
Abstract
N6-methyladenosine(m6A) is one of the most abundant modifications of mammalian cellular RNAs. m6A regulates various biological functions in epitranscriptomic ways, including RNA stability, decay, splicing, translation and nuclear export. Recent studies have indicated the growing importance of m6A modification in precancerous disease, influencing viral replication, immune escape, and carcinogenesis. Here, we review the role of m6A modification in HBV/HCV infection, NAFLD and liver fibrosis, and its function in liver disease pathogenesis. Our review will provide a new sight for the innovative treatment strategy for precancerous liver disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
Liver cancer remains the seventh most frequently occurring cancer and the third leading cause of cancer death worldwide, with approximately 906,000 new cases and 830,000 deaths in 2020 [1, 2]. Hepatocellular carcinoma (HCC) is the major form of liver cancer and accounts for more than 80% cases [3]. Hepatitis B virus (HBV) and Hepatitis C virus(HCV) infection are the main risk factors for HCC development and account for 80% cases globally [4]. Besides, nonalcoholic fatty liver disease (NAFLD) and liver fibrosis are well-established risk factors for HCC [5]. Recently, epitranscriptomic modifications, especially RNA methylations, are known to result in changes in gene expression and virus life-cycle, and have been shown to trigger HCC [6].
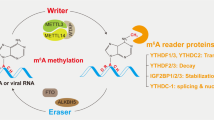
N6-methyladenosine(m6A) is the most abundant RNA methylation in eukaryotic RNA and widely occurs in mRNA and non-coding RNAs [ribosomal RNAs (rRNAs), tRNAs and circular RNAs (circRNAs)] [7, 8], and playing an essential role in regulating mammalian gene expression [7]. In mRNAs, most m6A sites are enriched in the 3′ untranslated region (UTR) and near stop codons [9, 10]. m6A most located in the consensus sequence RRACH (R = G or A and H = A, C, or U) [11]. The biological functions of m6A are mediated by corresponding enzymes, methyltransferases- “writers”, demethylases- “erasers” and “readers”. To date, an increasing number of m6A regulatory enzymes (writers, erasers and readers) have been discovered to involve in RNA stabilization, decay, splicing, translation, and nuclear export to influence liver disease development (Fig. 1).

Molecular mechanism of m6A modification of RNAs. N6-methyladenosine methylation is a dynamic process that occurs in the nucleus. The m6A of RNAs is catalyzed by the writer complex, including METTL3, METTL14, WTAP, VIRMA, RBM15/15B, ZC3H13, CBLL1 and METTL16, and demethylated by erasers including FTO and ALKBH5. The m6A reader proteins, including YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3, HNRNPC/G/A2B1 and eIF3/3 h, recognize the m6A sites to modulate RNA stability, degradation, splicing, translation and nuclear export. RBM15/15B means RBM15 and RBM15B, YTHDF1/2/3 means YTHDF1,YTHDF2 and YTHDF3, YTHDC1/2 means YTHDC1 and YTHDC2, IGF2BP1/2/3 means IGF2BP1, IGF2BP2 and IGF2BP3, HNRNPC/G/A2B1 means HNRNPC, HNRNPG and HNRNPA2B1, eIF3/3 h means eIF3 and eIF3h. Draw by Figdraw
1.1 Dynamic regulation of m6A
1.1.1 Writers
As shown in Fig. 1 and Table 1, m6A methylation is a dynamic process regulated by m6A methyltransferase complexes, which consist of methyltransferase-like 3/14/16 (METTL3/14/16) [11, 12], Wilms’ tumor 1-associating protein (WTAP) [13], vir like m6A methyltransferase associated (VIRMA, also called KIAA1429) [14], RNA-binding motif protein 15 (RBM15) [15], Cbl photo oncogene like 1(CBLL1) [16] and zinc finger CCCH-type containing 13(ZC3H13) [17]. METTL3, as the first identified component, is the catalytic subunit that binds to S-adenosylmethionine (SAM) and catalyzes methyl transfer [18,19,20]. METTL14 serves as structural support for METTL3 and assists to binds to the target RNA; WTAP ensures nuclear speckle localization for METTL3-METTL14 heterodimer [13, 18, 19]. VIRMA is critical for recruiting core components METTL3/METTL14/WTAP to mediate preferential mRNA methylation in 3′UTR and near stop codon [14, 17]. RBM15 and its paralogue RBM15B interact with METTL3 and WTAP, and recruit them to specific RNA sites [15]. Other proteins, such as CBLL1 and ZC3H13, bridge WTAP to mRNA-binding factor [21,22,23]. Besides, METTL16 catalyzes m6A modification in U6 snRNA and plays an important role in pre-RNA splicing [24].
1.1.2 Erasers
m6A methylation can be reversed via demethylases that convert m6A into A, and two demethylases, fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5), have been identified [25,26,27]. As a member of AlkB family, FTO was the first protein identified to catalyze m6A demethylation [25], and it can also demethylate m6Am (N6,2′-O-dimethyladenosine) [28]. Both FTO and ALKBH5 are located in nuclear speckles and belong to Fe2+/α- ketoglutarate dependent dioxygenases enzyme family, which recognize m6A in mRNA [29].
