
Abstract
Bladder cancer (BLCA) is one of the most frequent urothelium carcinoma, but with poor prognosis due to lack of reliable predictive biomarkers. Metabolic reprogramming involving in various nutrients, and is reported to be closely associated with malignant progression in BLCA. With the use of transcriptome sequencing data profiles of 349 patients from The Cancer Genome Atlas, we established a three-gene glycolysis-related signature to predict the prognosis of BLCA patients. Our signature constructed on the basis of AK3, GALK1 and NUP205 expression, detail features and interactions between these three genes were further explored. We established a nomogram by integrating clinical variables and the risk score. Glycolytic level and proliferation ability were detected to study the role and mechanisms of NUP205 on BLCA. The connections between three genes in our signature were independent. We found our signature gains more value for patients with highly malignant stage. The established nomogram also confirmed that the signature had a eligible clinically predict capacity. After inhibited NUP205 expression, we found the glycolysis level of BLCA cells decreased and proliferation ability suppressed, mainly through AMPK signaling pathway inactivation. Collectively, our study explored a three-gene glycolysis-related signature that predict the prognosis of patients with BLCA, and highlights NUP205 as a potential therapeutic target for inhibiting glycolytic processes and proliferation in BLCA cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Bladder cancer (BLCA) represents 7% of all cancer occurrences and 3% of all cancer-related deaths [1]. Patients with BLCA are divided into non-muscle invasive bladder cancer (NMIBC) and muscle invasive bladder cancer (MIBC) according to pathological grade. For BLCA especially MIBC, which is characterized by a high stage of invasion, metastasis and recurrence, the prognosis is still not optimistic, although there are constantly emerging diverse treatment methods and an evolving establishment of molecular pathology types [2, 3]. The reason for the poor prognosis might be attributed to the genomic, cellular heterogeneity of cancer cells and several environmental factors [4]. Therefore, it is crucial to develop an effective prediction model with the use of prognostic biomarkers to distinguish those patients at high risk and make a preliminary prognosis prediction.
Malignant tumors are not only genetic diseases but also energy metabolism diseases. The malignant behaviors of tumors, particularly solid tumors such as BLCA, typically consume more nutrients than necessary and divert metabolites to support biosynthetic platforms or alter metabolic patterns to adjust the tumor microenvironment, which often consists of hostile factors that result in cell starvation, inflammation, and hypoxia [5,6,7]. This process is also known as metabolic reprogramming. There are three main sources of nutrients, and metabolic reprogramming involving these nutrients has been found to be closely associated with malignant progression and poor prognosis in BLCA and other malignant tumors. It was reported that fatty acid metabolic reprogramming via the alteration of related gene expression, which could have an effect on metastatic progression and be associated with poor prognosis in human cancers [8]. Lee et al. [9] found that adjustment of lipid metabolism is essential for the establishment of cisplatin resistance in BLCA. Amino acids also play a crucial role in cancer and support the growth and reproduction of cancer cells. Several studies have discovered that, abnormal conversion or overexpression of some enzymes that correlate with amino acid metabolism, such as LAT1, is associated with poor patient prognosis and significant progression in BLCA [10,11,12]. The conversion of oxidative phosphorylation into glycolysis, even in the presence of sufficient oxygen is referred to as the Warburg effect, which is associated with malignant progression in most cancers [13]. There is a closely relationship between aerobic glycolysis and the malignant progression, therefore glycolysis-related genes might be potential predictors in prognostic prediction of BLCA. Tian et al. [14] established a three-gene glycolytic signature for risk stratification and prognosis prediction in pancreatic ductal adenocarcinoma. Liu et al. [15] also identified nine glycolysis-related genes that can predict the prognosis of endometrial cancer patients. Our group has successfully constructed a four glycolysis-related gene signature as a prognostic model, based on 19 pairs of BLCA patient samples and their adjacent normal tissue samples [16].
Here, we sought to identify a more representative prediction model, that may correlate with metabolic reprogramming based on the BLCA dataset from The Cancer Genome Atlas (TCGA), and we further divided BLCA patients with different clinical characteristics into matched subgroups, to improve the accuracy of our prediction model. Overall, we successfully established a three-gene glycolysis-related signature, that can predict and assess the prognosis of BLCA patients. The detail mechanisms and immune infiltration levels about our signature were further explored.
2 Materials and methods
2.1 Data collection
The transcriptome sequencing expression profiles and clinical information were downloaded from the TCGA database as the training cohort. In the aggregate, 349 samples consisting of 331 BLCA and 18 healthy samples were obtained for further study. The GEO dataset GSE13507 consisting of 165 BLCA patients was used as an external validation cohort, which was downloaded from the Gene Expression Omnibus database [17]. The details of the included cohorts’ clinical characteristics are listed in Table 1.
2.2 Differential expression analysis
The Limma algorithm was employed to extract the differentially expressed genes between BLCA and normal tissues. A significance threshold of adjusted P < 0.05 and |logFC|≥ 1 was applied to determine significant differences. The implementation of this algorithm was carried out using R software.
2.3 Gene set enrichment analysis (GSEA)
GSEA software 3.0 was used to identify the hallmark gene sets of different metabolic pathways, to predict the relevant biological processes in 331 bladder cancer and 18 normal samples from the TCGA cohort. The hallmark gene sets including “ACID AMINO ACID LIGASE ACTIVITY”, “GO FATTY ACID BETA OXIDATION”, “HALLMARK GLYCOLYSIS” and “REACTOME GLYCOLYSIS” were obtained from the Molecular Signatures Database. Gene set permutations were performed 1000 times to obtain a normalized enrichment score (NES) in each analysis. Normalized enrichment score (NES) > 1.6 and P < 0.05 were set as the cutoff and were used to select the core genes.
