pathways that could be affected in the pathogenesis of GC.
2 Materials and methods
2.1 Dataset selection
In whole GEO database, including 848 series of human gastric cancer, was searched to find datasets that fulfill these conditions: (1) data based on expression profiling by microarray, (2) affiliated projects on gastric cancer, (3) evaluation of both the normal and cancerous tissues, (4) the minimum sample size of two, and (5) CELL as the supplementary filter. The GSE54129 dataset was considered the preferred database which contained a total of 132 specimens, including 111 gastric tumor tissues as well as 21 gastric normal tissues, surgically resected from GC patients.
2.2 Selection of differentially expressed genes (DEGs)
GEO2R was used for the selection of the differentially expressed genes between gastric cancerous and normal tissues in the GSE54129 dataset. GEO2R is a web-based tool that provides users with the evaluation of the GEO Series [16]. A web-based program is applied in this tool that employs the Bioconductor packages [17], limma in R [18], and GEOQuery [19], with a default method of the Benjamini–Hochberg false-discovery rate method [20]. Adjusted p-value < 0.05 and log fold-change (LogFC) between − 1.5 and 1.5 were considered as filters for the selection of the differentially expressed genes. For further evaluation of the overexpressed genes, LogFC was set to > 1.5.
2.3 Network reconstruction and key genes selection
STRING is a database of protein interactions. The interactions among the significantly overexpressed genes were evaluated in STRING v.11.5 (http://string-db.org), and the PPI list was prepared. The PPI list was then inserted into Cytoscape (v.3.9.1), a platform to visualize the PPI networks. CytoHubba software (v.0.1) was chosen for identifying key proteins in the whole network [21]. Four different topological methods (MNC, DMNC, MCC, and Degree) were applied to illustrate the importance of the nodes in the biological network. The top four proteins were proposed as key nodes based on the mentioned four methods. Using the CytoHubba plugin, the subnetwork was finally drawn showing the interactions among hub nodes.
2.4 Enrichment analysis of the key genes
Gene ontology (GO) enrichment analysis, counting biological process (BP), molecular function (MF), and cellular components (CC), as well as the KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis was done using STRING [22].
2.5 Gene network clustering
CytoCluster (v.2.1.0) as a Cytoscape plugin was applied for gene network clustering. The IPCA (Identifying Protein Complex Algorithm) algorithm was selected for cluster analysis of the subnetwork. The threshold was set to 0.8, and the list of the genes in all clusters was imported into STRING for further analysis to achieve a better understanding of these genes' involvement in KEGG pathways.
2.6 in silico survival analysis
To identify the prognostic potential of the identified key genes, the Kaplan–Meier survival curves of GC patients with expression changes in these genes were extracted from GEPIA2 (Gene Expression Profiling Interactive Analysis) (http://gepia2.cancer-pku.cn).
2.7 Drug target analysis
The five key genes that showed decreased overall survival times of GC patients (p-value < 0.05) were investigated in DrugBank (https://go.drugbank.com/) to uncover the approved drugs targeting these hub genes.
3 Results
3.1 Selection of differentially expressed genes (DEGs)
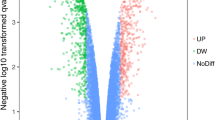
There were two groups in this study, gastric cancerous and normal tissues. Based on the analysis using GEO2R (adjusted p-value < 0.05 and |LogFC|> 1.5) the DEGs were selected. Figure 1 shows the results of GEO2R analysis on the selected dataset including the Volcano plot, Venn diagram, UMAP plot, and Boxplot of DEGs between human gastric cancerous and noncancerous tissues. A total of 2005 DEGs, including 1035 up-regulated and 970 down-regulated genes were identified in this study. To find drug targets, further analysis was done on the up-regulated genes.

