
Abstract
Ubiquitination-mediated proteolysis or regulation of proteins, ultimately executed by E3 ubiquitin ligases, control a wide array of cellular processes, including transcription, cell cycle, autophagy and apoptotic cell death. HECT-type E3 ubiquitin ligases can be distinguished from other subfamilies of E3 ubiquitin ligases because they have a C-terminal HECT domain that directly catalyzes the covalent attachment of ubiquitin to their substrate proteins. Deregulation of HECT-type E3-mediated ubiquitination plays a prominent role in cancer development and chemoresistance. Several members of this subfamily are indeed frequently deregulated in human cancers as a result of genetic mutations and altered expression or activity. HECT-type E3s contribute to tumorigenesis by regulating the ubiquitination rate of substrates that function as either tumour suppressors or oncogenes. While the pathological roles of the HECT family members in solid tumors are quite well established, their contribution to the pathogenesis of hematological malignancies has only recently emerged. This review aims to provide a comprehensive overview of the involvement of the HECT-type E3s in leukemogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
1.1 The ubiquitin system
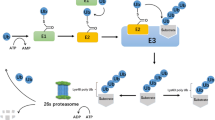
Ubiquitination is an essential posttranslational modification that involves the covalent attachment of the conserved 76-amino acid polypeptide ubiquitin to a target protein. The ATP-dependent and reversible reaction is a multistep process mediated by three classes of enzymes: E1 activating enzymes (E1), E2 conjugating enzymes (E2) and E3 ubiquitin proteins ligases (E3) [1,2,3]. In the first reaction, following ATP-dependent activation of ubiquitin, there is the formation of a thioester linkage between ATP-ubiquitin and the catalytic cysteine residue of the E1. Ubiquitin is then transferred to the E2 enzyme via a similar thioester linkage. In the final step of the ubiquitination enzymatic cascade, the E3s coordinate the transfer of ubiquitin from the E2 to a substrate or can directly promote the attachment of ubiquitin to the target polypeptide. Ubiquitin is transferred to the e-amino group of a lysine residue or to the amino-terminus of a polypeptide. The reverse reaction is carried out by deubiquitinases, proteases that remove ubiquitin chains from proteins and then release free ubiquitin from these polymers [4, 5].
The ubiquitination process can involve either the attachment of a single ubiquitin molecule (monoubiquitination) or the formation of a ubiquitin chain on a target protein (polyubiquitination). To form polyubiquitin chains, ubiquitins are linked via different internal lysine residues (K6, K11, K27, K29, K33, K48, K63) [6]. In addition, polyubiquitin chains can be generated through peptide bond formation between the C-terminal glycine of one ubiquitin and the amino-terminal alpha-amino group of the N-terminal methionine (M1) residue of the next ubiquitin [7,8,9]. Polyubiquitin chains linked through different lysine residues of the ubiquitin molecule can adopt different structures, dictating distinct fates of the modified proteins [6, 10]. Thus, ubiquitination can have different functional consequences, depending on the residue of the ubiquitin that is employed to form the chain. Protein modification by ubiquitin can result in proteasomal degradation of the target polypeptide, influence the activity, the subcellular localization and the interactions of proteins or promote the formation of signaling complexes [10]. In particular, K48-linked ubiquitin chains target proteins for proteolytic degradation by the 26S proteasome [11]. The ubiquitin proteasome system (UPS) is composed of the E1, E2 and E3 ubiquitin enzymes and the 26S proteasome.
Modification of proteins by ubiquitin is involved in the regulation of various relevant biological processes, including apoptosis, cell cycle progression, differentiation, transcription, signal transduction, DNA repair and autophagy [12,13,14,15,16,17,18,19]. Coherently, dysregulation of the UPS is associated with severe pathologies, such as neurodegenerative and immune disorders and cancer [20,21,22,23,24,25,26]. Indeed, the autophagic process, reviewed in [27,28,29,30], strongly relies on the protein degradation pathway.
