
Abstract
Mutations in BAP1 have been identified in a hereditary cancer predisposition syndrome and in sporadic tumours. Individuals carrying familiar BAP1 monoallelic mutations display hypersusceptibility to exposure-associated cancers, such as asbestos-driven mesothelioma, thus BAP1 status has been postulated to participate in gene-environment interaction. Intriguingly, BAP1 functions display also a high degree of tissue dependency, associated to a peculiar cancer spectrum and cell types of specific functions. Mechanistically, BAP1 functions as an ubiquitin carboxy-terminal hydrolase (UCH) and controls regulatory ubiquitination of histones as well as degradative ubiquitination of a range of protein substrates. In this article we provide an overview of the most relevant findings on BAP1, underpinning its tissue specific tumour suppressor function. We also discuss the importance of its epigenetic role versus the control of protein stability in the regulation of genomic integrity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Gene, environment and BAP1
The largest majority, if not the entirety, of the human diseases emerge from the interaction of at least an exogenous factor, including also microorganisms, and the genetics of the individual. How the cell responds to a stress and manages to maintain the stability of its molecular circuits determines whether there will be a disease. Ability to regulate epigenetic landscape and the integrity of the genome is especially important in cancer development, hence, here, the gene-environment (GxE) interactions play critical roles [1,2,3].
Epigenetic modifications as DNA methylation and histone posttranslational modifications (PTMs) regulate cellular processes. Epigenetic perturbations or even mutations in epigenetic enzymes can trigger changes in chromatin conformation leading to aberrant transcriptome which may support tumorigenesis [4,5,6]. The balance between euchromatin and heterochromatin is finely tuned by a number of chromatin modifying factors, including the Polycomb group (PcG) family, broadly classified into 3 complexes: Polycomb Repressive Complex 1 (PRC1), Polycomb Repressive Complex 2 (PRC2) and Polycomb Repressive Deubiquitinase Complex (PR-DUB) [7]. BRCA1-Associated Protein 1 (BAP1) is a ubiquitin carboxy-terminal hydrolase (UCH), which also functions as a member of Polycomb Repressive—Deubiquitinase complex (PR-DUB). PR-DUB removes monoubiquitin residue at lysine 119 of the Histone 2A (H2AK119ub), thus remodelling chromatin and maintaining functional epigenetic landscape. This enzymatic activity directly counteracts Polycomb Repressive Complex 1 (PRC1)-mediated histone ubiquitylation, modulating transcriptional programs and a variety of cellular processes including DNA repair, metabolism, cell proliferation, differentiation and cell death [8,9,10]
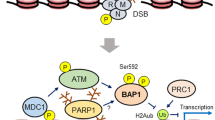
Originally, BAP1 was directly implicated in a mechanism of DNA repair following double strand break. BAP1 direct physical interaction with BRCA1-RING finger domain was associated to enhancement of BRCA1 tumour suppressor activity in breast cancer [11] (Fig. 1a). BAP1-BRCA1 interaction is however still controversial [12]. Several studies later demonstrated that BAP1 mainly interacts with BARD1 perturbing BARD1/BRCA1 complex [13]. Nevertheless, further works have highlighted that BAP1 enables the initial recruitment and accumulation of BRCA1 and other DNA damage repair proteins as RPA and RAD51, at the double strand breaks (DSBs), requisite for DNA repair via Homologous Repair (HR) system [14,15,16].

Source: cBioportal [75]
BAP1 structure and frequency of alteration in cancer. a Schematic representation of BAP1 structure and interacting partners. BAP1 protein can be divided in three regions: an N-terminal region (1–240 aa), where the catalytic triad is located and responsible for deubiquitylation; a middle region (241–596 aa), demonstrated to interact with BARD1, HCF1 through the HCF-1 binding motif (HBM) and FOXK1 and 2; the C-terminal region harbouring two nuclear localization sequences. The 2 NLS can be targeted by UBE2O allowing BAP1 retention in the cytoplasm. This region was demonstrated to interact with BRCA1, YY1 (Yin Yang 1) and ASLX1 and 2. b Frequency of genetic alteration of BAP1 in cancer. In red the most relevant alterations of BAP1 are shown. Mesothelioma, Intrahepatic Cholangiocarcinoma, Renal Clear Cell Carcinoma and Uveal Melanoma, display the highest frequency of BAP1 genetic alterations.