Volcano plot, Venn diagram, UMAP plot, and Boxplot of DEGs between human gastric cancerous and noncancerous tissues. a Volcano plot of the DEGs identified in gastric cancer tissues; Red: up-regulated genes, Blue: down-regulated genes, Black: no difference. b Venn diagram of the DEGs identified in GC tissues showing 2733 identified DEGs with adjusted p-value < 0.05. c UMAP plot of the human gastric cancerous and noncancerous tissues showing a complete difference between these two groups. d Boxplot of the human gastric cancerous and noncancerous tissues
3.2 Network reconstruction and key genes selection
The upregulated genes (LogFC > 1.5) were selected in this study to be analyzed more precisely with the STIRING online tool and the Cytoscape platform. To identify key proteins in the biological network, we utilized the CytoHubba software (v.0.1). We applied four different topological methods (MNC, DMNC, MCC, and Degree) to determine the importance of the nodes. Based on these methods, the top four proteins were identified as key nodes. We then visualized the interactions among these hub nodes by drawing a subnetwork using the CytoHubba plugin. Figure 2 shows the PPI networks of the key genes. Based on four topological methods, the top four proteins were identified as key nodes. Ten key genes identified in this study based on the previously mentioned four topological methods are introduced in Table 1.

PPI network of the key genes. The overexpressed genes (LogFC > 1.5) in GC and their known neighbors
3.3 Enrichment analysis of the key genes
GO analysis is one of the most practical and known methods to illustrate typical biological aspects of high-throughput genome or transcriptome data. The enrichment analysis results are shown in Figs. 3, 4, 5, 6.

Gene Ontology enrichment analysis (biological process) of the determined subnetwork of hub genes in GC using STRING v.11.5 (http://string-db.org)

Gene Ontology enrichment analysis (Cellular Component) of the determined subnetwork of hub genes in GC using STRING v.11.5 (http://string-db.org)

Gene Ontology enrichment analysis (Molecular Function) of the hub genes subnetwork in GC using STRING

KEGG pathways enrichment analysis of the hub genes of the subnetwork in GC using STRING v.11.5 (http://string-db.org)
GO terms that are commonly observed in at least 50% of genes for BP (Fig. 3) are highly enriched in various areas, including cellular and biological processes, response to stimuli, organismal and anatomical development, regulation of metabolic processes, and cell communication.
As shown in Fig. 4, the most common GO terms for CC were enriched in various regions such as cellular anatomical entity, intracellular, organelle, membrane-bounded organelle, cytoplasm. Extracellular region, cell periphery, plasma membrane, vesicle, and endomembrane system among others.
As shown in Fig. 5, the predominant GO terms found for MF are significantly enriched in binding, ion binding, protein binding, cation binding, metal ion binding, molecular function regulator, anion binding, signaling receptor binding, carbohydrate derivative binding, identical protein binding, protein-containing complex binding, and enzyme binding.
According to KEGG pathway enrichment analysis (Fig. 6), the significant enriched pathways include PI3K-Akt focal adhesion, signaling pathways, pathways in cancer, and human papillomavirus infection.
3.4 Gene network clustering
To find the functional modules in the reconstructed subnetwork cluster analysis was done using the CytoCluster tool with the IPCA algorithm. Two-hundreds and seventeen clusters were found in the cluster analysis process of the subnetwork. Clusters with ranks 1 to 4 were selected to be discussed in this paper (Fig. 7).

PPI network of the genes in clusters rank 1 to 4 using CytoCluster (v.2.1.0)
As illustrated in Table 2, the PI3K-Akt signaling pathway, focal adhesion, protein digestion and absorption, proteoglycans in cancer, human papillomavirus infection, pathways in cancer, ECM-receptor interaction, AGE-RAGE signaling pathway in diabetic complications, Relaxin signaling pathway, and microRNAs in cancer were the common pathways among all ranks.
3.5 in silico survival analysis
The Kaplan–Meier overall survival curves of patients with GC are shown in Fig. 8. These curves indicate that overexpression of CTGF, FN1, IL-6, THBS1, and WISP1 is associated with decreased overall survival times of GC patients (p-value < 0.05).