1.1.3 Readers
m6A reader proteins recognize m6A modification and affect the fate of mRNAs to exert biological functions. To date, readers include the YT521-B homology (YTH) domain family proteins (YTHDF1/2/3) [30, 31], YTH domain containing proteins (YTHDC1/2) [32,33,34], heterogeneous nuclear ribonucleoprotein (HNRNPC, HNRNPG, and HNRNPA2B1) [30, 35, 36], eukaryotic translation initiation factor 3 (eIF3) [37], insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) [38]. The YTHDF family proteins have three similar paralogues, and with different effect on mRNA fate:YTHDF1 recognize m6A-modified mRNA and promotes mRNA translation [31], YTHDF2 selectively binding m6A-containing mRNA to promote degradation of mRNA [39], YTHDF3 cooperate with YTHDF1 to enhance protein synthesis, and affects methylated mRNA decay mediated via YTHDF2 [40]. YTHDC1 locates in nuclear and recruits pre-mRNA splicing factor SRSF3 to regulate mRNA splicing [32], and it also facilitates nuclear export of m6A-containing mRNA via interacting with SRSF3 and NXF1 [41]. YTHDC2 plays critical roles in mammalian germline by augmenting the translation efficiency of target methylated mRNA and diminishing their levels [33, 42]. Meanwhile, YTHDC2 regulate the stability of m6A-containing mRNAs by recruiting 5′–3′ exoribonuclease XRN1 [43]. m6A alters RNA structure to mediate the accessibility of RNA binding motifs, referred as ‘m6A-switch’ mechanism, which enhances binding of HNRNPC, an abundant nuclear RNA-binding protein responsible for pre-mRNA processing [30]. Besides, other RNA-binding proteins are also mediated via m6A-switch: HNRNPG is involved in mRNA alternative splicing, and HNRNPA2B1 is involved in primary microRNA processing [36, 44]. eIF3 is identified as an m6A reader, recognize m6A in the 5′UTR and initiates protein translation in a cap-independent manner [37]. METTL3 interacts with eIF3h and recognize the m6A sites close to the stop codon, and promote oncogenic mRNAs translation [45]. IGF2BPs, including IGF2BP1/2/3, recognize the consensus GG(m6A)C sequence and promote mRNA stability and translation [38].
1.2 m6A modification in HBV infection
HBV, a member of the Hepadnaviridae family, contains a partially double-stranded DNA genome, which leads to chronic hepatitis and results in HCC development. Recent studies discovered that m6A modification could directly or indirectly regulate HBV replication (Fig. 2, Table 2).

The functional role of m6A modification in HBV infection. A The m6A sites in HBV RNAs. HBV RNA is methylated at the single position (A1907) within the epsilon stem-loop region. The m6A site present in the 3′ terminus of all HBV RNAs and at both the 5′ and 3′ termini of the pgRNA. B The dynamic methylation of m6A in HBV RNAs. HBx recruits METTL3/METTL14 to the HBV cccDNA to catalyze m6A modification of HBV RNA/pgRNA, as well as promote nuclear import of m6A writers. C The m6A modification regulates HBV life cycle. YTHDF2/3 recognize the m6A site within 5′ epsilon stem-loop to increase the reverse transcription of pgRNAs and recognizes the m6A site within 3′ epsilon stem-loop to decrease the stability of HBV RNAs. Meanwhile, YTHDC1 promotes nuclear export of HBV pgRNAs and IGF2BP3 enhance the stability, reduce proliferation and stemness of HBV pgRNAs in an m6A-dependent manner. D The m6A modification regulates host immune responses. YTHDF2/3 hider RIG-I recognition of HBV RNAs to suppress immune response via interacting with m6A-modified HBV RNAs. ISG20 degrades HBV RNAs via interacting with YTHDF2 to recognize the m6A-modified HBV RNAs. YTHDF2/3 means YTHDF2 and YTHDF3. Draw by Figdraw
Imam and colleagues discovered that there is a conserved m6A consensus motif (A1907) located within the epsilon stem-loop region [46]. The m6A site is present at the 3′ terminus of all HBV RNAs and at both the 5′ and 3′ termini of the pgRNA [46]. Depleting of METTL3/METTL14 or YTHDF2/YTHDF3 results in increased expression of HBcAg and HBsAg; conversely, knocking down of FTO/ALKBH5 reduces the expression of HBV proteins [46]. Besides, researchers discovered that the m6A modification at the 5′ stem-loop plays positive roles for reverses transcription of pgRNA while m6A at 3′ stem-loop lower the stability of HBV RNAs and viral protein production [46]. Recently, another pivotal role of m6A modification in HBV life cycle was discovered[47]. The methyltransferases (METTL3/METTL14) influenced the encapsidation of HBV pgRNA, and depleting of methyltransferases increase HBV-RNA and protein levels but decrease levels of packaged pgRNA, rcDNA and extracellular HBV-DNA [47]. The m6A modification of 5′ epsilon stem-loop region increased the viral RNA packaging efficiency and facilitated the interaction with core proteins, though m6A modification of 3′ stem was unessential for viral encapsidation [47]. A recent study showed that HBV X (HBx) protein binds to m6A methyltransferases and mediated m6A modification, which reduces the stability of HBV RNAs and promotes the nuclear import of METTL3/ METTL14 [48]. Recently, a targeted RNA demethylation by SunTag system (TRADES) was established [49]. This system is based on CRISPR/Cas and recruits RNA demethylase to demethylate the target m6A site [49]. Researchers targeted HBV RNA via TRADES and lead to ~ threefold increase of HBV cccDNA copy numbers [49]. Decreased m6A levels of pgRNA reduce HBV DNA levels and promote viral protein production, including the HBx, which may promote the nuclear import of METTL3/14 and reverse the demethylase function. Another study uncovered that YTHDC1 and FMRP recognize m6A-methylated HBV RNAs and facilitate their nuclear export [50]. Besides, depletion of YTHDC1 or FMRP could reduce viral DNA synthesis in the core particles, as well as the cccDNA levels [50]. Another study demonstrated that IGF2BP3, an m6A reader, binds to pgRNA and enhances the stability of HBV-pgRNA, meanwhile, pgRNA up-regulates IGF2BP3 expression at the posttranscriptional level via miR-let-7e-5p [51]. Silencing IGF2BP3 could significantly abrogate the proliferation, stemness and tumorigenicity of pgRNA [51].