BAP1 has attracted strong interest in the recent decades since the identification of the “BAP1 cancer syndrome”. Individuals carrying germline monoallelic mutation in BAP1 show a high frequency of malignant mesothelioma (MM), uveal melanoma (UM) and clear cell renal cell carcinoma (ccRCC) [15, 17, 18]. The germline mono-allelic mutation of BAP1 appears to play a role predisposition to exposure-induced cancers. This is particularly relevant in the context of asbestos-associated mesothelioma and UV-associated melanomas. Hence, such specific pattern of human cancers associated to BAP1 inactivation suggest a role in the response to environmental stressors and indicate a cooperation of predisposing gene mutations and environmental factors in cancer onset and progression. Thus, BAP1 mutation was proposed as key prototype of Gene-Environment interaction (GxE) [19]. Intriguingly, alike the canonical tumour suppressors such as p53 [20], sporadic mutations of BAP1 are found a peculiar spectrum of tumours, that recall the genetic predispositions (Fig. 1b). Hence, BAP1 inactivation emerged as directly linked to the tumorigenesis process of these cancers.
Despite the evidence that BAP1 enforces control of the epigenetic landscape and influences genomic integrity, it is still unclear whether the intersection of these two processes underlies a role of BAP1 in cancer. In this perspective, we discuss classic and more recent literature regarding BAP1, aiming to provide a unifying view of its role in epigenetic stability and genomic integrity.
2 BAP1/PR-DUB: the fine regulation of histone ubiquitination
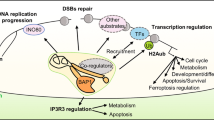
The PR-DUB is a multiprotein complex constituted by BAP1, HCF1, FOXK1/2, OGT, MBD5/6, LSD2 associated with ASXL1, 2 or 3 [7, 21]. In this complex, BAP1 is the catalytic subunit and its activity is strongly dependent on the interaction with conserved DEUBiquitinase Adaptor (DEUBAD) domain of ASXLs that induces conformational changes able to increase BAP1 affinity for ubiquitin [22]. Moreover, FOXK1 and FOXK2 seem to have a role in site-specific recruitment of BAP1 across the genome [7] (Fig. 2).

BAP1 in the PRC-DUB complex. BAP1 is directly involved in controlling the dynamics of chromatin as a component of PR-DUB complex. Loss of BAP1 directly impacts of cell transcriptome due to the altered deposition of H2AK119ub and H3K27me3 histone marks
The interplay between PRC1 and PRC2 complexes control the chromatin dynamic. Polycomb Repressive complex 1 monoubiquitylates H2A at lysine 119 (H2AK119ub), while Polycomb Repressive complex 2 catalyse mono-, di- and trimethylation of H3 at lysine K27 (H3K27me1-2-3), both posttranslational modifications of histone tails required for modulating chromatin architecture, cellular stemness and differentiation. An additional level of complexity is given by PR-DUB complex, which reshapes the epigenetic landscape by counteracting PRC1 activity through the removal of H2Aub from chromatin and, thus, indirectly influencing the PRC2-mediated H3K27me [7, 8, 23].
H2Aub is particularly enriched at specific silent genomic regions, notably Polycomb Target Genes (PcG) [22]. In mammalian, this group includes 39 Hox genes located in 4 clusters, involved in vertebrate development and organogenesis and their deregulation is commonly observed in cancer [24, 25]. Generally, Hox genes are silenced via two different mechanisms: the methylation of CpG islands in their promoters and PRC1/PRC2-driven chromatin repression by ubiquitination of H2A and trimethylation of histone H3 at lysine 27. RNF1/RNF2-mediated ubiquitylation of histone H2A is a required step for the recruitment of PRC2 on chromatin thereby leading to the subsequent deposition of H3K27me3 [22]. BAP1/PR-DUB associates to chromatin at active gene promoters and removes ubiquitylation mainly deposited by PRC1.3/5 (non-canonical PRC1 complex) while it is excluded from the canonical PRC1 (c-PRC1) and PRC2 Polycomb repressive regions [8]. Overall, BAP1 maintains the spatial distribution of both H2AK119ub and H3K27me3 on Polycomb regions, preserving gene repression. Indeed, loss of BAP1 catalytic function titrates away both c-PRC1 and PRC2 complexes from their genomic loci causing derepression of Polycomb genes, the spreading and accumulation of H2AK119ub and H3K27me3 sustained by PRC1.3/5 at intergenic sites across the genome [8]. This results in a global chromatin compaction, reshaping of epigenetic landscape and aberrant transcriptome that associates with loss of cellular identity, oncogenesis, immune evasion and poor tumour response to immunotherapy [26,27,28] (Fig. 2). Indeed, Yan and colleagues have recently demonstrated that the unbalanced activity of chromatin modifying factors like that caused by PRC2 inactivation, can strongly contributes to epigenetic reprogramming and transcriptional downregulation of genes involved in the immune cell recruitment, driving immune-desert tumour microenvironment [28]. Thus, identification and targeting of tumour-specific epigenetic dysregulations represent a possible therapeutic approach via administration of epidrugs as EZH2 (inhibitors of histone methyltransferase, core component of PRC2 complex), DNMT (DNA Methyltransferase), HDAC (Histone deacetylase) and BAP1 inhibitors [18, 29,30,31].