The Kaplan–Meier overall survival curves of patients with GC based on differential expression of the ten hub genes identified in this study were extracted from the Gene Expression Profiling Interactive Analysis (GEPIA2) database. Kaplan–Meier estimates found that overexpression of (b) CTGF, (e) FN1, (f) IL-6, (h) THBS1, and (j) WISP1 was significantly associated with decreased overall survival times of GC patients (p-value < 0.05). The other panels (a) ACTB, (c) CXCL5, (d) ELN, (g) MMP2, and (i) TP53 show no statistically significant association with overall survival (p-value > 0.05)
3.6 Drug target analysis
Five hub genes (CTGF, FN1, IL-6, THBS1 and WISP1) significantly associated with decreased overall survival times of GC patients (p-value < 0.05) were searched in DrugBank (https://go.drugbank.com/) to find the drugs that target these genes. Currently, the FDA has not authorized any medications, targeting CTGF, THBS1, or WISP1. However, as shown in Table 3, a few approved drugs do target FN1 and IL-6, such as Ocriplasmin (targeting FN1), Foreskin fibroblast, Binimetinib, and Siltuximab (targeting IL-6). Among these approved drugs, only Siltuximab has a single target (IL-6), while the other drugs have multiple targets. A list of the drugs that target these genes is provided in Error! Reference source not found.
4 Discussion
In recent years, different investigations in multi-center genomics research from gene to systems level and next-generation sequencing, clarified the various mechanisms specifically involved in tumor progression [23]. This study used bioinformatics analysis to further investigate the genes involved in GC pathogenesis. In our study, we focused on up-regulated genes in gastric cancer to identify potential targets for drug repurposing, aiming to utilize existing approved drugs for novel therapeutic strategies. This approach is motivated by the potential to significantly reduce the time and cost of drug development, leveraging the overexpression of certain genes in the tumor environment that are pivotal for cancer progression. By concentrating on genes that are likely overexpressed and play crucial roles in cell proliferation, survival, invasion, and metastasis, our goal was to disrupt key signaling pathways and biological processes essential for tumor growth. This strategy also allows for the expedited clinical application of our findings, harnessing the safety and efficacy data of already approved pharmacological inhibitors to target these overexpressed genes.
Ten hub genes were identified in this study, including FN1, CTGF, CXCL5, IL6, ELN, ADAMTS2, MMP2, TP53, WISP1, and THBS1. The relationship between the overexpression of these genes and GC is an area of active scientific investigation. These genes have been implicated in various molecular and cellular pathways relevant to cancer initiation, progression, and metastasis.
CXCL5 is a protein that influences immune responses and angiogenesis, playing a significant role in the progression and metastasis of GC, particularly by enhancing tumor spread and being associated with advanced disease stages [24,25,26]. The presence of immune cells and inflammation in the microenvironment can potentially contribute to the development of GC, promoting tumor growth and metastasis [27]. Up-regulation of ELN, which encodes the elastin protein, may affect tissue integrity, elasticity, and angiogenesis, potentially impacting the invasive behavior of GC cells [28]. ADAMTS2, involved in extracellular matrix remodeling, might influence tumor cell invasion and angiogenesis in GC [29]. MMP2, a matrix metalloproteinase, has implications in extracellular matrix degradation, which is essential for tumor invasion and metastasis in GC [30]. The preservation of genomic stability depends greatly on the TP53 gene, which functions as a tumor suppressor. Mutations or dysregulation of TP53 are commonly associated with GC, contributing to uncontrolled cell growth and reduced DNA repair [31]. In conclusion, the overexpression of these genes is interconnected with various cellular processes and molecular pathways relevant to GC. However, the specific roles of each gene in the context of GC initiation, progression, and metastasis are complex and multifaceted. Further comprehensive research is essential to elucidate the precise contributions of these genes, potentially leading to the development of targeted therapeutic strategies and the identification of biomarkers for GC management.
The results of our study showed that the PI3K-Akt signaling pathway is the prominent pathway affected in GC. The PI3K-Akt pathway is commonly disrupted in GC, with multiple essential components showing alterations. Activation of this pathway leads to enhanced tumor growth, survival, metastasis, angiogenesis, and resistance to treatments in GC [32]. The primary genomic changes causing abnormal activation of the PI3K-Akt pathway involve PIK3CA amplifications/mutations, loss of PTEN, and the overexpression of proteins that activate chemical signaling pathways called receptor tyrosine kinases (such as EGFR, HER2, cMET) [33]. Clinical investigations have linked active PI3K-Akt signaling to aggressive disease, advanced tumor stages, metastasis, and unfavorable prognoses in patients with GC. Experimental research shows that blocking the PI3K-Akt signaling pathway can reduce the proliferation of gastric cancer cells, trigger apoptosis, and increase their sensitivity to chemotherapy and radiotherapy [34]. Preliminary results from early-stage trials of PI3K/Akt/mTOR inhibitors, either alone or combined, have demonstrated potential efficacy against advanced GC [35]. Noteworthy challenges encompass treatment-related toxicity, acquired resistance, and the subset of patients who respond positively to PI3K pathway inhibition, thus emphasizing the need to explore predictive biomarkers.