2.4 Establishment of a metabolism-related gene signature as a prognostic model
A univariate Cox regression analysis was used to screen out prognosis-associated genes related to metabolism. A hazard ratio (HR) < 1 represents good overall survival (OS) outcomes, and a HR > 1 represents poor OS outcomes. We chose genes with P < 0.05 as prognosis-related metabolic genes and utilized them for the multivariate Cox regression analysis. There were 6 glycolysis-related genes primary identified by univariate Cox analysis that were closely related to the overall survival (OS) of BLCA patients. Subsequently, multivariate Cox regression analysis was employed and a signature of three glycolysis-related genes were created, genes with P < 0.05 were regarded as independent prognosis factors. The risk score formula was as follows: \(\left(\text{RiskScore}\right)={\sum }_{\text{i}=1}^{\text{n}}\left(\text{Expi}*\text{Coei}\right)\) [18]. In this formula, “n” is the number of prognostic genes, “Exip” is the expression of gene i, and “Coei” is the regression coefficient of the corresponding gene obtained by the multivariate Cox regression model. BLCA patients were divided into high-risk or low-risk subgroups by the median risk score.
2.5 Validation of the efficacy of the prognostic three-gene glycolysis-related signature
The prognostic three-gene glycolysis-related signature was tested in the external validation cohort and in the two cohorts combined. Patients in the validation cohort and the whole cohort were divided into high- or low-risk groups depending on the median risk score obtained from the training cohort. Kaplan–Meier curve analysis, 3- and 5-year survival time-dependent ROC analysis, and risk scores for each patient were generated for each clinical characteristic, and the stratified analysis of BLCA patients was performed using the “survival” and “survminer” packages of R software.
2.6 Protein–protein interaction (PPI) network construction
We used the GeneMANIA (http://www.genemania.org/) website to predict the interactions and functions of the genes, build the network, and identify hub genes in the regulatory network, including physical interaction and coexpression.
2.7 Estimation of clinical independence and construction of the nomogram
We eliminated BLCA patients who lacked exact clinicopathological information, such as TNM stage or American Joint Committee on Cancer (AJCC) stage, from the training cohort. We utilized the “rms” R package (version 3.5.1) to integrate the clinical features with the risk scores obtained with our three-gene glycolysis-related signature, to construct a nomogram for further clinical prediction. Then, we built calibration plots and time-dependent receiver operating characteristic (ROC) curves to evaluate the performance of our nomogram.
2.8 Immune cells infiltration analysis
The R software’s CIBERSORT algorithm was employed to determine the profile of Tumor-Infiltrating Immune Cells (TIICs) (including 22 immune cells) in all tumor samples of training cohort. The relationship of TIICs and risk score were analyzed by limma package. We detected the correlation of each gene in our signature with levels of immune cell infiltration in BLCA using the TIMER database.
2.9 Cell culture and treatment
BLCA cells (T24) were obtained from American Type Culture Collection (Manassas, VA). Normal treated groups were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, MA) at 37℃ in 5% CO2. Small interfering RNAs (siRNAs) targeting human NUP205 or scrambled negative control (si-NC) designed and synthetized by GenePharma (Shanghai, China) (sequences listed in Table 2), transfected into the cell lines with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions.
2.10 Quantitative real‐time polymerase chain reaction
Total RNA was drawn out from T24 cells WITH the use of TRIzol (Invitrogen, California, USA). PrimeScript RT reagent kit (TaKaRa, Osaka, Japan) were used to reverse‐transcribe RNA to cDNA. Real-time qPCR was performed with SYBR Green (TaKaRa) on an ABI 7500 Real‐Time PCR System (Applied Biosystems), the entire process lasted for 40 cycles following the manufacturer’s instructions. Each sample was repeated three times detection. Data were standardized to β‐actin using the 2−ΔΔCt method. The primer sequences used for the real-time qPCR are in Table 2.
2.11 Immunoblots
Total protein was drawn out using radioimmunoprecipitation assay lysis buffer (Beyotime, China) containing 1 mmol/L phenylmethylsulfonyl fluoride (PMSF) and 1 mmol/L β-Glycerophosphate sodium salt hydrate (Selleck, Houston, TX). The concentration of protein samples was detected by a BCA protein quantitative kit (Beyotime, China). 10% sodium dodecyl sulfate‐polyacrylamide gel was chosen for total protein separation and transferred to nitrocellulose membranes (Millipore, USA). Membranes were blocked by skimmed milk for 2 h at room temperature and then incubated with primary antibodies including: anti-nup205 (1:2000, ab157090; Abcam), anti-β-actin (1:5000, 20536-1-AP; Proteintech), anti-Cdk1 (1:3000, 19532-1-AP; Proteintech), anti-AMPKα (1:1000, A5008; Selleck), anti-phospho-AMPKα (S496) (1:1000, A5194; Selleck), anti-HIF-1α (1:2000, K009156P; Solarbio). These antibodies were added and incubated overnight at 4 ℃. Then bands washed by Tris‐buffered saline three times and enhanced chemiluminescence reagents (Millipore, USA) were used to assess protein expression, which were normalized to the corresponding bands for β-actin.