1.2 HECT E3 protein ubiquitin ligases
Homologous to E6AP C-Terminus (HECT)-type enzymes are a subfamily of E3s that comprises 28 members [31, 32]. HECT E3s directly catalyze substrate ubiquitination in a two-step process. They first load activated ubiquitin from the E2 on themselves, through a ubiquitin-thioester catalytic intermediate with a catalytic cysteine located in the HECT domain; then, they transfer ubiquitin to the substrate. The HECT domain is composed of two lobes connected by a short unstructured linker: the N-terminal lobe containing the E2 binding site, and the smaller C-terminal lobe including the catalytic cysteine (Fig. 1). The flexibility of the linker region is required to juxtapose the catalytic residues of the E2 and E3 [33].

A Structural models of ITCH (PDB: 3TUG) [114] showing the two lobes, with the hinge region in two orthogonal views. B Comparison of the HECT structure among different members of the sub-family, to highlight the high structural similarity between WWP1 (PDB: 1ND7) [33], WWP2 (PDB: 4Y07) [115], HUWE1 (PDB: 3G1N) [116], and NEDD4 (PDB: 2XBF) [117]
HECT E3s can either directly recruit their targets through specific protein–protein interaction modules located within their N-terminal region or require accessory or adaptor proteins to recognize the substrate [34]. HECT-type E3s assemble poly-ubiquitin chains by employing all the ubiquitin linkage types, with each family member displaying different chain type specificity [35, 36]. Based on the distinct structural features of the N-terminal protein–protein interaction modules, the HECT E3s have been further classified into three sub-families: the NEDD4-like (also referred as the C2-WW-HECT), the HERCs and the “Other”-HECT E3s (Fig. 2). The NEDD4-like enzymes share a modular architecture that consists of an N-terminal protein kinase C-related C2 domain, and two to four tryptophan-tryptophan (WW) protein interacting domains, preceding the HECT domain (Fig. 2). In humans, this subfamily is composed of nine members: NEDD4-1, NEDD4-2, ITCH, SMURF1, SMURF2, WWP1, WWP2, NEDL1, and NEDL2 [37]. WW domains are protein modules that mediate protein–protein interactions through the recognition of proline-rich peptide motifs and phosphorylated serine/threonine-proline sites. The six members of the HERC sub-family of E3s bind substrates through one or more regulator of chromosome condensation 1 (RCC1)-like domains (RLDs) (Fig. 2). They are further classified into small and large HERCs, containing a single or multiple RLDs, respectively. The third subfamily includes enzymes that contain neither WW nor RLDs domains. They recruit substrates through a variety of substrate-binding modules located at their N-terminus, with many members having more than one domain (Fig. 2).

Structural features of HECT‐type E3 enzymes. All the family members share the presence of the catalytic HECT domain, which is located at the C-terminus. According to their N‐terminal protein–protein interaction domains, the HECT E3s have been divided into three subgroups. The NEDD4-like members contain an N‐terminal protein kinase C‐related C2 domain and two‐four central WW domains that mediate substrate recruitment. HERC E3s possess a single (small HERCs) or more (large HERCs) RCC [regulator of chromatin condensation 1]-like domain (RLDs) preceding the HECT domain. Large HERCs contain additional domains, such as SPRY and WD40 domains. The SI(ngle)‐HECT subfamily is composed of varied number and types of domains such as armadillo repeat‐containing domain (ARM), amino‐terminal Zn‐finger of Ube3a ligase domain (AZUL), WWE domain (WWE), Bcl-2 homology 3 domain (BH3), ankyrin repeat‐containing domain (ANK), polyadenylate‐binding protein C‐terminal domain (PABC), ubiquitin-associated domain (UBA), and zinc finger domain (ZnF)
Aberrant expression, mutations, or deregulated activity of the HECT enzymes have been associated with tumorigenesis [31, 32]. HECT E3s can contribute to tumour development by controlling the ubiquitination of substrates that function as either tumour suppressors or oncogenes. The pathological roles of the HECT family members in hematological malignancies, particularly in myeloid related neoplasms, have only recently emerged. In this review, we will highlight the contribution of the HECT-type E3s in leukemogenesis.