Recent years, m6A modifications were confirmed to regulate innate immunity and antiviral response [52]. It has recently been established that m6A modification site (A1907) of HBV pgRNA 5′ stem-loop was involved in regulating the innate immune response [53]. Mutant of m6A modification site (A1907C) enhances retinoic acid–inducible gene I (RIG-I) sensing, stimulating IRF-3 activation and IFN signal. Besides, depletion of METTL3 and METTL14, as well as YTHDF2/3, promotes HBV RNA recognition by RIG-I, and leads to an increase in interferon production [53]. Notably, YTHDF2/3 hider RIG-I recognition of HBV RNAs via interacting with m6A-modified HBV RNAs, resulting in inhibiting RIG-I mediated immune response [53]. Furthermore, the HBV transcripts can be bound and degraded by the IFN-induced interferon-stimulated gene 20 (ISG20) [54]. Moreover, ISG20 degrades HBV transcripts via interacting with YTHDF2 to recognize the m6A modification of HBV RNAs [53]. The mutant HBV RNAs, which lack m6A methylation at both termini of HBV RNAs, were resistant to ISG20-mediated suppression [54]. PTEN, a tumor suppressor protein, mediates dephosphorylation of IRF-3 at Ser-97 to induce IRF-3 nuclear import and IFN synthesis [55]. Kim et all found that HBV infection suppressed IFN signaling via increasing the m6A modification of PTEN mRNA which resulted in RNA instability [55].
1.3 m6A modification in HCV infection
HCV, belonging to the Flaviviridae family, is a positive single-stranded RNA virus and cause chronic Hepatitis C, which is another major etiology factor of HCC. The RNA genomes of Flaviviridae family, including HCV, dengue, Zika, yellow fever, and West Nile virus, were modified by m6A in conserved regions [56].
Harnessing methyl-RNA immunoprecipitation (MeRIP) sequencing, Gokhale and colleagues identified that HCV RNA is modified by m6A and there are ~ 19 m6A peaks at the HCV RNA genome (Fig. 3) [56]. Remarkably, m6A modification in HCV RNA has been observed to negatively regulate extracellular viral RNA levels and viral particle production, without any impact on HCV translation or RNA replication [56]. Notably, all three YTHDF proteins bind to m6A-methylated HCV RNA, leading to the relocation of HCV RNA to lipid droplets to suppress HCV particle production [56]. Besides, there are several m6A modification sites within HCV RNA genome, and each m6A modification may have different functions depending on its location. For example, the m6A modification in HCV E1 gene regulates viral RNA packaging via recognizing by YTHDF proteins [56], the m6A modification in the internal ribosome entry site (IRES) influences translation [57]. A recent study discovered that there is an m6A site at HCV nucleotide(nt) 331, located about 10 nt upstream of initiation codon (AUG) [57]. YTHDC2 recognizes m6A methylation at nt 331 to regulate viral life cycle by inducing the HCV IRES translation activity [57]. Similar to HBV infection, depleting of METTL3 and METTL14,as well as YTHDF2 and YTHDF3, significantly increased IFN-β synthesis and IRF-3 phosphorylation in HCV infection cell models [53]. The pathogen-associated molecular patterns (PAMPs), nucleotides 8872–9616 in HCV genome, recognized by RIG-I affect IFN-β level and activation of IRF-3 [58]. There is no m6A modification in the HCV PAMP RNA, but an m6A modification site (nucleotide 8766) was identified at ~ 100 bp upstream of PAMP RNA. Via constructing A8766C-mutated models, authors discovered that m6A modification of HCV 8766 nucleotide affects the function of PAMP by reducing their sensitivity to RIG-I [53].

The functional role of m6A modification in HCV infection. The HCV genome contains several m6A-modified regions (~ 19 regions), including E1 region, IRES element and 3′ end genome region. YTHDC2 recognizes the m6A modification in IRES element influence HCV translation initiation. YTHDFs recognize the m6A modification in HCV E1 gene to regulate viral RNA packaging. The m6A site at 3′ end HCV genome (~ 100 bp upstream of PAMP), recognized by YTHDF2/3, affects the function of PAMP by reducing its sensitivity to RIG-I. YTHDF2/3 means YTHDF2 and YTHDF3. Draw by Figdraw
Another study spotlighted on the mechanisms of m6A modifications in HCV infection [59]. Depletion of both WTAP and METTL3 + 14 led to decreased m6A levels on HCV RNA and m6A modified transcript during HCV infection [59]. METTL3 directly binds with HCV RNA in a WTAP-dependent manner to reduce the production of infectious viral particles. Like METTL3 + 14, WTAP declines infections of HCV virion in a METTL3-dependent manner but does not alter HCV RNA replication in the cytoplasm [59].