2.12 Cell proliferation and viability
Cell proliferation was demonstrated by Cell Counting Kit-8 (CCK8, Boster Biotechnology). Cells transfected different sequences were inoculated onto 96-well plates at the initial density of 5000 cells per well. Then CCK8 reagent was added 10 μL to each well at 0, 24, 48 and 72 h. After 1 h incubation at 37 ℃, absorbance values of different groups were measured at 450 nm.
2.13 Lactate production and glucose consumption measurement
Lactate and glucose concentrations in the culture supernatants were detected using Lactate Assay kit (Solarbio, Beijing, China) and Glucose Assay kit (Solarbio) respectively, according to the manufacturer’s instructions and absorbance values were measured at the corresponding absorbance. The results were normalized by the number of cells in each sample in the culture plates, lactate production and glucose consumption were calculated by comparison with normal medium.
2.14 Statistical analysis
The data was processed via PERL programming langue. GraphPad Prism 8 software and R software were used for statistical analyses and demonstration. Analysis of variance, Mann–Whitney test or students T test were used for different statistical comparisons. P < 0.05 was considered statistically significant.
3 Results
3.1 Identification metabolism-related genes using GSEA
The flow chart of the entire study is shown in Fig. 1. In the aggregate 349 samples including 331 BLCA samples and 18 healthy samples with their clinical feature information were obtained from the TCGA database as the training cohort, and 165 BLCA samples were retrieved from the GEO database as the external validation cohort (Table 1). We integrated mRNA expression profiles and clinical information. The limma algorithm was used to identify the mRNAs that were differentially expressed in the BLCA samples relative to the healthy samples in the training cohort. There were 132 upregulated and 33 downregulated differentially expressed genes in the BLCA samples compared with the healthy samples by setting adjusted P < 0.05 and |logFC|≥ 1 as the selection criteria. GSEA was used to explore whether there were significant genomic differences between BLCA and normal tissues in different kinds of metabolic phenotypes. Finally, we found that these genes were significantly enriched in amino acid (ACID AMINO ACID LIGASE ACTIVITY), fatty acid (GO FATTY ACID BETA OXIDATION) and glycolysis metabolism. Glycolysis was the most obvious pathway in the two gene sets (HALLMARK GLYCOLYSIS and REACTOME GLYCOLYSIS) (Fig. 2). Then, we analyzed differentially expressed mRNAs and found 151 participating genes, including 109 genes in the HALLMARK_GLYCOLYSIS gene set (NES = 1.71, nominal P < 0.005, FDR < 0.005) and 42 in the REACTOME_GLYCOLYSIS gene set (NES = 1.62, nominal P < 0.05, FDR < 0.05).

Flowchart for construction and validation of the risk model

GSEA of metabolism-related gene sets. Gene set enrichment analysis (GSEA) of four metabolism-related gene sets including a HALLMARK_GLYCOLYSIS, b REACTOME_GLYCOLYSIS, c GO_FATTY_ACID_BETA_OXIDATION, d ACID_AMINO_ACID_LIGASE_ACTIVITY) that differentiated bladder cancer (BLCA) and normal tissues based on The Cancer Genome Atlas (TCGA).
3.2 Construction of glycolysis related genes and BLCA prognosis models
Univariate Cox regression analysis was used to preliminarily investigate the relationship between glycolysis and the prognosis of BLCA patients, and six genes with a cut off P < 0.05 were initially selected. Then, multivariate Cox regression analysis was used to examine the relevance of the expression of these six genes to patient survival. We constructed a prognostic model with three genes, a protective gene (arginine kinase 3, AK3) with HR < 1 and lower expression and two risk genes (galactokinase 1, GALK1 and nucleoporins 205, NUP205) with HR > 1 and higher expression (Fig. 3a). The risk score formula was as follows: − 0.33227* AK3 + 0.462604* GALK1 + 0.539478* NUP205.

Risk score and prognostic analysis of the signature in the TCGA training cohort. a Expression heatmap of the 3-mRNA signature in each patient. b The risk score and c survival status of the prognostic model for the training cohort. d Kaplan–Meier curves of OS rate and e time-dependent ROC curves for the prediction of 3- and 5-year OS rates. f, g Univariate and multivariate Cox regression analyses for the risk score and each clinical characteristic
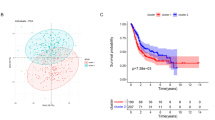
BLCA patients were ranked by their risk scores and split into a high-risk group (n = 165) and a low-risk group (n = 166) using the median risk score of the series (Fig. 3b). As shown in Fig. 3c, a higher death rate was notable for the high-risk group. In the model, the low-risk group had a significantly longer overall survival time as shown by the Kaplan–Meier survival curve and log-rank test (Fig. 3d). Then, we used our prognostic model to perform a ROC curve analysis (Fig. 3e) of the 3- and 5-year survival rate and obtained the area under the receiver operating characteristic curve (AUC) values, which were 0.651 and 0.709, respectively, to verify the sensitivity and specificity of our model for predicting the survival rate of BLCA patients. We used both univariate and multivariate Cox regression analyses to assess whether our prognosis model was an independent predictor of BLCA patient prognosis. The results revealed moderate and independent prognostic power for the risk score (Fig. 3f, g), which further validated the dependability of our previous prognostic model.