1.3 Dysregulation of the UPS in hematopoietic malignancies
Leukemias are a group of life-threatening malignant disorders of the blood and bone marrow characterized by extensive biologic diversity. They are classified according to the type of blood cells affected, the morphology and immunophenotype of the leukemic blasts, and how quickly the disease progresses (Table 1). The dominant leukemia cells can be either mature cells, such as in chronic lymphocytic leukemia (CLL) or precursor (stem/progenitor) cells of various lineages, such as in the acute leukemias, or both precursor and mature cells, such as in chronic myeloid leukemia (CML). The UPS is the main cellular degradation machinery that regulates the turnover of crucial proteins involved in tumorigenesis. Dysregulation of the UPS can therefore contribute to the initiation and progression of neoplastic transformation. Three common types of alterations in the UPS may occur in hematopoietic malignancies: (1) mutations in E3s leading to impaired ubiquitylation of their own substrates [38, 39], (2) altered expression of E3s (overexpression/deletion) resulting in the aberrant degradation or accumulation of their targets [40,41,42], and (3) mutations in substrates that impair their recruitment by E3s [43]. Several evidences strongly link leukemogenesis to pathways where proteostasis is essential, like for example NRF2 and IAP [44,45,46,47], and where BH3 profiling may offer therapeutic venues [48,49,50,51].
1.4 Regulation of leukemic oncoproteins turnover by the UPS
Chromosome translocations can lead to the expression of chimeric proteins that act as oncogenic drivers in leukemia [52]. Deregulated protein stability, as a result of chromosomal translocations, can be a driving pathogenic factor in leukemias. Over 95% of cases of CML and about 15% of acute lymphocytic leukemia (ALL) are associated with the Philadelphia chromosome. This genetic abnormality results from the (9;22)(q34;q11) reciprocal chromosomal translocation that creates the constitutively active tyrosine kinase BCR (breakpoint cluster region)-ABL (Abelson proto-oncogene) fusion protein. BCR-ABL activates several oncogenic signalling pathways leading to neoplastic transformation, tumor growth and proliferation. The tyrosine kinase activity of BCR-ABL largely derives from the ABL tyrosine kinase. BCR-ABL is the ideal therapeutic target for CML. Indeed, imatinib, a potent and selective inhibitor of ABL kinase activity, is widely used for CML treatment. However, disease progression is associated with decreased responsiveness to imatinib, due to amplification or point mutations in the BCR-ABL gene. BCR-ABL-dependent resistance remains a major challenge in the field, and novel strategies are still required in CML therapy. The stability of the BCR-ABL protein is regulated by the ubiquitin–proteasome pathway [53]. A number of E3s, including CHIP, c-CBL, and SH2-U-box have been involved in BCR-ABL polyubiquitination and subsequent degradation [54, 55]. Importantly, ABL undergoes protein degradation significantly faster than the BCR-ABL oncoprotein [56], indicating that a different regulation of ABL and BCR-ABL protein stability may contribute to BCR-ABL–mediated tumorigenesis.
Degradation of fusion oncoproteins can also offer a therapeutic strategy for leukemia eradication, as for acute promyelocytic leukemia (APL), a clinical subtype of acute myeloid leukemia (AML). This leukemia comprises a clinically heterogeneous group of hematological malignancies, with varying karyotypic, genetic and epigenetic abnormalities. The disease is characterized by uncontrolled proliferation and altered differentiation of myeloid progenitor/stem cells. APL is curable by using a combination of all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO) that restores the ability of leukemic cells to differentiate [57]. Mechanistically, this treatment triggers the disposal of the promyelocytic leukemia protein (PML)/retinoic acid receptor-alpha (RARalpha) chimeric oncoprotein, which results from the t(15;17) chromosomal translocation and is responsible for the differentiation block of APL blasts. PML-RARalpha binds to RA responsive elements on DNA with high affinity and recruits transcriptional repressive complexes that inhibit the expression of RARalpha target genes necessary for myeloid differentiation. ATRA and ATO induce degradation of PML-RARalpha through several cooperating proteolytic mechanisms including caspase-3-mediated cleavage, proteasomal degradation and autophagy [58]. Similarly, elimination of the BCR-ABL fusion protein through autophagy has been shown in CML, where imatinib has the dual effects of inhibiting the tyrosine kinase activity of BCR-ABL, and inducing its degradation [59].