1.4 m6A modification in NAFLD
Nonalcoholic fatty liver disease (NAFLD), which is rapidly emerging among children and adolescents, has become another significant risk factor foe HCC [60]. NAFLD, characterized by excessive triglyceride accumulation and steatosis in hepatocytes, develops to nonalcoholic steatohepatitis (NASH) with hepatic inflammation and hepatocytic injury, ultimately leading to hepatic cirrhosis and/or HCC [61]. Recent studies have investigated the m6A methylation plays a crucial role in hepatic lipid metabolism and the development of NAFLD (Fig. 4) [62].

The functional role of m6A modification in NAFLD. In NAFLD patients, the lipogenesis, metabolism-associated and lipid transport genes are regulated by m6A effectors in an m6A-dependent manner to affect NAFLD development. Draw by Figdraw
The m6A writers and readers also participate in the regulation of NAFLD development. In a high fat diet (HFD)-induced mouse model of NAFLD, the m6A level and the methyltransferases METTL3 were found to be significantly upregulated [63]. METTL3-mediated m6A modification suppressed autophagic flux by targeting Rubicon mRNA and upregulating its expression to accelerate lipid droplet accumulation [63]. Meanwhile, YTHDF1 enhances the stability of Rubicon mRNA by binding to it, leading to inhibition of autophagic flux and accelerating lipid droplet accumulation [63]. In another study, loss of METTL3 leads to a reduction in the activity of mTOR and NF-κB signaling pathways, resulting from the stabilization of m6A-mediated DNA Damage Inducible Transcript 4 (DDIT4) mRNA in macrophages, which regulated metabolic reprogramming in NAFLD and obesity [64]. Besides, the circadian clock regulates hepatic lipid metabolism in an m6A-dependent manner. Knockdown of METTL3 decreased PPaRα m6A abundance, and prolonged PPaRα mRNA lifetime via recognized by YTHDF2, which reduced lipid accumulation [62]. Recently, YTHDC2 was identified as a regulator of hepatic lipogenesis through recognition of m6A sites and reduce mRNA stability of lipogenesis genes [65]. Authors observed a downregulation of YTHDC2 in both obese mice models and NAFLD patients. They also found that the overexpression of YTHDC2 suppresses lipogenic genes (SREBF1, FASN, ACACA, SCD and GPAM) mRNA stability to reduce cellular TG contents [65]. Yang and colleagues established a new NAFLD model and discovered lipid metabolism was regulated by ACLY and SCD via m6A-modified manner [66]. Overexpression of METTL3 or METTL14 significantly enhanced lipid droplet accumulation by elevating the m6A levels of ACLY and SCD gene transcripts, finally resulting in up-regulated expression [66].
Apart from being an m6A demethylase, FTO is also a crucial regulator in glucose and lipid metabolism, and it may regulate NAFLD in an m6A-dependent manner [67]. Overexpressed FTO enhances lipid accumulation by increasing the expression of metabolism-associated genes (FASN, SCD, and MGAT1), while simultaneously downregulating the expression of lipid transport genes (MTTP, APOB, and LIPC) [68]. Meanwhile, mutant FTO (R136A) with no demethylation activity did not have these functions [68]. Using corticosterone (CORT)-induced fatty liver models, authors discovered that the m6A level of mRNA was decreased and FTO was increased in liver. Besides, interleukin-17A (IL-17A), one isoform of IL-17 family cytokines, plays pivotal roles in the pathogenesis of NASH, which regulated by m6A modulator FTO [69]. Researchers revealed decreased IL-17RA m6A levels and increased IL-17RA protein levels in the progression of HCC [69]. In murine NAFLD models, FTO facilitates liver injury and inflammation via decreased m6A enrichment of IL-17RA mRNA [69]. Meanwhile, the levels of m6A on the lipogenic genes (SREBF1, FASN, ACACA and SCD), were significantly decreased under dexamethasone combined with oleic acid (OA/ DEX) treatment and resulted in the upregulation of proteins [70]. Notably, meclofenamic acid (MA), an FTO inhibitor, could alleviate OA/DEX–induced m6A hypomethylation on the lipogenic genes. Besides, knockdown of FTO alleviates DEX-induced fatty liver in mice as well [70].
1.5 m6A modification in hepatic fibrosis
Hepatic fibrosis increases the risk of HCC, and ~ 4% of cirrhosis cases develop to HCC per year [71]. As a critical post-transcriptional modification, m6A methylation plays a vital role in liver fibrosis progress (Fig. 5).