This study showed that overexpression of the CTGF, FN1, IL-6, THBS1, and WISP1 genes correlate with decreased survival rates in individuals diagnosed with GC. Further investigations are warranted to elucidate their potential as therapeutic targets. CTGF, a growth factor involved in fibrosis and tissue remodeling, plays a role in the development of gastric cancer (GC) by affecting the tumor microenvironment, angiogenesis, and cell proliferation. Its overexpression in GC enhances tumor growth, invasion, and chemotherapy resistance, largely through activating the PI3K-Akt signaling pathway, a process driven by CTGF’s direct binding to PI3K’s p85 subunit [36, 37]. FN1, an essential component of the extracellular matrix, is linked to enhanced tumor cell adhesion, migration, and invasion, thus increasing the metastatic capability of cancer cells, including in GC [38]. This is supported by bioinformatics analyses by Sun et al., who identified FN1 as a crucial gene in GC pathogenesis [39], aligning with our findings. The upregulation of FN1 activates FAK/Src signaling in GC cells, which in turn interacts with and intensifies PI3K-Akt signaling, further promoting cellular migration and invasion processes [40]. In addition, IL-6 overexpression in GC triggers the GP130/JAK/STAT3 cascade, leading to the activation of PI3K-Akt signaling, which confers increased advantages in cell proliferation and survival [41]. As a pro-inflammatory cytokine, IL6 can activate various signaling pathways that contribute to cell survival, proliferation, and immune modulation in GC, potentially playing a significant role in tumor progression [42]. THBS1, an adhesive glycoprotein, is involved in angiogenesis and cell–matrix interactions, and its overexpression may contribute to tumor angiogenesis and metastasis in GC [43]. Its upregulation in GC leads to PI3K-Akt signaling activation through integrin-mediated pathways, fostering increased tumor growth and resistance to therapy [44]. WISP1, which regulates the Wnt signaling pathway, may have a part in encouraging cell growth and movement in GC, which could help in the progression of the tumor [45]. WISP1 partially activates the PI3K-Akt/mTOR axis, consequently intensifying proliferation, invasion, and angiogenesis [46]. Importantly, a reciprocal relationship emerges, wherein PI3K-Akt signaling can upregulate the expression of certain genes among this group, forming positive feedback loops that reinforce cancer-related phenotypes.
The overexpression of these genes and activation of the PI3K-Akt pathway are interconnected in promoting gastric tumorigenesis and progression through their abilities to upregulate each other and mediate downstream oncogenic effects. Targeting this signaling connection may have therapeutic potential in GC. The results of our investigation into these genes using DrugBank indicate that there are currently no drugs approved by the FDA targeting CTGF, THBS1, or WISP1. However, a few approved drugs do target FN1 and IL-6, such as Ocriplasmin (targeting FN1), Foreskin fibroblast, Binimetinib, and Siltuximab (targeting IL-6). These drugs are suggested for further investigation as potential new therapeutic options for EC patients. Among these approved drugs, only Siltuximab has a single target (IL-6), indicating specific action and potentially fewer side effects. Therefore, Siltuximab could be considered a suitable choice for EC patients.
The exploration of pharmacotherapeutic agents targeting hub genes holds promise for personalized treatment strategies. While limited FDA-approved drugs targeting FN1 and IL6 were identified, the lack of approved agents for CTGF, THBS1, and WISP1 underscores an unmet therapeutic need. The multitarget nature of the drugs introduced in this study necessitates in-depth evaluation to assess potential off-target effects. Our results support the need for further research to investigate the underlying mechanisms of the key hub genes and their related pathways identified in our study. The integration of multi-omics data, coupled with experimental validation, could shed light on their roles in GC initiation, progression, and therapeutic response. Developing targeted therapies addressing these hub genes could potentially revolutionize treatment strategies for GC patients. Despite the comprehensive nature of our study, limitations such as the reliance on bioinformatics predictions and the lack of experimental validation merit consideration. Future investigations should encompass clinical validation to confirm the roles of these hub genes in GC pathogenesis and treatment response.