More broadly, prognosis of patients with AML is strongly influenced by the type of chromosomal or genetic alteration, as well by changes in gene expression. Approximately one-third of AML patients do not achieve complete remission in response to chemotherapy and, even when complete remission is achieved, about 70% of patients relapse within 5 years. Thus, despite great progress in defining molecular determinants involved in AML pathogenesis, much of the variability of the alterations characterising this haematological disorder is still a relevant open question in the field, and there is urgent need to identify novel therapeutic targets.
2 Role of the HECT E3s in the pathogenesis of leukemia
2.1 WW domain-containing E3 ubiquitin protein ligase 1 (WWP1)
WWP1 (WW domain-containing E3 ubiquitin protein ligase 1) possesses a C2 N-terminal domain, followed by four WW domains that precede the HECT domain. The C2 domain mediates WWP1 localization at cellular membranes. Indeed, WWP1 is predominantly localized to plasma membrane, endosomes and Golgi apparatus, though it is also partially nuclear. Its expression is frequently deregulated in tumors, in which WWP1 plays an oncogenic role as a positive modulator of cancer cell proliferation and survival [42, 60,61,62]. High expression of WWP1 is generally associated with poor prognosis. In AML, WWP1 overexpression confers a proliferative advantage to leukemic blasts, by limiting the stability of p27Kip and, ultimately, promoting cell cycle entry [42] (Fig. 3). In addition, WWP1 overexpression represses basal autophagy thus increasing cell viability and inhibiting differentiation of leukemic blasts. In particular, in APL cells, re-establishment of the autophagic flux via WWP1 silencing is responsible for myeloid maturation of leukemic blasts expressing oncoproteins that are sensitive to autophagy-dependent proteolysis (e.g. PML-RARalpha and FLT3/ITD) (Fig. 3). Autophagy-dependent destabilization of PML-RARalpha upon WWP1 inactivation results in a partial restoration of ATRA target-gene expression, including genes involved in granulocytic differentiation. From a molecular perspective, WWP1 seems to prevent autophagy activation by affecting ATG7 and LC3 stability, thus interfering with the formation and elongation of autophagosomes. Though still speculative, data from Sanarico and colleagues [42] suggest that WWP1 would favor the degradation of autophagic proteins that are involved in the elongation and closure of the autophagosomal membranes, thus ultimately inhibiting the autophagic flux. The prevalent subcellular localization of WWP1 to membrane compartments, which represent the relevant nucleation sites for autophagosome formation, further implies WWP1 as a regulator of the early steps of autophagy.

A model for the outcome of WWP1 overexpression in AML blasts. Elevated amounts of WWP1 may limit the stability of the cell cycle inhibitor p27Kip, thus promoting cell cycle progression and proliferation. WWP1 can also interfere with the basal autophagic flux, possibly by negatively regulating ATG7 and LC3 levels, hence preventing autophagy and increasing blast survival. In leukemias bearing oncoproteins (e.g. (PML-RARalpha) susceptible to autophagy-mediated disposal, WWP1 overexpression would counteract their proteolysis. As a consequence, WWP1 would prevent myeloid differentiation
2.2 ITCH
ITCH was initially identified through genetic studies examining the mutation of the agouti locus, which causes coat-colour alterations and itching of the skin in mice [63, 64]. The expression of both agouti and itch genes is affected by the 18H mutation, which characterizes Itchy mice with severe immune and inflammatory defects [64, 65]. ITCH has been additionally shown to be required for the development of immune cells. In particular, it regulates the differentiation of T lymphocytes via its Jun family protein substrates, c-Jun and JunB [66].
ITCH is interestingly emerging to have a role in tumorigenesis, and in haematological malignancies as well. Most of its binding partners and targets are indeed involved in signalling, differentiation and apoptosis [66,67,68,69]. ITCH itself is a substrate of apoptotic effectors, such as caspases, which cleave it in CLL primary samples [70] (Fig. 4). The enzymatic cleavage (Asp240) causes the loss of an N-terminal ITCH fragment that contains a JNK1 regulatory region [71, 72]. Phosphorylation of this regulatory domain by JNK1 is necessary for disrupting an intramolecular inhibitory interaction, and to induce a conformational change that enhances the catalytic activity of ITCH [72]. The cleavage by caspases might therefore interfere with ITCH ubiquitination function. In addition, since the N-terminal C2 domain of the NEDD4-like E3s is important for their subcellular localization [73], the caspase activity on ITCH may affect it as well [70].