The functional role of m6A modification in liver fibrosis. The activation, proliferation, migration and ferroptosis ability of HSCs are regulated by m6A effectors in an m6A-dependent manner to influence liver fibrosis process. Draw by Figdraw
Recent study discovered that m6A modification not only involved in the progression and reversal of hepatic fibrosis, but also participated the dynamic regulation of fibrosis [72]. For example, the m6A level of CCR2 (a chemokine receptor) is markedly elevated in the progression of liver fibrosis, while decreased in the reversal of liver fibrosis, indicating its potential as a therapeutic target [72]. ASIC1a was discovered upregulated in liver fibrosis, mediated m6A regulation of miR-350 mature to participate in liver fibrosis via PI3K/AKT and ERK pathways [73]. Mechanistically, METTL3 was associated with ASIC1a promoting liver fibrosis and subsequently regulated the DGCR8-mediated synthesis of miR-350 by m6A modification [73]. In the liver fibrosis mice, the m6A methylation abundance was significantly decreased as well as the expressions of WTAP, ALKBH5 and YTHDF1, and the decreased expression of WTAP has been shown to induce the development of liver fibrosis and promote hepatic stellate cell (HSC) activation [74]. Likewise, in liver fibrosis models, METTL3 and MALAT1 were upregulated in Kupffer cells and macrophages cells. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis stimulated pyroptosis and inflammation of macrophages, leading to the aggravation of liver fibrosis [75]. Overexpressed METTL3 increased MALAT1 expression through m6A modification, and then MALAT1 directly bound with PTBP1 to down-regulated USP8, which resulted in reduced ubiquitination of TAK1 [75].
The activation of HSCs is a key process of liver fibrosis, and HSC-specific knockout of METTL3 inhibits HSC activation and significantly alleviates liver fibrosis [76]. Via reducing m6A levels of LATS2 mRNA and suppressing its degradation, upregulated LATS2 promoted the phosphorylation of the downstream transcription factor YAP. This process led to the downregulation of pro-fibrotic gene expression and affected the Hippo/YAP signaling pathway to mitigate liver fibrosis progress [76]. YTHDF3 directly regulated the mRNA translation of PRDX3 in an m6A-dependent manner to alleviate liver fibrosis [77]. PRDX3 acted as a master regulator of mitochondrial oxidative stress and suppressed HSC activation via the (ROS)/TGF-β1/Smad2/3 pathway [77]. Besides, the activation of HSCs was attenuated under dihydroartemisinin (DHA) treatment via the ferroptosis pathway in an m6A-mediated manner [78]. The DHA downregulated FTO and boosted m6A abundance of BECN1 mRNA [79]. Subsequently, YTHDF1 recognized the m6A modification to stabilize the BECN1 mRNA, thus leading to autophagy activation and inducing HSC ferroptosis [79]. ALKBH5 regulated liver fibrosis by affecting the stability of PTCH1 mRNA in an m6A-mediated manner [80].
1.6 m6A modification in other liver diseases
Besides, m6A modification has been uncovered associated with liver regeneration. Researchers observed that the m6A level and METTL3 were significantly upregulated in the 2/3 partial hepatectomy (PHx) mice. By constructing liver-specific METTL3 knock-out mouse model, researchers discovered hepatocyte proliferation was suppressed, and the hepatocyte cell cycle progression was delayed through the SOCS6/STAT3 pathway [81]. Moreover, perinatal deletion of METTL3 triggers apoptosis, steatosis and fibrosis, resulting in severe liver damage and ultimately culminating in postnatal death within 7 weeks [82]. Deletion of METTL3 inducing a significant decrease of m6A deposition on transcription factor, Hnf4a, via reducing the half-life of Hnf4a mRNA in an IGF2BP1-dependent manner [82]. Smilarly, loss of METTL14 has been shown to impair liver regeneration after PHx, with extensive parenchymal necrosis [83, 84]. The METTL14-deficented hepatocytes are arrested in the G1 phase of cell cycle, as well as inducing excessive endoplasmic reticulum (ER) stress [83]. Alpha-1 antitrypsin deficiency (AATD) was a prevalent inherited liver disease characterized by misfolded protein accumulation in the endoplasmic reticulum (ER), ultimately leading to inflammation, fibrosis,cirrhosis and HCC. However, it is noteworthy that the majority of patients do not develop liver toxicity [85]. Wei and his collegues discovered that misfolded proteins in AATD are involved in suppressing the ubiquitination of METTL14 via an interaction with HRD1. Consequently, the expression level of METTL14 is up-regulated, leading to the alleviation of ER-stress induced liver damage via promoting CHOP mRNA decay in an m6A-dependent manner [85].
2 Conclusion and perspectives
RNA methylation, especially m6A modification, has been discovered that play vital roles in various biological functions [34, 37, 37, 39]. RNA biology, including splicing, stability, editing, translational and degradation, is involved in liver diseases in an m6A-dependent manner [46, 56, 73]. In this review, we have delineated the dynamic m6A modification process, and described the functional effects of methylation in various liver diseases, including HBV infection, HCV infection, NAFLD and liver fibrosis. It has become clear that the m6A modification plays an outsized role in premalignant disease of HCC and participated in the progress to HCC.
Recent studies have demonstrated that the HCV RNA, as well as the RNA transcripts of HBV, are administrated by m6A modification to regulate viral life cycle, virus particle production and the process of pathogenesis [46, 51, 53, 54, 56]. Here, we discussed the functions of m6A modification in HBV and HCV infections. Notably, m6A modifications regulate the HBV and HCV life cycles, packaging and nuclear import in complex ways depending on their location in the HBV RNAs and HCV genome. Additionaly, it has been observed that m6A methylation plays a critical role in regulating host RNAs during HBV and HCV infections, thereby affecting the immune escape of the virus and contribute to viral persistence and chronicity [48]. Eventually, the regulation of m6A modification during HBV and HCV infections has a significant impact on the development of liver disease and promotes liver carcinogenesis.