We understand that gastric cancer exhibits spatiotemporal heterogeneity, which makes genetic analysis challenging due to its diverse pathological types. However, our study aimed to identify common molecular pathways and hub genes that are overexpressed across a wide range of gastric cancer samples. By focusing on these commonalities, we aimed to provide insights that can be applied to multiple subtypes of the disease, potentially guiding more personalized therapeutic approaches in the future. Despite the limitations imposed by this heterogeneity, we believe that our findings contribute to a foundational understanding of gastric cancer's molecular biology. They can serve as a valuable resource for further research aimed at unraveling the disease's complex genetic landscape and informing the development of targeted treatments.
5 Conclusion
The study employed bioinformatics analysis to unveil ten hub genes—FN1, CTGF, CXCL5, IL6, ELN, ADAMTS2, MMP2, TP53, WISP1, and THBS1—in GC pathogenesis, expanding the understanding of their overexpression and its implications across critical molecular and cellular pathways for cancer initiation, progression, and metastasis. The study demonstrated the PI3K-Akt pathway’s central involvement in GC, frequently disrupted in GC, with pronounced alterations in essential components. Survival analysis revealed significant correlations between CTGF, FN1, IL-6, THBS1, and WISP1 overexpression and reduced overall survival times in GC patients. A mutual interplay emerged, where PI3K-Akt signaling could upregulate certain genes, forming feedback loops and intensifying cancer phenotypes. The interconnected overexpression of genes and the PI3K-Akt pathway fosters gastric tumorigenesis, suggesting therapeutic potential. DrugBank analysis identified limited FDA-approved drugs, advocating for further exploration while targeting these hub genes could reshape GC treatment. While comprehensive, the study acknowledges limitations such as bioinformatics reliance and calls for additional clinical validation. We understand the complexity of gastric cancer's heterogeneity, but our study was focused on identifying common molecular pathways and hub genes across various samples. This approach aimed to offer insights applicable to multiple subtypes, potentially aiding the development of personalized treatments. Despite the challenges of heterogeneity, we believe our findings provide a valuable foundation for further research into the molecular biology of gastric cancer and the advancement of targeted therapies.
6 Limitation section
Our study on identifying prognostic and therapeutic targets in gastric cancer, while offering valuable insights, is subject to limitations including reliance on a single dataset, which may not fully represent the genetic diversity of gastric cancer worldwide. The inherent heterogeneity of gastric cancer, with its varied pathological subtypes, poses additional challenges to the generalizability of our findings. Furthermore, the bioinformatics approach necessitates experimental validation to confirm the therapeutic potential of the identified genes and pathways. Thus, while our findings contribute to the foundational understanding of gastric cancer’s molecular landscape, they should be interpreted with caution, serving as a basis for future, more detailed investigations.
Data availability
The data supporting the conclusions of the study are all provided in the manuscript. The GSE54129 dataset is extracted from the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
Code availability
Not applicable.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin. 2021;71(3):209–49.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635–48.
Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99.
Boussioutas A, Li H, Liu J, Waring P, Lade S, Holloway AJ, Taupin D, Gorringe K, Haviv I, Desmond PV. Distinctive patterns of gene expression in premalignant gastric mucosa and gastric cancer. Can Res. 2003;63(10):2569–77.
Nagini S. Carcinoma of the stomach: a review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J Gastrointest Oncol. 2012;4(7):156.
Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5(10):588–99.
Tavassoly I, Hu Y, Zhao S, Mariottini C, Boran A, Chen Y, Li L, Tolentino RE, Jayaraman G, Goldfarb J, et al. Genomic signatures defining responsiveness to allopurinol and combination therapy for lung cancer identified by systems therapeutics analyses. Mol Oncol. 2019;13(8):1725–43.
Mehrpooya A, Saberi-Movahed F, Azizizadeh N, Rezaei-Ravari M, Saberi-Movahed F, Eftekhari M, Tavassoly I. High dimensionality reduction by matrix factorization for systems pharmacology. Brief Bioinform. 2021. https://doi.org/10.1093/bib/bbab410.