3.3 Hierarchical analysis between the clinical characteristics and the three-gene glycolysis-related signature
The results of the univariate and multivariate Cox regression analyses revealed obvious differences between the high- and low-risk groups, that were associated with the risk score, age, clinicopathology and cancer stage according to the AJCC (Fig. 3f, g). Based on our univariate Cox regression analysis results, we used stratification analysis to explore the relationship between overall survival rates and different clinical features. And we found that age and tumor status, including T stage, N stage and M stage and stage according to our three-gene glycolysis-related signature, were significantly related to the survival outcomes as determined by the Kaplan–Meier survival curve and log-rank test (Fig. 4a–e). Next, we classified the patients into different subgroups based on the median risk score identified with our signature according to age (≤ 65 to > 65), T stage (T0-2 to T3-4), N stage (N0 to N1-3), M stage (M0 to M1-3) and AJCC stage (stage I–II to stage III–IV). As shown in the Fig. 4f–j and Figure S1a–e, Kaplan–Meier curves showed that in the training cohort, patients with high-risk scores had a poorer prognosis in the T3-4 subgroup, N0 subgroup, M0 subgroup, higher stage group and older (age > 65) group (P < 0.05) than in the other groups, which suggested that our three-gene glycolysis-related signature may have a better prognostic value for BLCA patients with higher malignancy.

Survival analysis for separate clinical characteristics. Hierarchical survival analysis with clinical characteristics based on Kaplan–Meier analysis of a age, b–d TNM stage and e American Joint Committee on Cancer (AJCC) stage. Survival time of patients in the high-risk and low-risk groups in different subgroups; f age, g T3-4 stage, h N0 stage, i M0 stage and j AJCC stage
3.4 Validation of the three-gene glycolysis-related signature for survival prediction
Next, we verified our prognostic model in the validation cohort and the training mixed validation cohorts to further confirm its reliability. We analyzed the expression levels of the three glycolysis-related genes in both the validation cohort and the combined cohort. GALK1 and NUP205 were expressed at higher levels in the high-risk group, while AK3 was expressed at lower levels (Fig. 5a, b). To validate the survival prediction of the three-gene glycolysis-related signature, risk scores were calculated for each patient in both the validation cohort and the combined cohort. As previously described, BLCA patients were ranked and divided into a high-risk group or a low-risk group using the median risk score from the training cohort as the cutoff point (Fig. 5c, d). A higher death rate was noted for BLCA patients with high-risk scores than for those with low-risk scores in each cohort (Fig. 5e, f). In accordance with our training cohort results, low-risk patients had longer overall survival times in both the validation and overall cohort (Fig. 5g, h) with P < 0.001 in each cohort, and ROC curve analysis also confirmed the independent prognostic power of our model (Fig. 5i, j). Then, we further analyzed the association between our three-gene glycolysis-related signature and the clinicopathological features of BLCA in the validation cohort. The results indicated that our model is notably related to older age, higher clinicopathological stage and AJCC stage, suggesting that the signature may play a crucial role in predicting BLCA malignant progression (Fig. 5k–o). We analyzed the association between various clinical features and survival rates in our validation group. The Kaplan–Meier curves also showed that in the validation cohort, patients with progression had clinicopathological features that were associated with poorer prognosis in the T3-4 subgroup, N1-3 subgroup, M1-3 subgroup, higher stage group and older (age > 65) age group (P < 0.05) (Figure S1f–j), which also confirmed that our model possesses a more accurate prognostic value for higher malignancy BLCA patients.

Validation of the efficacy of the risk model in the validation cohort and entire cohort. a, b The expression heatmap, c, d survival status, e, f risk score, g, h Kaplan–Meier survival, and i, j time-dependent ROC curves of the prognostic model. Stratified analysis of different risk scores with clinical characteristics based on k age, l–n TNM stage and o AJCC stage
3.5 Genetic information of the three glycolysis related genes and validation of the gene expression levels
The frequently altered or mutated sites of the three glycolysis-related genes and their frequencies of mutation were analyzed by cBioPortal software (Fig. 6a, b). The network of AK3, GALK1, NUP205 the neighboring genes they most frequently interact with was constructed by GeneMANIA (Fig. 6c). As shown in the figure, a network of 23 genes, including the three target genes, was automatically generated by GeneMANIA to visualize physical interactions and coexpression. To eliminate the interrelationship of the three hub genes, we analyzed the correlation among the three genes and found that the absolute values of the correlation coefficient among them were all less than 0.3, indicating that there is no correlation among these three genes (Fig. 6d).

The regulatory network and gene expression of the signature. a, b The frequency and site of alteration of the three genes were explored with the use of the cBioPortal database. c From GeneMANIA, we successfully built a protein–protein interaction network including these three genes. d The interaction between the three genes in the dataset from the TCGA was analyzed with the cutoff of 0.3. e–g The expression of these three genes from training cohort BLCA tissue was compared with normal tissues. h, i IHC staining of both AK3 and GALK1 protein in normal urothelial and bladder cancer tissues from the Human Protein Atlas database
Subsequently, we explored the expression patterns of these three genes in both normal and BLCA tissues. Within the training cohort, AK3 exhibited overexpression in normal tissues, while GALK1 and NUP205 demonstrated elevated expression levels in BLCA tissues (Fig. 6e–g). Immunohistochemistry (IHC) staining images were downloaded from the Human Protein Atlas database (https://www.proteinatlas.org/) to verify the expression levels. These images revealed that the expression of AK3 in urothelial normal tissue was stronger than that in urothelial carcinoma tissue, while the intensity of GALK1 expression was the opposite (Fig. 6h, i), which also confirmed that our three-gene glycolysis-related signature comprises both risk factors and protective markers and represents an accurate prognostic model.