3 BAP1 in cancer: more than epigenetic?
Several studies have highlighted that BAP1 tumour suppressor activity is strictly cell type- and context-dependent: inactivation of BAP1 or catalytically inactive mutants (i.e. C91A) can drive opposite phenotypes in different tissues [12, 32, 33].
BAP1 conditional knock out mice showed hematopoietic defects as anaemia, thrombocytopenia, leucocytosis, liver damage and atrophy of pancreas. In these organs, increases in cleaved caspase-3 levels suggest that the loss of this protein triggers apoptotic events. Furthermore, BAP1 loss-associated lethality can be observed in several cell types including embryonic stem cells, primary keratinocytes and E1A-immortalized embryo fibroblasts [31]. Conversely, BAP1 genetic deletion in mouse primary melanocytes and mesothelial cells induces proliferation and the expression of pro-survival genes. The differential activity in cell types resides into the different selectivity of BAP1 in regulating gene expression [32, 34]. The tumour suppressor function of BAP1 is also described in prostate and kidney cancers in which BAP1-dependent deubiquitinase activity stabilizes the tumour suppressor Phosphatase and Tensin homolog (PTEN) and Death Inducer-Obliterator 1 (DIDO1), a protein of the centrosome involved in spindle assembly and correct chromosome segregation [35,36,37]. In pancreas, BAP1 inactivation causes organ atrophy while triggers the inactivation of tumour suppressor Hippo pathways in pancreatic KRAS mutated cancer [38, 39]. Hence the interplay between BAP1 deficiency and oncogenic KRAS leads to pancreatic tumour progression. In breast cancer cell lines, BAP1 plays an oncogenic function by directly deubiquitinating and stabilizing KLF5 (Kruppel-like factor 5). Protein stabilization of KLF5 promotes cell proliferation, migration and tumour growth [38]. Moreover, in these KLF5-positive cell lines, BAP1 knock-down inhibits the DNA synthesis reducing cell viability, while it has no effect on the cell growth in KLF-5 negative MDA-MB-231 breast cancer cells [21, 41]. In small cell lung carcinoma (SCLC), BAP1 promotes oncogenic roles inducing the expression of ASCL1 (Achaete-Scute Family BHLH Transcription Factor 1), a key lineage-specific oncogenic driver in SCLC. BAP1 inhibitors and CRISPR-cas9 knock-out in NCI-H1963, NCI-H748 and NCI-H1882 cells abrogate ASCL1 chromatin occupancy at the promoter region of its target genes reducing cell growth [18]. In leukaemia the gain-of-function of ASXL1 mutants increase PR-DUB activity; the stabilization of BAP1 and its undue ASXL1 mutant-dependent chromatin recruitment leads to aberrant oncogenic pattern of gene expression [42, 43]. In this context, the reduction of BAP1 catalytic activity with iBAP (BAP1 inhibitors) might represent a therapeutic strategy [40]. In contrast, BAP1 function is required to avoid the onset of myeloproliferative disorder since BAP1 KO mice showed hematopoietic defects as myeloid progenitor expansion [44, 45].
BAP1 exerts its function predominantly in the nuclear compartment as the two NLS can direct the translocation of this protein in the nucleus. Notably, BAP1 can be sequestered in the cytoplasm by the ubiquitin-conjugating enzyme UBE2O that ubiquitylates its nuclear localization signals leading to its cytoplasmatic retention. Exogenous and endogenous stimuli trigger the activation of the self-deubiquitylation activity of BAP1. This allows the translocation from cytoplasm into nucleus where BAP1 regulates the cell biological response [44]. Furthermore, BAP1 exerts cytoplasmatic functions as it can localize in the endoplasmic reticulum for modulating intracellular Ca 2+ release and the activation of apoptosis [45]. BAP1 regulates stabilization of type-3 inositol-1,4,5-trisphosphate-receptor (IP3R3), ER calcium channel that controls the release of Ca2 + from endoplasmic reticulum into cytosol and mitochondria. Changes in mitochondrial permeability are required for the activation of the apoptotic process and the loss of BAP1 protects the cells from caspase-induced cell death as a consequence of IP3R3 level reduction and Ca2 + signalling decrease [45]. In addition to apoptosis, BAP1 also modulates the activation of cysteine-dependent cell death, ferroptosis, by downregulating the expression of SLC7A11, the major transporter for extracellular cysteine uptake. Also in this context, the inactivation of BAP1 triggers cell resistance to ferroptosis [10]. Moreover, BAP1 deficiency drives the reprogramming of cell metabolism, promoting anaerobic glycolysis for energy production rather than mitochondrial respiration and increasing extracellular lactate secretion which induces immune evasion, tumour growth and cell malignant transformation.