Implications of ITCH in apoptotic cell death of CLL cells. A Upon apoptotic stimuli, Caspase-3 and Caspase-7-mediated cleavage of ITCH on Asp240 residue may affect its function and localization by releasing an N-terminal fragment that contains the JNK1 regulatory domain and the C2 domain, respectively. B Both DNA damage (e.g. chemotherapeutic drugs) and HDAC inhibitors promote ITCH downregulation. HDAC inhibition activates E2F1- and Myc-mediated transcription of miR106b, promotes ITCH downregulation. ITCH reduces p73 levels by targeting it for poly-ubiquitination and proteasomal degradation, thus inhibiting apoptosis
ITCH involvement in cell death regulation is further demonstrated by the crucial role of one of its substrates, the tumour suppressor p73. The latter is rarely mutated in cancer [74, 75], differently from p53, which instead accumulates mutations also in CLL [76]. p73 efficiently binds p53 responsive elements for transcription activation of proapoptotic target genes [77]. Under basal conditions, ITCH maintains p73 at low levels, by binding the tumor suppressor via its C-terminal PPPY motif and targeting it for polyubiquitination and subsequent proteasomal degradation [68]. In response to genotoxic stress, ITCH expression is downregulated, leading to the stabilization and concomitant transcriptional activation of p73 (Fig. 4).
A similar mechanism has been described in CLL, a disorder characterized by dysfunctional apoptosis and low miRNAs expression, which are thought to be responsible for the resistance of leukemic cells to cell death [78]. Sampath and collaborators [79] have reported that treatment of CLL primary cells with deacetylase inhibitors reactivates E2F1- and MYC-mediated transcription of miR106b, which, in turn, promotes ITCH downregulation and p73 stabilization (Fig. 4). As a result, p73-mediated PUMA transcription is induced, leading to mitochondrial dysfunction, activation of caspases and apoptosis of CLL cells [79]. Such body of evidence suggests that the use of deacetylase inhibitors for ITCH inhibition may be considered as a novel antitumor approach for CLL therapy.
2.3 Neuronal precursor cell-expressed developmentally down-regulated (NEDD4)
NEDD4, the prototypic member of the NEDD4-like subgroup, plays a role in different cellular functions including inflammation, adaptive immunity, proliferation, apoptosis and carcinogenesis [80]. Abnormal expression of NEDD4, has been reported in several human cancers including, prostate, bladder and colon carcinomas [81], and its overexpression is frequently associated with disease progression [82]. However, dual roles for NEDD4 in carcinogenesis have been described. The majority of the studies converge towards an oncogenic role for NEDD4. Its oncogenic functions are mainly the result of its ability to activate the PI3K/AKT signalling pathway [83, 84]. Mono-ubiquitination of PTEN by NEDD4, instead, appears to have a tumour suppressive outcome, because it drives PTEN cytoplasmic/nuclear shuttling [85].
The extent of NEDD4 contribution to haematological malignancies is still far to be understood. Shukla et al. [86] have investigated the expression of NEDD4 in CLL cells and focused on its possible upstream regulatory factors. Characterization of Interferon Regulatory Factor (IRF4) deficient mice that develop spontaneous CLL revealed a dramatic reduction of NEDD4 and consequent hyperactivation of the NOTCH signaling, which is indispensable for leukemia development. Indeed, abnormal activation of the NOTCH signaling pathway is one of the most recurrent molecular alterations in human CLL [87]. Being a transcriptional target of IRF4 [86] and acting as an E3 for NOTCH [88], NEDD4 provides a functional link between IRF4 and the NOTCH signaling pathway in CLL cells.