3.6 Nomogram establishment and validation
We built a nomogram to predict the 3- and 5-year OS rate and establish a clinical method to predict the survival probability of BLCA patients based on the training cohort. Seven independent predictors, including age, grade, AJCC stage, T stage, N stage, M stage and risk score, were incorporated to build the nomogram (Fig. 7a). The calibration plots indicated satisfactory coherence between the nomogram predictions and actual observations (Fig. 7b, c). The ROC curve analysis also showed that the AUCs of the nomogram for the 3- and 5-year survival rates were 0.707 and 0.76, respectively (Fig. 7d). The proportional hazard assumption analysis was employed to validate data that constructing this nomogram, which was realized by The R software’s “survminer” and “survival” package (Figure S2a–g). Results demonstrated that neither global schoenfeld test (P = 0.8767) nor schoenfeld individual tests P < 0.05, except schoenfeld individual test on age (P = 0.038), indicating that data used to construct the nomogram conform to proportional hazard assumption.

Nomogram establishment and validation. a Nomogram containing clinical factors and risk scores incorporated with the 3-mRNA signature for predicting 3- and 5-year OS rates based on the TCGA dataset. b, c Calibration plots for predicting patient OS at 3 and 5 years. The probability of survival predicted by the nomogram is plotted on the X-axis, while actual survival is plotted on the Y-axis. d Time-dependent ROC curves rely on the nomogram from the training cohort for the 3- and 5-year OS rate
3.7 Functional enrichment and validation of three-gene glycolysis-related signature
Next, we made an enrichment on different expression genes of three-gene glycolysis-related signature with KEGG pathways and GO terms. The KEGG pathways enrichment demonstrated that different expression genes were mainly enriched in metabolic pathways of different substances, AMPK signaling pathway, HIF-1 pathway and so on (Fig. 8a). Next, gene set enrichment analysis (GSEA) was conducted to identify glycolysis-related biological processes of GO terms involved in carcinogenesis. Results demonstrated that material or energetic metabolism, cell cycle process and modification on different substances (NES > 1, P < 0.05) were enriched significantly in high-risk group (Fig. 8b). There were limited reports on NUP205 in BLCA, but several studies indicated its potential regulatory role in cancer cells metabolic reprogramming [16]. In order to investigate the involvement of NUP205 in BLCA, we firstly detected its expression in urothelial cells. After collecting normal urothelial and BLCA tissues from clinical patients, IHC results demonstrated the overexpression of NUP205 in bladder urothelial carcinoma (Fig. 8c). Next, we designed two sequences of siRNA, aimed at suppressing the expression of NUP205. Results indicated that NUP205 expression were significantly inhibited at both transcriptional and translational level by our siRNA-1 and siRNA-2 in T24 cells (Fig. 8d, e). The glucose consumption and lactate production rate were decreased a lot after transfected siRNAs (Fig. 8f, g), indicated the aberrantly glycolysis of T24 cells were significantly inhibited after NUP205 silenced. According to forementioned KEGG and GSEA enrichment results, we further detected the molecular mechanisms that NUP205 regulated. After intervening NUP205 expression, we found HIF-1α were slightly downregulated but not as significantly as AMPK inactivation, Cdk1 expression were also suppressed, suggesting NUP205 could influence the cell cycle through AMPK signaling pathway (Fig. 8h). Based on above results, we chose sequence 2 to testify NUP205 impacts on cell proliferation. The CCK-8 assay demonstrated that the proliferation ability of T24 cells were inhibited a lot by silencing NUP205, in contrast to negative control (Fig. 8i). In summary, we found silencing NUP205 could significantly inhibit the glycolysis process and proliferation of BLCA cells, mainly through AMPK signaling pathway.

Functional enrichment and role of NUP205 validation. a KEGG pathway enrichment results. b Terms of GO-BP enriched in high-risk group including metabolic process, cell cycle, cell cycle process, peptidyl amino acid modification, peptidyl lysine modification, protein sumoylation, pyruvate metabolic process and RNA location (NES > 1, P < 0.05). c IHC staining of NUP205 protein in normal urothelial and BLCA tissues from clinical patient with BLCA. d Real-time qPCR was performed relative expression levels of NUP205 expression in T24 cells transfected siRNAs or not (n = 3, unpaired students T test, Pns > 0.05, P* < 0.05 and P** < 0.01). e Western-blot results demonstrated NUP205 expression in T24 cells transfected siRNAs. f, g Glucose consumption and lactate production ratios were measured in T24 cells after been transfected with different siRNAs, homogenized by cell counting as 104 cells. h Western-blot were employed to detect the expression of HIF-1α, AMPK and CDK1 after intervening NUP205 in T24 cells. i CCK-8 assay was demonstrated the viabilities of T24 cells transfected with negative control or siRNA-2 (n = 3, Mann–Whitney test, P* < 0.05)
3.8 Expression of three-gene glycolysis-related signature is correlated with immune infiltration levels in BLCA
The CIBERSORT algorithm was employed to explore the correlation of our three-gene glycolysis-related signature with TIICs in BLCA. The proportion of each BLCA patient’s TIICs were detected (Fig. 9a). A comparison of the TIIC levels between the high- and low-risk group demonstrated elevated levels of M0 macrophages, neutrophils and decreased levels of regulatory T cells, resting mast cells in the high-risk group (Fig. 9b). Based on the TIMER database, we further detected the correlation of each gene in our signature with levels of immune cell infiltration in BLCA. Results indicated that AK3 (Fig. 9c) was associated with M1 macrophages (r = 0.115, P = 2.67e−02) and neutrophils (r = 0.258, P = 5.06e−07). GALK1 (Fig. 9d) was significantly associated with M0 macrophages (r = 0.228, P = 1.03e−05) and M2 macrophages (r = 0.118, P = 2.32e−02). NUP205 (Fig. 9e) was significantly associated with regulatory T cells (r = 0.16, P = 2.04e−03), M0 macrophages (r = 0.254, P = 8.08e−07), M1 macrophages (r = 0.273, P = 9.98e−08) and resting mast cells (r = 0.218, P = 2.52e−05).