BAP1 emerges as a highly tissue-specific and context-specific tumour suppressor participating to the biology of the tumour with multiple mechanisms and different levels (summary in Table 1).
4 The BAP1 cancer syndrome
Germline monoallelic inactivation of BAP1 is a prototype of GxE predisposing to tumorigenesis. Carriers of BAP1 mutations have high frequency of mesothelioma, cutaneous and uveal melanoma, clear cell renal cell carcinoma. In carrier individuals, tumour onset is accompanied by the loss of heterozygosity with the inactivation of the second wild-type allele [12]. More than 80% of gene carriers are affected by at least one type of cancer and 90% of the affected individuals have at least two close first-degree relatives affected by a cancer. BAP1 families require genetic and oncological counselling to handle cancer risk management and undergo routine testing for at-risk family members.
Tumour onset in carriers is accompanied by the loss of heterozygosity with the inactivation of the second wild-type allele [12]. Mutations frequently occur in the N-terminal catalytic UCH domain within Gly185, Arg227, impacting the affinity of BAP1 for ubiquitin, and within Cys91, His169, Asp184 by inactivating the catalytic domain. Missense mutations are also found in BAP1 interacting domains and in C-terminal region, interfering with its nuclear localization, auto-deubiquitination and recruitment on chromatin [12, 46, 47].
Therapeutic approaches have been suggested for the treatment of BAP1-deficient cancers such as the epigenetic drugs that inhibit EZH2, ther platinum-based compounds and PARP-1 inhibitors. EZH2 inhibitors reduces proliferation of BAP1-mutant mesothelioma cell lines, while platinum-based drugs and PARP-1 inhibitors should be able to target cancer cells with defective DNA repair mechanisms [9, 44, 48].
Here, we discuss the current knowledge about the contribution of inactivating BAP1 mutations in development and progression of inherited cancers as Mesothelioma, Uveal melanoma and Clear Cell Renal Cell Carcinoma in which BAP1 is recurrently lost.
4.1 Mesothelioma
Malignant Mesothelioma (MM) is a tumour arising from mesothelial cell transformation mainly of pleura and peritoneum and it is correlated to persistent exposure to environmental carcinogen such as asbestos that includes 6 natural fibres (crocidolite, actinolite, tremolite, anthophyllite, amosite and chrysotile) [17]. After inhalation, asbestos is phagocytized by macrophages and mesothelial cells of pleura where accumulates forming deposits and hence exerts its cytotoxic effects [47]. The initiation of carcinogenesis process is attributed to HMGB1 extracellular release by necrotic mesothelial cells that activates chronic inflammation and ROS production triggering an inflammatory microenvironment [48]. In addition, these fibres could mechanically interfere with chromosomal segregation during mitosis leading to DNA damage, genome instability, thus contributing to mesothelial cell oncogenic transformation [49, 50]. In addition to mutations of BAP1, frequent deletion of tumour suppressors Cdkn2a/b and Nf2 were observed in malignant mesothelioma [51,52,53]. The functional interaction between BAP1 inactivation and these other genetic events in the development of MM has not been fully elucidated.
4.2 Uveal melanoma
Uveal Melanoma (UM) is the most widespread primary intraocular malignant tumour in adult arising from melanocytes of pigmented uveal tissues as the iris in the anterior chamber of the eye and ciliary body and choroid in the posterior chamber of the eye [54, 55]. UM incidence shows a south to north increasing gradients as it ranges from < 1 (Africa) up to 9 (Norway and Denmark) per million population per years depending on the countries [54]. Despite the treatment with radiotherapy until the ocular enucleation in most advanced cases, the half of patients affected by UM develop metastases within 5-years in liver, lung, skin and brain reducing the survival at less of one year from the onset of symptoms [55]. Genetic features as fair-skin, light-coloured eyes, ocular melanocytosis besides germline mutations in BAP1 gene, increase the odds of developing uveal melanoma [12, 56, 57]. Moreover, loss of chromosome 3 or BAP1 deficiency are predictors of metastatic UM since BAP1 biallelic inactivation correlates with the most aggressive phenotype of melanocytes, characterized by driver mutations in G-protein-α subunits GNAQ or QNA11 [25, 30, 58, 59] that are not sufficient alone to induce malignant transformation but sustain cell growth by downstream activation of YAP/TAZ [60,61,62]. However, the molecular mechanisms through which BAP1 promotes UM metastasis is still unclear.