NAFLD is another important risk factor for HCC, and NAFLD-related HCC accounts for 1% to 38% of the HCC burden in different countries/regions [60]. The m6A demethyltransferase, FTO, is involved in the lipid metabolism. The downstream molecules (FASN, ACACA, SCD et al.), regulated by FTO in the m6A-dependent manner, are implicated in lipogenesis, lipid transport to participate in the process of NAFLD. Meanwhile, METTL3 and METTL14 regulate the m6A sites at mRNAs of various lipid metabolism genes to promote lipid accumulation, which further leads to the progression of NAFLD and HCC [66].
Liver fibrosis is an important premalignant disease leading to HCC, and effective treatment remains absent. The activation of HSCs promotes the development of liver fibrosis, which is regulated by m6A modification in complex ways. In this review, we detailed that MALAT1, LATS2, PRDX3, PTCH1 and BECN1 genes affect the HSCs activation, proliferation, migration and ferroptosis in an m6A-dependent manner, which regulates the progress of liver fibrosis.
In liver regeneration, m6A methyltransferase plays an important function as well. In the PTx mice models, knocking out METTL3 results in suppressed hepatocyte proliferation delayed cell cycle as well as METTL14. Noteably, loss of METTL14 in PTx mice leads to excessive ER stress [83], and the misfolded proteins in AATD patients upregulates METTL14 to protect against ER stress-induced liver damage [85]. These results indicate that m6A modification is critical for maintaining endoplasmic reticulum homeostasis.
Thus, m6A modification is a dynamic and reversible process that plays diverse functions in premalignant liver disease. Understanding the mechanisms and clinical implications of m6A modification is crucial for preventing or intervening in the occurrence and development of liver diseases and HCC. Further research on the methylation mechanism and its role in liver disease will provide valuable insights into the therapy strategy for the treatment of liver diseases and HCC. Overall, comprehensively understanding the m6A modification and its clinical implications can transform the field of liver disease and HCC research.
Data availability
Not applicable.
Abbreviations
- m6A:
-
N6-methyladenosine
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- NAFLD:
-
Nonalcoholic fatty liver disease
- HCC:
-
Hepatocellular carcinoma
- METTL3:
-
Methyltransferase-like 3
- WTAP:
-
Wilms’ tumor 1-associating protein
- VIRMA:
-
Vir like m6A methyltransferase associated
- RBM15:
-
RNA-binding motif protein 15
- CBLL1:
-
Cbl photo oncogene like 1
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- SAM:
-
S-adenosylmethionine
- UTR:
-
Untranslated region
- FTO:
-
Fat mass and obesity-associated protein
- ALKBH5:
-
AlkB homolog 5
- m6Am :
-
N6,2′-O-dimethyladenosine
- YTHDFs:
-
YT521-B homology domain family proteins
- YTHDCs:
-
YTH domain containing proteins
- HNRNPC:
-
Heterogeneous nuclear ribonucleoprotein C
- eIF3:
-
Eukaryotic translation initiation factor 3
- IGF2BPs:
-
Insulin-like growth factor 2 mRNA-binding proteins
- SRSF3:
-
Serine and arginine rich splicing factor 3
- NXF1:
-
Nuclear RNA export factor 1
- XRN1:
-
5′–3′ Exoribonuclease 1
- pgRNA:
-
Pregenomic RNA
- HBcAg:
-
Hepatitis B core antigen
- HBsAg:
-
Hepatitis B surface antigen
- rcDNA:
-
Relaxed circular DNA
- RIG-I:
-
Retinoic acid–inducible gene I
- IRF-3:
-
Interferon regulatory factor 3
- IFN:
-
Interferon
- ISG20:
-
Interferon-stimulated gene 20
- PAMP:
-
Pathogen-associated molecular patterns
- DDIT4:
-
Damage inducible transcript 4
- PPaRa:
-
Peroxisome proliferator activated receptor alpha
- SREBF1:
-
Sterol regulatory element binding transcription factor 1
- FASN:
-
Fatty acid synthase
- ACACA:
-
Acetyl-CoA carboxylase alpha
- SCD:
-
Stearoyl-CoA desaturase
- GPAM:
-
Glycerol-3-phosphate acyltransferase, mitochondrial
- ACLY:
-
ATP citrate lyase
- MGAT1:
-
Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase
- MTTP:
-
Microsomal triglyceride transfer protein
- APOB:
-
Apolipoprotein B
- LIPC:
-
Lipase C, hepatic type
- ASIC1a:
-
Acid-sensing ion channel 1a
- CCR2:
-
C–C motif chemokine receptor 2
- DGCR8:
-
DGCR8 microprocessor complex subunit
- MALAT1:
-
Metastasis associated lung adenocarcinoma transcript 1
- HSC:
-
Hepatic stellate cell
- LATS2:
-
Large tumor suppressor kinase 2
- PRDX3:
-
Peroxiredoxin 3
- PTCH1:
-
Patched 1
- BECN1:
-
Beclin 1
- PHx:
-
Partial hepatectomy
- ER:
-
Endoplasmic reticulum
- AATD:
-
Alpha-1 antitrypsin deficiency
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6.
Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604.
McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73(Suppl 1):4–13.
Nagaraju GP, Dariya B, Kasa P, Peela S, El-Rayes BF. Epigenetics in hepatocellular carcinoma. Semin Cancer Biol. 2021;S1044-579X(21)00211-X.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–9.
Wang X, Ma R, Zhang X, Cui L, Ding Y, Shi W, et al. Crosstalk between N6-methyladenosine modification and circular RNAs: current understanding and future directions. Mol Cancer. 2021;20:121.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169:824-835.e14.
Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–96.
Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73.
Růžička K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, et al. Identification of factors required for m6 A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017;215:157–72.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306–17.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–8.
Wei X, Huo Y, Pi J, Gao Y, Rao S, He M, et al. METTL3 preferentially enhances non-m6A translation of epigenetic factors and promotes tumourigenesis. Nat Cell Biol. 2022;24:1278–90.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32:415–29.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69:1028-1038.e6.
Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, et al. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013;288:33292–302.
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, et al. Human METTL16 is a N6-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18:2004–14.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, et al. Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42:4741–54.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541:371–5.
Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10:927–9.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–308.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–63.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Roundtree IA, Luo G-Z, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife. 2017;6: e31311.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m6A transcripts by the 3′→5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68:374-387.e12.
Kretschmer J, Rao H, Hackert P, Sloan KE, Höbartner C, Bohnsack MT. The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′-3′ exoribonuclease XRN1. RNA. 2018;24:1339–50.
Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. 2018;9:420.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–60.
Imam H, Khan M, Gokhale NS, McIntyre ABR, Kim GW, Jang JY, et al. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc Natl Acad Sci U S A. 2018;115:8829–34.
Kim G-W, Moon J-S, Siddiqui A. N6-methyladenosine modification of the 5′ epsilon structure of the HBV pregenome RNA regulates its encapsidation by the viral core protein. Proc Natl Acad Sci U S A. 2022;119: e2120485119.
Kim G-W, Siddiqui A. Hepatitis B virus X protein recruits methyltransferases to affect cotranscriptional N6-methyladenosine modification of viral/host RNAs. Proc Natl Acad Sci U S A. 2021;118: e2019455118.
Mo J, Chen Z, Qin S, Li S, Liu C, Zhang L, et al. TRADES: targeted RNA demethylation by suntag system. Adv Sci (Weinh). 2020;7:2001402.
Kim G-W, Imam H, Siddiqui A. The RNA binding proteins YTHDC1 and FMRP regulate the nuclear export of N6-methyladenosine-modified hepatitis B virus transcripts and affect the viral life cycle. J Virol. 2021;95: e0009721.
Ding W-B, Wang M-C, Yu J, Huang G, Sun D-P, Liu L, et al. HBV/pregenomic RNA increases the stemness and promotes the development of HBV-related HCC through reciprocal regulation with insulin-like growth factor 2 mRNA-binding protein 3. Hepatology. 2021;74:1480.
Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol. 2017;18:1094–103.
Kim G-W, Imam H, Khan M, Siddiqui A. N 6-Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J Biol Chem. 2020;295:13123–33.
Imam H, Kim G-W, Mir SA, Khan M, Siddiqui A. Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified Hepatitis B Virus transcripts. PLoS Pathog. 2020;16: e1008338.
Kim G-W, Imam H, Khan M, Mir SA, Kim S-J, Yoon SK, et al. HBV-induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC. Hepatology. 2021;73:533–47.
Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, et al. N6-methyladenosine in flaviviridae viral RNA genomes regulates infection. Cell Host Microbe. 2016;20:654–65.
Kim G-W, Siddiqui A. N6-methyladenosine modification of HCV RNA genome regulates cap-independent IRES-mediated translation via YTHDC2 recognition. Proc Natl Acad Sci U S A. 2021;118: e2022024118.
Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–7.
Sacco MT, Bland KM, Horner SM. WTAP targets the METTL3 m 6 A-methyltransferase complex to cytoplasmic hepatitis C virus RNA to regulate infection. bioRxiv. 2022;2022.06.27.497872.
Shah PA, Patil R, Harrison SA. NAFLD-related hepatocellular carcinoma: the growing challenge. Hepatology. 2022;77:323.
Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411–28.
Zhong X, Yu J, Frazier K, Weng X, Li Y, Cham CM, et al. Circadian clock regulation of hepatic lipid metabolism by modulation of m6A mRNA methylation. Cell Rep. 2018;25:1816-1828.e4.
Peng Z, Gong Y, Wang X, He W, Wu L, Zhang L, et al. METTL3-m6A-Rubicon axis inhibits autophagy in nonalcoholic fatty liver disease. Mol Ther. 2021;S1525-0016(21)00471-8.
Qin Y, Li B, Arumugam S, Lu Q, Mankash SM, Li J, et al. m6A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell Rep. 2021;37: 109968.
Zhou B, Liu C, Xu L, Yuan Y, Zhao J, Zhao W, et al. N6 -methyladenosine reader protein YT521-B homology domain-containing 2 suppresses liver steatosis by regulation of mRNA stability of lipogenic genes. Hepatology. 2021;73:91–103.