Dorvash M, Farahmandnia M, Tavassoly I. A systems biology roadmap to decode mTOR control system in cancer. Interdiscip Sci: Comput Life Sci. 2020;12(1):1–11.
Tavassoly I, Parmar J, Shajahan-Haq A, Clarke R, Baumann W, Tyson J. Dynamic modeling of the interaction between autophagy and apoptosis in mammalian cells. CPT: Pharmacom Syst Pharmacol. 2015;4(4):263–72.
Mottaghi-Dastjerdi N, Ghorbani A, Montazeri H, Guzzi PH. A systems biology approach to pathogenesis of gastric cancer: gene network modeling and pathway analysis. BMC Gastroenterol. 2023;23(1):248.
Mottaghi-Dastjerdi N, Soltany-Rezaee-Rad M, Sepehrizadeh Z, Roshandel G, Ebrahimifard F, Setayesh N. Gene expression profiling revealed overexpression of vesicle amine transport protein-1 (VAT-1) as a potential oncogene in gastric cancer. New Delhi: NISCAIR-CSIR; 2016.
Mottaghi-Dastjerdi N, Soltany-Rezaee-Rad M, Sepehrizadeh Z, Roshandel G, Ebrahimifard F, Setayesh N. Identification of novel genes involved in gastric carcinogenesis by suppression subtractive hybridization. Hum Exp Toxicol. 2015;34(1):3–11.
Mottaghi-Dastjerdi N, Soltany-Rezaee-Rad M, Sepehrizadeh Z, Roshandel G, Ebrahimifard F, Setayesh N. Genome expression analysis by suppression subtractive hybridization identified overexpression of Humanin, a target gene in gastric cancer chemoresistance. DARU J Pharm Sci. 2014;22:1–7.
Lin Y-H, Lim S-N, Chen C-Y, Chi H-C, Yeh C-T, Lin W-R. Functional role of mitochondrial DNA in cancer progression. Int J Mol Sci. 2022;23(3):1659.
Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93–110.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80.
Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004. https://doi.org/10.2202/1544-6115.1027.
Davis S, Meltzer PS. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–7.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B. 1995;57(1):289–300.
Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2010;27(3):431–2.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–52.
Chong X, Peng R, Sun Y, Zhang L, Zhang Z. Identification of key genes in gastric cancer by bioinformatics analysis. BioMed Res Int. 2020. https://doi.org/10.1155/2020/7658230.
Zhang W, Wang H, Sun M, Deng X, Wu X, Ma Y, Li M, Shuoa SM, You Q, Miao L. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun. 2020;40(2–3):69–80.
Chen C, Xu Z-Q, Zong Y-P, Ou B-C, Shen X-H, Feng H, Zheng M-H, Zhao J-K, Lu A-G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019;10(3):178.
Zhou Z, Xia G, Xiang Z, Liu M, Wei Z, Yan J, Chen W, Zhu J, Awasthi N, Sun X, et al. A C-X-C chemokine receptor type 2–dominated cross-talk between tumor cells and macrophages drives gastric cancer metastasis. Clin Cancer Res. 2019;25(11):3317–28.
Deng J, Jiang R, Meng E, Wu H. CXCL5: a coachman to drive cancer progression. Front Oncol. 2022;12:944494.
Fang T, Zhang L, Yin X, Wang Y, Zhang X, Bian X, Jiang X, Yang S, Xue Y. The prognostic marker elastin correlates with epithelial–mesenchymal transition and vimentin-positive fibroblasts in gastric cancer. J Pathol: Clin Res. 2023;9(1):56–72.
Jiang C, Zhou Y, Huang Y, Wang Y, Wang W, Kuai X. Overexpression of ADAMTS-2 in tumor cells and stroma is predictive of poor clinical prognosis in gastric cancer. Hum Pathol. 2019;84:44–51.
Łukaszewicz-Zając M, Mroczko B, Szmitkowski M. Gastric cancer—the role of matrix metalloproteinases in tumor progression. Clin Chim Acta. 2011;412(19–20):1725–30.