An important open question is whether the sunlight exposure could cooperate with BAP1 inactivation in UM development and progression. Interestingly, few evidences indicate a direct link between ultraviolet radiation exposure, a common environmental risk factor for cutaneous melanoma onset, and the occurrence or progression of uveal melanoma [56, 63]. Nevertheless, the melanoma of the iris, the part of the eye directly exposed to sunlight, shares ultraviolet radiation (UVR) mutational signature, suggesting an association between UV exposure and this malignancy [64, 65].
4.3 Clear cell renal cell carcinoma
Clear cell renal cell carcinoma (ccRCC) represents 70–80% of all kidney tumours and arises from epithelial cells of renal tubular. ccRCC is characterized by genetic features as loss of chromosome 3p, mutations in Protein Polybromo-1 gene (PBRM1), Von Hippel–Lindau (VHL), Set domain-containing 2 (SETD2) histone methyltransferase and BAP1 genes [66, 67]. BAP1 inactivation status is strongly associated to high tumour grade and worse clinical outcomes in ccRCC patients [68,69,70]. However, BAP1 mutations increase the susceptibility of renal cancer cells to the treatment with CCR5 inhibitor. This compound reduces CCR5+ T-reg cells in the tumour microenvironment increasing the immune response and cancer regression [71, 72].
5 Conclusion
The last decades have seen the accumulation of a huge amount of cancer genomics data, that have supported the development of predictive models of cancer [71]. Despite this data have undoubtedly pointed out a role for BAP1 in tumorigenesis of a specific spectrum of cancer, the determination of the molecular underlying mechanisms has not yet led to development of therapeutic strategies. Important questions remain elusive; the selectivity of the cancer spectrum has not yet an explanation. Remarkably the relevance of the epigenetic role of BAP1 versus the other described functions has still no answer. Finally, we argue that in cancer development the maintenance of genomic integrity is a pivotal aspect of the pathogenesis and whether and BAP1 dependent regulation of the cellular epigenome impact the genomic integrity remain an eluded critical question.
References
Aristizabal MJ, et al. Biological embedding of experience: a primer on epigenetics. Proc Natl Acad Sci USA. 2020;117(38):23261–9. https://doi.org/10.1073/pnas.1820838116.
Liston A, Humblet-Baron S, Duffy D, Goris A. Human immune diversity: from evolution to modernity. Nat Immunol. 2021;22(12):1479–89. https://doi.org/10.1038/s41590-021-01058-1.
Poczai P, Santiago-Blay JA. Principles and biological concepts of heredity before Mendel. Biol Direct. 2021;16(1):1–17. https://doi.org/10.1186/s13062-021-00308-4.
Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19(11):776–800. https://doi.org/10.1038/s41573-020-0077-5.
Butera A, Melino G, Amelio I. Epigenetic ‘Drivers’ of cancer. J Mol Biol. 2021;433(15):167. https://doi.org/10.1016/j.jmb.2021.167094.
Sun L, Lv S, Song T. O-GlcNAcylation links oncogenic signals and cancer epigenetics. Discov Oncol. 2021. https://doi.org/10.1007/s12672-021-00450-5.
Kolovos P, et al. PR-DUB maintains the expression of critical genes through FOXK1/2- A nd ASXL1/2/3-dependent recruitment to chromatin and H2AK119ub1 deubiquitination. Genome Res. 2020;30(8):1119–30. https://doi.org/10.1101/gr.261016.120.
Conway E, et al. BAP1 enhances Polycomb repression by counteracting widespread H2AK119ub1 deposition and chromatin condensation. Mol Cell. 2021;81(17):3526-3541.e8. https://doi.org/10.1016/j.molcel.2021.06.020.
Louie BH, Kurzrock R. BAP1: not just a BRCA1-associated protein. Cancer Treat Rev. 2020;90:102091. https://doi.org/10.1016/j.ctrv.2020.102091.