Relationship between risk model and tumor-infiltrating immune cells (TIICs). a Relative percentage of 22 kinds of TIICs infiltrating in high-risk or low-risk group calculated by the CIBERSORT algorithm and demonstrated in the bar plot. b Higher infiltrated levels of TIICs including neutrophils and M0 macrophages were observed in high-risk group (students T test, pns > 0.05, P* < 0.05, P** < 0.01, P*** < 0.001). c Correlation analysis between each gene expression and single immune cell infiltrated levels
4 Discussion
BLCA is a solid tumor and one of the most common malignant tumors. Within the hostile tumor environment, starvation and hypoxia can induce alterations in the metabolic patterns of tumor cells and adjust to tumor microenvironment in a process known as metabolic reprogramming [19, 20]. It is believed that metabolic reprogramming is a hallmark of cancer cells [21]. Several researchers have investigated the mechanism of metabolic reprogramming and its effect on the malignant behaviors of tumors [22, 23]; here, we draw attention to the prognostic role of metabolic reprogramming. After integrating the transcriptional expression data of the TCGA cohort and analyzing enrichment via GSEA of different metabolic gene sets, we determined that glycolysis was most significantly correlated with the prognosis of BLCA. Glycolysis is a common metabolic pathway in cells and is potentially associated with the development of BLCA [24]. In our study, we performed a bioinformatic analysis to identify metabolism-related genes based on the differentially expressed genes between BLCA samples and normal tissues. By focusing on a limited number of genes with biological rationale and robust associations with bladder cancer prognosis, we aimed to develop a parsimonious model that minimizes complexity while maximizing predictive accuracy and clinical utility. We finally developed a three-gene glycolysis-related signature (AK3, GALK1 and NUP205) for BLCA prognosis.
We found that these three genes have been associated with BLCA or other tumors respectively by reviewing the existing literature. The AK3 gene is located on chromosome 9 and mainly functions in the mitochondrial matrix; AK3 has been demonstrated to have a tumor suppressor function in many other solid tumors other than BLCA [25]. Melle et al. [26] found that AK3 could play a tumor suppressive role in hepatocellular carcinoma, and its low expression was associated with tumor development and progression. Chang et al. [27] reported that in urothelial cells, AK3 could help to enhance the sensitivity of BLCA cells to cisplatin, while downregulation of AK3 expression induced by cigarette smoke condensate contributes to the establishment of drug resistance. The suppressive role of AK3 in BLCA was also validated in our study, but further exploration is needed to determine the mechanism of AK3 and what facilitates AK3 expression downregulation. GALK1 is associated with the metabolism of galactose absorbed from digested food, as well as that produced endogenously. Tang et al. [28] discovered the role of GALK1 in hepatocellular carcinoma and reported that inhibition of GALK1 via siRNA could suppress the growth of cancer cells, and they also found that the galactose metabolic and PI3K/AKT pathways potentially interact. However, there have been no reports on the function of GALK1 in BLCA, and there remains enormous potential to characterizes its role as an oncogene. NUP205 is a widely researched oncogene in other cancers that could modulate the timing of mitotic onset [29]. Xiong et al. [30] reported that NUP205 is associated with the clinical outcomes of hepatocellular carcinoma patients via crosstalk between hsa_circ_0088364 and hsa_circ_0090049. But there was little study reported the role NUP205 played and how it works in BLCA. In our study, we found the value of NUP205 not only in prognosis prediction, but also effecting the glycolysis process and proliferation of BLCA cells. While we preliminary found NUP205 functions through AMPK signaling pathway, the detail mechanisms need further exploration.
Glycolysis-related prognostic prediction models based on core glycolysis-related genes with different clinical characteristics have been indicated to serve as reliable prognostic predictors in several solid tumors [14]. Clinical outcome is an important factor on which we focused with respect to, and we determined the possible glycolysis-related genes that correlated with the differentiation of cancer samples from normal tissue to generate a more credible model. To validate the accuracy of our model, we obtained the clinical feature information from the GEO database for using as the external validation cohort.
In the present study, we also found that our model was more valuable for patients with worse clinical characteristics, such as older age or higher malignancy, which was also confirmed in our validation group. Our three-gene glycolysis-related signature could accurately categorize BLCA patients into high- or low-risk subgroups, which accurately represented patient outcomes. We constructed a comprehensive nomogram that incorporated our model with different clinical features, including age, sex, TNM stage and AJCC stage, to effectively predict the survival rate of BLCA patients.