Busuttil RA, Zapparoli GV, Haupt S, Fennell C, Wong SQ, Pang J-MB, Takeno EA, Mitchell C, Di Costanzo N, Fox S. Role of p53 in the progression of gastric cancer. Oncotarget. 2014;5(23):12016.
Matsuoka T, Yashiro M. The role of PI3K/Akt/mTOR signaling in gastric carcinoma. Cancers. 2014;6(3):1441–63.
Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol. 2016;2(12):1565–73.
Ying J, Xu Q, Liu B, Zhang G, Chen L, Pan H. The expression of the PI3K/AKT/mTOR pathway in gastric cancer and its role in gastric cancer prognosis. Onco Targets Ther. 2015. https://doi.org/10.2147/OTT.S88592.
Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol. 2019;30(10):x12–20.
Sang Q, Dai W, Yu J, Chen Y, Fan Z, Liu J, Li F, Li J, Wu X, Hou J. Identification of prognostic gene expression signatures based on the tumor microenvironment characterization of gastric cancer. Front Immunol. 2022;13:983632.
Mao Z, Ma X, Rong Y, Cui L, Wang X, Wu W, Zhang J, Jin D. Connective tissue growth factor enhances the migration of gastric cancer through downregulation of E-cadherin via the NF-κB pathway. Cancer Sci. 2011;102(1):104–10.
Zhou Y, Cao G, Cai H, Huang H, Zhu X. The effect and clinical significance of FN1 expression on biological functions of gastric cancer cells. Cell Mol Biol. 2020;66(5):191–8.
Sun C, Yuan Q, Wu D, Meng X, Wang B. Identification of core genes and outcome in gastric cancer using bioinformatics analysis. Oncotarget. 2017;8(41):70271.
Wang H, Zhang J, Li H, Yu H, Chen S, Liu S, Zhang C, He Y. FN1 is a prognostic biomarker and correlated with immune infiltrates in gastric cancers. Front Oncol. 2022;12:918719.
Ni Y, Low JT, Silke J, O’Reilly LA. Digesting the role of JAK-STAT and cytokine signaling in oral and gastric cancers. Front Immunol. 2022;13:835997.
Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2021;33(3):127–48.
Zhang S, Xiang X, Liu L, Yang H, Cen D, Tang G. Bioinformatics analysis of hub genes and potential therapeutic agents associated with gastric cancer. Cancer Manag Res. 2021. https://doi.org/10.2147/CMAR.S341485.
Zhang X, Huang T, Li Y, Qiu H. Upregulation of THBS1 is related to immunity and chemotherapy resistance in gastric cancer. Int J Gen Med. 2021;14:4945–57.
Jia S, Qu T, Feng M, Ji K, Li Z, Jiang W, Ji J. Association of Wnt1-inducible signaling pathway protein-1 with the proliferation, migration and invasion in gastric cancer cells. Tumor Biol. 2017;39(6):1010428317699755.
Lei ZN, Teng QX, Tian Q, Chen W, Xie Y, Wu K, Zeng Q, Zeng L, Pan Y, Chen ZS, et al. Signaling pathways and therapeutic interventions in gastric cancer. Signal Transduct Target Ther. 2022;7(1):358.
Acknowledgements
Our sincere appreciation goes out to the Iran University of Medical Sciences for their support. This article is extracted from a thesis with the ethical code of IR.IUMS.REC.1401.699.
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
N.M.D. and M.S.R.R. made the concept. N.M.D. and F.K. and H.M. and B.G. provide the Data curation. N.M.D. and F.K. conducted the investigation and formal analysis. N.M.D. supervised the manuscript. N.M.D. and F.K. wrote the original draft of the manuscript. A.G., and H.M., and F.Y., and M.S.R.R., and S.S. reviewed and edited the manuscript. N.M.D. and F.K. prepared the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable to this study.
Consent for publication
Not applicable to this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khoshdel, F., Mottaghi-Dastjerdi, N., Yazdani, F. et al. CTGF, FN1, IL-6, THBS1, and WISP1 genes and PI3K-Akt signaling pathway as prognostic and therapeutic targets in gastric cancer identified by gene network modeling. Discov Onc 15, 344 (2024). https://doi.org/10.1007/s12672-024-01225-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01225-4