Zhang Y, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–92. https://doi.org/10.1038/s41556-018-0178-0.
Jensen DE, et al. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16(9):1097–112. https://doi.org/10.1038/sj.onc.1201861.
Carbone M, et al. Biological mechanisms and clinical signifi cance of BAP1 mutations in human cancer. Cancer Discov. 2020;10(8):1103–20. https://doi.org/10.1158/2159-8290.CD-19-1220.
Nishikawa H, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009;69(1):111–9. https://doi.org/10.1158/0008-5472.CAN-08-3355.
Ismail IH, Davidson R, Gagné JP, Xu ZZ, Poirier GG, Hendzel MJ. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res. 2014;74(16):4282–94. https://doi.org/10.1158/0008-5472.CAN-13-3109.
Masclef L, et al. Roles and mechanisms of BAP1 deubiquitinase in tumor suppression. Cell Death Differ. 2021;28(2):606–25. https://doi.org/10.1038/s41418-020-00709-4.
Liu X, et al. BAP1 is a novel target in HPV-negative head and neck cancer. Clin Cancer Res. 2018;24(3):600–7. https://doi.org/10.1158/1078-0432.CCR-17-1573.
Carbone M, et al. Mesothelioma: scientific clues for prevention, diagnosis, and therapy. CA Cancer J Clin. 2019;69(5):402–29. https://doi.org/10.3322/caac.21572.
Tsuboyama N, et al. Therapeutic targeting of BAP1/ASXL3 sub-complex in ASCL1-dependent small cell lung cancer. Oncogene. 2022. https://doi.org/10.1038/s41388-022-02240-x.
Carbone M, et al. Tumour predisposition and cancer syndromes as models to study gene–environment interaction. Nat Rev Cancer. 2020;20(9):533–49. https://doi.org/10.1038/s41568-020-0265-y.
Panatta E, Zampieri C, Melino G, Amelio I. Understanding p53 tumour suppressor network. Biol Direct. 2021;16(1):10–6. https://doi.org/10.1186/s13062-021-00298-3.
Szczepanski AP, Wang L. Emerging multifaceted roles of BAP1 complexes in biological processes. Cell Death Discov. 2021. https://doi.org/10.1038/s41420-021-00406-2.
Barbour H, Daou S, Hendzel M, Affar EB. Polycomb group-mediated histone H2A monoubiquitination in epigenome regulation and nuclear processes. Nat Commun. 2020;11(1):1–16. https://doi.org/10.1038/s41467-020-19722-9.
Wu X, et al. Tumor suppressor ASXL1 is essential for the activation of INK4B expression in response to oncogene activity and anti-proliferative signals. Cell Res. 2015;25(11):1205–18. https://doi.org/10.1038/cr.2015.121.
Mallo M. Reassessing the role of Hox genes during vertebrate development and evolution. Trends Genet. 2018;34(3):209–17. https://doi.org/10.1016/j.tig.2017.11.007.
Miao Y, Zhang W, Liu S, Leng X, Hu C, Sun H. HOXC10 promotes growth and migration of melanoma by regulating Slug to activate the YAP/TAZ signaling pathway. Discov Oncol. 2021. https://doi.org/10.1007/s12672-021-00408-7.
Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–71. https://doi.org/10.1038/nrc2826.
Sun Q, Melino G, Amelio I, Jiang J, Wang Y, Shi Y. Recent advances in cancer immunotherapy. Discov Oncol. 2021. https://doi.org/10.1007/s12672-021-00422-9.
Yan J, et al. Tumor-intrinsic PRC2 inactivation drives a context-dependent immune-desert microenvironment and is sensitized by immunogenic viruses. J Clin Invest. 2022;132(17):1–16. https://doi.org/10.1172/jci153437.
Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14(6):1–25. https://doi.org/10.1371/journal.pgen.1007362.
Jin B, et al. Verification of EZH2 as a druggable target in metastatic uveal melanoma. Mol Cancer. 2020;19:1–15.
He M, et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science. 2019;364(6437):283–5. https://doi.org/10.1126/science.aav4902.
Basar MA, Beck DB, Werner A. Deubiquitylases in developmental ubiquitin signaling and congenital diseases. Cell Death Differ. 2021;28(2):538–56. https://doi.org/10.1038/s41418-020-00697-5.
Badhai J, et al. Combined deletion of Bap1, Nf2, and Cdkn2ab causes rapid onset of malignant mesothelioma in mice. J Exp Med. 2020. https://doi.org/10.1084/jem.20191257.