Among three genes in our signature, NUP205 attracted our attention due to its highest risk coefficient as a risk factor, and its role in the malignant progression of bladder cancer still remains unclear. In our study, we found its overexpression in BLCA tissues. After silencing NUP205 transcriptional expression, we noticed the impaired glycolysis process in BLCA cells. For the first time, we confirmed blocking its expression could suppress proliferation of BLCA cells through AMPK signaling pathway.
As exploring the relationship between our three-gene glycolysis-related signature and immune infiltration levels in TME, we also found the elevated levels of M0 macrophages, neutrophils and decreased levels of regulatory T cells, resting mast cells in the high-risk group, indicating the closely relationship between BLCA cells and immune microenvironment. Immune microenvironment plays crucial roles especially in tumor immunotherapy, which holds antitumor immune response by restarting and maintaining the tumor-immune cycle to eliminate tumor cells [31]. Immunotherapy has evolved in recent years and has been proven to treat a variety of cancers, including BLCA. It is of great value to classify patients by promising biomarkers and to administer appropriate immunotherapy to sensitive patients as early as possible. Several studies found different immunocompetent subtypes in BLCA, and specific immunotherapy based on subgroups could obtain more accurate and effective therapeutic outcomes [32]. We have conducted a preliminary exploration of our constructed signature on relevant immune cells, but it is far from enough compared with other related studies in BLCA.
Our three-gene glycolysis-related signature has the potential to guide clinical decision-making, optimize treatment approaches, and improve patient outcomes according to survival analysis and nomogram construction. The integration of our signature into clinical application could enhance prognostic accuracy and facilitate personalized medicine in BLCA management. Beyond prognostication, our findings highlight the intricate interplay between metabolic reprogramming and BLCA progression. Strategies aimed at targeting glycolytic pathways, such as small molecule inhibitors or immunotherapies, may offer new avenues for therapeutic intervention and improved treatment outcomes in BLCA. Our investigation into the immune microenvironment's association with our glycolysis-related signature provides intriguing insights into potential immunotherapeutic strategies in BLCA. However, we acknowledge the limitations of our study. Our study lacks enough clinical cohort to test the efficiency and accuracy of our signature, which should be validated in our future works. Detail mechanisms and more reliable validation need further explored in NUP205 effecting AMPK induced proliferation of BLCA cells, we will conduct these explorations in our future research endeavors.
5 Conclusion
In summary, our study identified a three-gene glycolysis-related signature to predict the prognosis of BLCA patients based on the expression of AK3, GALK1 and NUP205. The signature was validated in an external cohort from GEO database and combined cohort. Our findings reveal novel potential molecular therapeutic targets as there have been few studies on the role of these three genes in BLCA, which also play crucial roles in tumor immune microenvironment regulation. For the first time, we explored NUP205 as an oncogene and involved in the proliferation of BLCA, which might be attributed to its crucial role in glycolysis and cell cycling through AMPK signaling pathway.
Data availability
All data analyzed during this study are included in this published article.
References
Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström PU, Choi W, Guo CC, Lotan Y, Kassouf W. Bladder cancer. Lancet. 2016;388(10061):2796–810.
Dalangood S, Zhu Z, Ma Z, Li J, Zeng Q, Yan Y, Shen B, Yan J, Huang R. Identification of glycogene-type and validation of ST3GAL6 as a biomarker predicts clinical outcome and cancer cell invasion in urinary bladder cancer. Theranostics. 2020;10(22):10078–91.
Lian P, Wang Q, Zhao Y, Chen C, Sun X, Li H, Deng J, Zhang H, Ji Z, Zhang X, et al. An eight-long non-coding RNA signature as a candidate prognostic biomarker for bladder cancer. Aging. 2019;11(17):6930–40.
Meeks JJ, Al-Ahmadie H, Faltas BM, Taylor JA 3rd, Flaig TW, DeGraff DJ, Christensen E, Woolbright BL, McConkey DJ, Dyrskjøt L. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nat Rev Urol. 2020;17(5):259–70.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20.
Lozy F, Karantza V. Autophagy and cancer cell metabolism. Semin Cell Dev Biol. 2012;23(4):395–401.
Wu X, Zhou Z, Xu S, Liao C, Chen X, Li B, Peng J, Li D, Yang L. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett. 2020;478:93–106.
Nath A, Chan C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep. 2016;6:18669.
Lee MY, Yeon A, Shahid M, Cho E, Sairam V, Figlin R, Kim KH, Kim J. Reprogrammed lipid metabolism in bladder cancer with cisplatin resistance. Oncotarget. 2018;9(17):13231–43.
Kaira K, Sunose Y, Arakawa K, Ogawa T, Sunaga N, Shimizu K, Tominaga H, Oriuchi N, Itoh H, Nagamori S, et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br J Cancer. 2012;107(4):632–8.
Maimaiti M, Sakamoto S, Yamada Y, Sugiura M, Rii J, Takeuchi N, Imamura Y, Furihata T, Ando K, Higuchi K, et al. Expression of L-type amino acid transporter 1 as a molecular target for prognostic and therapeutic indicators in bladder carcinoma. Sci Rep. 2020;10(1):1292.
Bacci M, Lorito N, Ippolito L, Ramazzotti M, Luti S, Romagnoli S, Parri M, Bianchini F, Cappellesso F, Virga F, et al. Reprogramming of amino acid transporters to support aspartate and glutamate dependency sustains endocrine resistance in breast cancer. Cell Rep. 2019;28(1):104-118.e8.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9.