Yang Y, Cai J, Yang X, Wang K, Sun K, Yang Z, et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol Ther. 2022;S1525-0016(22)00105-8.
Yang Z, Yu G-L, Zhu X, Peng T-H, Lv Y-C. Critical roles of FTO-mediated mRNA m6A demethylation in regulating adipogenesis and lipid metabolism: Implications in lipid metabolic disorders. Genes Dis. 2022;9:51–61.
Kang H, Zhang Z, Yu L, Li Y, Liang M, Zhou L. FTO reduces mitochondria and promotes hepatic fat accumulation through RNA demethylation. J Cell Biochem. 2018;119:5676–85.
Gan X, Dai Z, Ge C, Yin H, Wang Y, Tan J, et al. FTO promotes liver inflammation by suppressing m6A mRNA methylation of IL-17RA. Front Oncol. 2022;12: 989353.
Hu Y, Feng Y, Zhang L, Jia Y, Cai D, Qian S-B, et al. GR-mediated FTO transactivation induces lipid accumulation in hepatocytes via demethylation of m6A on lipogenic mRNAs. RNA Biol. 2020;17:930–42.
Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–62.
Cui Z, Huang N, Liu L, Li X, Li G, Chen Y, et al. Dynamic analysis of m6A methylation spectroscopy during progression and reversal of hepatic fibrosis. Epigenomics. 2020;12:1707–23.
Zhu Y, Pan X, Du N, Li K, Hu Y, Wang L, et al. ASIC1a regulates miR-350/SPRY2 by N6 -methyladenosine to promote liver fibrosis. FASEB J. 2020;34:14371–88.
Fan C, Ma Y, Chen S, Zhou Q, Jiang H, Zhang J, et al. Comprehensive analysis of the transcriptome-wide m6A methylation modification difference in liver fibrosis mice by high-throughput m6A sequencing. Front Cell Dev Biol. 2021;9: 767051.
Shu B, Zhou Y-X, Li H, Zhang R-Z, He C, Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Discov. 2021;7:368.
Li Y, Kang X, Zhou Z, Pan L, Chen H, Liang X, et al. The m6A methyltransferase Mettl3 deficiency attenuates hepatic stellate cell activation and liver fibrosis. Mol Ther. 2022;S1525–0016(22):00441–5.
Sun R, Tian X, Li Y, Zhao Y, Wang Z, Hu Y, et al. The m6A reader YTHDF3-mediated PRDX3 translation alleviates liver fibrosis. Redox Biol. 2022;54: 102378.
Shen M, Li Y, Wang Y, Shao J, Zhang F, Yin G, et al. N6-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021;47: 102151.
Shen M, Guo M, Li Y, Wang Y, Qiu Y, Shao J, et al. m6A methylation is required for dihydroartemisinin to alleviate liver fibrosis by inducing ferroptosis in hepatic stellate cells. Free Radic Biol Med. 2022;182:246–59.
Yang J-J, Wang J, Yang Y, Yang Y, Li J, Lu D, et al. ALKBH5 ameliorated liver fibrosis and suppressed HSCs activation via triggering PTCH1 activation in an m6A dependent manner. Eur J Pharmacol. 2022;922: 174900.
Meng J, Zhao Z, Xi Z, Xia Q. Liver-specific Mettl3 ablation delays liver regeneration in mice. Genes Dis. 2022;9:697–704.
Xu Y, Zhou Z, Kang X, Pan L, Liu C, Liang X, et al. Mettl3-mediated mRNA m6A modification controls postnatal liver development by modulating the transcription factor Hnf4a. Nat Commun. 2022;13:4555.
Cao X, Shu Y, Chen Y, Xu Q, Guo G, Wu Z, et al. Mettl14-mediated m6A modification facilitates liver regeneration by maintaining endoplasmic reticulum homeostasis. Cell Mol Gastroenterol Hepatol. 2021;12:633–51.
Yang I, Oh SY, Jang S, Kim IY, Sung YM, Seong JK. Mettl14 mutation restrains liver regeneration by attenuating mitogens derived from non-parenchymal liver cells. BMB Rep. 2022;55:633–8.
Wei J, Harada BT, Lu D, Ma R, Gao B, Xu Y, et al. HRD1-mediated METTL14 degradation regulates m6A mRNA modification to suppress ER proteotoxic liver disease. Mol Cell. 2021;81:5052-5065.e6.
Gonzales-van Horn SR, Sarnow P. Making the mark: the role of adenosine modifications in the life cycle of rna viruses. Cell Host Microbe. 2017;21:661–9.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81902141), the Natural Science Foundation of Hubei Province (2022CFB127), the Interdisciplinary Innovative Talents Foundation from Renmin Hospital of Wuhan University(JCRCFZ-2022-023) and the Knowledge Innovation Program of Wuhan -Basic Research (No.2022020201010402).
Author information
Authors and Affiliations
Contributions
JH provided conception and design of this manuscript. ZL and RR contributed to the writing of the manuscript. YY prepared the tables and figures. JH, MX and HS reviewed, edited and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lv, Z., Ran, R., Yang, Y. et al. The interplay between N6-methyladenosine and precancerous liver disease: molecular functions and mechanisms. Discov Onc 14, 78 (2023). https://doi.org/10.1007/s12672-023-00695-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-023-00695-2