Deng R, et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol Oncol. 2021;15(1):279–98. https://doi.org/10.1002/1878-0261.12844.
Xiao J, Zhang R, Peng J, Yang Z. BAP1 maintains chromosome stability by stabilizing DIDO1 in renal cell carcinoma. Am J Cancer Res. 2020;10(5):1455–66.
Lee HJ, et al. The tumor suppressor BAP1 regulates the hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res. 2020;80(8):1656–68. https://doi.org/10.1158/0008-5472.CAN-19-1704.
Perkail S, Andricovich J, Kai Y, Tzatsos A. BAP1 is a haploinsufficient tumor suppressor linking chronic pancreatitis to pancreatic cancer in mice. Nat Commun. 2020. https://doi.org/10.1038/s41467-020-16589-8.
Wang X, et al. Arginine methyltransferase PRMT5 methylates and stabilizes KLF5 via decreasing its phosphorylation and ubiquitination to promote basal-like breast cancer. Cell Death Differ. 2021;28(10):2931–45. https://doi.org/10.1038/s41418-021-00793-0.
Qin J, et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat Commun. 2015;6:1–12. https://doi.org/10.1038/ncomms9471.
Wang L, et al. Epigenetic targeted therapy of stabilized BAP1 in ASXL1 gain-of-function mutated leukemia. Nat Cancer. 2021;2(5):515–26. https://doi.org/10.1038/s43018-021-00199-4.
Guo Y, et al. Reduced BAP1 activity prevents ASXL1 truncation-driven myeloid malignancy in vivo. Leukemia. 2018;32(8):1834–7. https://doi.org/10.1038/s41375-018-0126-9.
Lafave LM, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2016;21(11):1344–9. https://doi.org/10.1038/nm.3947.Loss.
Dey A, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012. https://doi.org/10.1126/science.1221711.Loss.
Mashtalir N, et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell. 2014;54(3):392–406. https://doi.org/10.1016/j.molcel.2014.03.002.
Bononi A, et al. BAP1 regulates IP3R3-mediated Ca 2+ flux to mitochondria suppressing cell transformation. Nature. 2017;546(7659):549–53. https://doi.org/10.1038/nature22798.
Foglizzo M, et al. A bidentate Polycomb Repressive-Deubiquitinase complex is required for efficient activity on nucleosomes. Nat Commun. 2018. https://doi.org/10.1038/s41467-018-06186-1.
Zolondick AA, Gaudino G, Xue J, Pass HI, Carbone M, Yang H. Asbestos-induced chronic inflammation in malignant pleural mesothelioma and related therapeutic approaches—a narrative review. Precis Cancer Med. 2022. https://doi.org/10.21037/pcm-21-12.
Yang K, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29(1):133–46. https://doi.org/10.1038/s41418-021-00841-9.
Sazonova EV, Petrichuk SV, Kopeina GS, Zhivotovsky B. A link between mitotic defects and mitotic catastrophe: detection and cell fate. Biol Direct. 2021;16(1):1–11. https://doi.org/10.1186/s13062-021-00313-7.
Kato S, Tomson BN, Buys TPH, Elkin SK, Carter JL, Kurzrock R. Genomic landscape of malignant mesotheliomas. Mol Cancer Ther. 2016;15(10):2498–507. https://doi.org/10.1158/1535-7163.MCT-16-0229.
Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016;35(5):537–48. https://doi.org/10.1038/onc.2015.125.
Yu L, et al. Co-occurrence of BAP1 and SF3B1 mutations in uveal melanoma induces cellular senescence. Mol Oncol. 2022;16(3):607–29. https://doi.org/10.1002/1878-0261.13128.
Amaro A, et al. The biology of uveal melanoma. Cancer Metastasis Rev. 2017;36(1):109–40. https://doi.org/10.1007/s10555-017-9663-3.
Kaliki S, Shields CL. Uveal melanoma : relatively rare but deadly cancer. Eye. 2016;31(2):241–57. https://doi.org/10.1038/eye.2016.275.
Jager MJ, et al. Uveal melanoma. Nat Rev Dis Prim. 2020;6(1):18–20. https://doi.org/10.1038/s41572-020-0158-0.
Pandiani C, et al. Single-cell RNA sequencing reveals intratumoral heterogeneity in primary uveal melanomas and identifies HES6 as a driver of the metastatic disease. Cell Death Differ. 2021;28(6):1990–2000. https://doi.org/10.1038/s41418-020-00730-7.