Tian G, Li G, Liu P, Wang Z, Li N. Glycolysis-based genes associated with the clinical outcome of pancreatic ductal adenocarcinoma identified by the cancer genome atlas data analysis. DNA Cell Biol. 2020;39(3):417–27.
Liu J, Li S, Feng G, Meng H, Nie S, Sun R, Yang J, Cheng W. Nine glycolysis-related gene signature predicting the survival of patients with endometrial adenocarcinoma. Cancer Cell Int. 2020;20:183.
Zhang C, Gou X, He W, Yang H, Yin H. A glycolysis-based 4-mRNA signature correlates with the prognosis and cell cycle process in patients with bladder cancer. Cancer Cell Int. 2020;20:177.
Riester M, Taylor JM, Feifer A, Koppie T, Rosenberg JE, Downey RJ, Bochner BH, Michor F. Combination of a novel gene expression signature with a clinical nomogram improves the prediction of survival in high-risk bladder cancer. Clin Cancer Res. 2012;18(5):1323–33.
Chen K, He Y, Liu Y, Yang X. Gene signature associated with neuro-endocrine activity predicting prognosis of pancreatic carcinoma. Mol Genet Genomic Med. 2019;7(7): e00729.
Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett. 2015;368(1):7–13.
Xue M, Chen W, Xiang A, Wang R, Chen H, Pan J, Pang H, An H, Wang X, Hou H, et al. Hypoxic exosomes facilitate bladder tumor growth and development through transferring long non-coding RNA-UCA1. Mol Cancer. 2017;16(1):143.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22.
Logotheti S, Marquardt S, Gupta SK, Richter C, Edelhäuser BAH, Engelmann D, Brenmoehl J, Söhnchen C, Murr N, Alpers M, et al. LncRNA-SLC16A1-AS1 induces metabolic reprogramming during bladder cancer progression as target and co-activator of E2F1. Theranostics. 2020;10(21):9620–43.
Giacomini I, Ragazzi E, Pasut G, Montopoli M. The pentose phosphate pathway and its involvement in cisplatin resistance. Int J Mol Sci. 2020;21(3):937.
Tao T, Su Q, Xu S, Deng J, Zhou S, Zhuang Y, Huang Y, He C, He S, Peng M, et al. Down-regulation of PKM2 decreases FASN expression in bladder cancer cells through AKT/mTOR/SREBP-1c axis. J Cell Physiol. 2019;234(3):3088–104.
Qin WY, Feng SC, Sun YQ, Jiang GQ. MiR-96-5p promotes breast cancer migration by activating MEK/ERK signaling. J Gene Med. 2020;22(8): e3188.
Melle C, Ernst G, Scheibner O, Kaufmann R, Schimmel B, Bleul A, Settmacher U, Hommann M, Claussen U, von Eggeling F. Identification of specific protein markers in microdissected hepatocellular carcinoma. J Proteome Res. 2007;6(1):306–15.
Chang X, Ravi R, Pham V, Bedi A, Chatterjee A, Sidransky D. Adenylate kinase 3 sensitizes cells to cigarette smoke condensate vapor induced cisplatin resistance. PLoS ONE. 2011;6(6): e20806.
Tang M, Etokidem E, Lai K. The Leloir pathway of galactose metabolism—a novel therapeutic target for hepatocellular carcinoma. Anticancer Res. 2016;36(12):6265–71.
Hachet V, Busso C, Toya M, Sugimoto A, Askjaer P, Gönczy P. The nucleoporin Nup205/NPP-3 is lost near centrosomes at mitotic onset and can modulate the timing of this process in Caenorhabditis elegans embryos. Mol Biol Cell. 2012;23(16):3111–21.
Xiong DD, Feng ZB, Lai ZF, Qin Y, Liu LM, Fu HX, He RQ, Wu HY, Dang YW, Chen G, et al. High throughput circRNA sequencing analysis reveals novel insights into the mechanism of nitidine chloride against hepatocellular carcinoma. Cell Death Dis. 2019;10(9):658.
Lu X, Meng J, Su L, Jiang L, Wang H, Zhu J, Huang M, Cheng W, Xu L, Ruan X, Yeh S, Liang C, Yan F. Multi-omics consensus ensemble refines the classification of muscle-invasive bladder cancer with stratified prognosis, tumour microenvironment and distinct sensitivity to frontline therapies. Clin Transl Med. 2021;11(12): e601.
Meng J, Lu X, Zhou Y, Zhang M, Ge Q, Zhou J, Hao Z, Gao S, Yan F, Liang C. Tumor immune microenvironment-based classifications of bladder cancer for enhancing the response rate of immunotherapy. Mol Ther Oncolytics. 2021;20:410–21.
Acknowledgements
The authors gratefully acknowledge the assistance of the Department of Urology Surgery, Chongqing Medical University, China.
Publication of clinical datasets
Not applicable.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
WYX and LTH conceived and designed the study. WYX interpreted and analyzed the data. LY and WYX wrote the manuscript. LTH revised the manuscript critically.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article meets the conformances to requirements of TCGA and GEO websites of publishment, and does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Y., Luo, Y. & Li, T. A metabolic reprogramming-related gene signature correlates with prognosis and proliferation of BLCA. Discov Onc 15, 338 (2024). https://doi.org/10.1007/s12672-024-01219-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01219-2