Souri Z, et al. Loss of BAP1 is associated with upregulation of the NFkB pathway and increased HLA class I expression in Uveal Melanoma. Cancers (Basel). 2019;11(8):1–16. https://doi.org/10.3390/cancers11081102.
Bakhoum MF, et al. Loss of polycomb repressive complex 1 activity and chromosomal instability drive uveal melanoma progression. Nat Commun. 2021;12(1):1–16. https://doi.org/10.1038/s41467-021-25529-z.
Smit KN, Jager MJ, de Klein A, Kiliҫ E. Uveal melanoma: towards a molecular understanding. Prog Retin Eye Res. 2019. https://doi.org/10.1016/j.preteyeres.2019.100800.
Liu Y, Zhuang Y, Fu X, Li C. LncRNA POU3F3 promotes melanoma cell proliferation by downregulating lncRNA MEG3. Discov Oncol. 2021. https://doi.org/10.1007/s12672-021-00414-9.
Pandiani C, Béranger GE, Leclerc J, Ballotti R, Bertolotto C. Focus on cutaneous and uveal melanoma specificities. Genes Dev. 2017;31(8):724–43. https://doi.org/10.1101/gad.296962.117.
Johansson PA, et al. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat Commun. 2020;11(1):1–8. https://doi.org/10.1038/s41467-020-16276-8.
Chalada M, Ramlogan-Steel CA, Dhungel BP, Layton CJ, Steel JC. The impact of ultraviolet radiation on the aetiology and development of uveal melanoma. Cancers (Basel). 2021;13(7):1–17. https://doi.org/10.3390/cancers13071700.
Singh D. Current updates and future perspectives on the management of renal cell carcinoma. Life Sci. 2021;264:118632. https://doi.org/10.1016/j.lfs.2020.118632.
Jonasch E, Walker CL, Rathmell WK. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol. 2021;17(4):245–61. https://doi.org/10.1038/s41581-020-00359-2.
Wi YC, et al. Loss of nuclear BAP1 expression is associated with high WHO/ISUP grade in clear cell renal cell carcinoma. J Pathol Transl Med. 2018;52(6):378–85. https://doi.org/10.4132/jptm.2018.09.21.
Peng J, et al. Stabilization of MCRS1 by BAP1 prevents chromosome instability in renal cell carcinoma. Cancer Lett. 2015;369(1):167–74. https://doi.org/10.1016/j.canlet.2015.08.013.
Gu YF, et al. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer Discov. 2017;7(8):900–17. https://doi.org/10.1158/2159-8290.CD-17-0292.
Zhou Q, et al. CCR5 blockade inflames antitumor immunity in BAP1-mutant clear cell renal cell carcinoma. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2019-000228.
Wang C, et al. Targeting a positive regulatory loop in the tumor-macrophage interaction impairs the progression of clear cell renal cell carcinoma. Cell Death Differ. 2021;28(3):932–51. https://doi.org/10.1038/s41418-020-00626-6.
Amelio I, et al. Cancer predictive studies. Biol Direct. 2020;15(1):1–7. https://doi.org/10.1186/s13062-020-00274-3.
Chen XX, et al. BAP1 acts as a tumor suppressor in intrahepatic cholangiocarcinoma by modulating the ERK1/2 and JNK/c-Jun pathways. Cell Death Dis. 2018. https://doi.org/10.1038/s41419-018-1087-7.
Field MG, et al. BAP1 loss is associated with DNA methylomic repatterning in highly aggressive Class 2 uveal melanomas. Clin Cancer Res. 2019. https://doi.org/10.1158/1078-0432.CCR-19-0366.BAP1.
Novelli F, et al. BAP1 forms a trimer with HMGB1 and HDAC1 that modulates gene × environment interaction with asbestos. Proc Natl Acad Sci USA. 2021;118(48):1–11. https://doi.org/10.1073/pnas.2111946118.
Cerami E, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012. https://doi.org/10.1158/2159-8290.CD-12-0095.
Acknowledgements
This work has been supported by the Carl Zeiss Stiftung to IA (Endowed Professorship, #15972218, 2022-2027; Prisma Programme, #P2022-5-003 2022-2023) and by the Carl Zeiss Stiftung and German Scholars Organization with the Fund for international researchers to IA (#15978021, 2022-2024).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
The authors confirm contribution to the paper as follows: SC wrote the manuscript draft; AB prepared the figures; AI reviwed the manuscript and supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caporali, S., Butera, A. & Amelio, I. BAP1 in cancer: epigenetic stability and genome integrity. Discov Onc 13, 117 (2022). https://doi.org/10.1007/s12672-022-00579-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-022-00579-x