2.4 SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1)
SMURF1 acts as an E3 for ABL that is rapidly degraded by the proteasome upon ubiquitination [56, 89]. ABL modification by SMURF1 is dependent on the first 45 amino acids of the kinase that, as a result of the chromosomal translocation, are not present in the BCR-ABL chimera [56]. This N-terminal domain of ABL contains a degradation signal that is required for its interaction with SMURF1 and contains four lysine residues, whose modification regulates ABL turnover. Coherently, both BCR-ABL and a deletion mutant of ABL (ABLD45), lacking the first 45 amino acids, but not ABL, drive growth factor-independent proliferation and survival of the human erythroleukemic cell line TF-1 [56]. Similarly, ABLD45 expression promotes tumor development and growth in xenotrasplantation models [56]. In conclusion, the study by Yan and colleagues [56] demonstrates that loss of the N-terminal ABL domain, caused by the (9;22)(q34;q11) chromosomal translocation, prevents ABL degradation thus contributing to the oncogenic potential of the BCR-ABL fusion protein.
2.5 HERC1
HERC1 is one of the large HERC enzymes, whose contribution to tumorigenesis has only recently began to be explored. Accumulating evidences have highlighted a role for HERC1 as a tumor suppressor. Point mutations in HERC1 have been detected in AML, T-ALL and T-cell prolymphocytic leukemia [90, 91]. Somatic deletions of HERC1 have been observed in newly diagnosed ALL, in which they have been associated to an increased resistance to antileukemic agents [92]. These leukemias express high mRNA levels of the DNA mismatch repair enzyme MSH2, while its protein levels are low or undetectable, suggesting the existence of a post-transcriptional regulatory mechanism. HERC1 functions as a positive regulator of MSH2 protein stability; thus, reduction of MSH2 protein amount and chemoresistance are possibly achieved as a result of HERC1 deletion/inactivation in ALL cells. In addition, though very rare, an atypical HERC1-PML transcript fusion mRNA has been described in APL, though the functional outcome of this chimera has not been yet investigated [93]. Ali and collaborators [94] have recently reported that HERC1 expression is dysregulated in myeloid related neoplasms. At diagnosis, HERC1 transcript levels are markedly down-regulated in AML, CML and primary myelofibrosis, an aggressive form of myeloproliferative neoplasm (MPN), relatively to bone marrow and peripheral blood control cells. In particular, in AML patients, HERC1 deregulation was not found associated with any molecular or chromosomal alteration. In CML patients, molecular remission was associated with restoration of HERC1 expression to the levels of control subjects, while, at relapse, the amount of the E3 underwent a further decline. At the molecular level, HERC1 is a protein interactor and a substrate for BCR-ABL1-mediated phosphorylation. To date, whether HERC1 may act as a BCR-ABL1 negative modulator, or if its phosphorylation by BCR-ABL1 may regulate its enzymatic activity, as for other family members, remain open questions. Similarly, the specific contribution of HERC1 to normal hematopoiesis and leukemogenesis as well as the identification of the involved substrates require further investigation.
2.6 E6-associated protein (E6AP)
E6AP is a member of the “Other”-HECT E3 sub-group and was originally identified as a protein involved in the human papillomavirus E6-oncoprotein-induced degradation of p53 [95]. However, a number of E6-independent targets of E6AP have been identified [32]. Amongst these, PML has been reported as a target for E6AP-mediated ubiquitin/proteasomal degradation in normal lymphoid cells [96] and in B-cell lymphomas [97]. The E6AP-PML axis plays a role in B-cell lymphomagenesis. E6AP is overexpressed in Burkitt lymphoma tumor cells, in which it counteracts the ability of PML to induce cellular senescence [97].
Another relevant target of E6AP in leukemogenesis is C/EBPα [98], a transcription factor that regulates myeloid differentiation and is mutated or deregulated in AML. E6AP-dependent disposal of C/EBPα negatively affects its transcriptional activity. By targeting C/EBPα for degradation, E6AP contributes to inhibit granulopoiesis of AML cells. Furthermore, E6AP prevents myeloid maturation inducing the proteolysis of MAX-binding protein MNT [99]. MNT is a member of the MYC/MAX/MAD network of transcription factors that acts as a MYC antagonist. It is up-regulated in response to differentiating agents and is a key mediator of ATRA-induced myeloid growth arrest and granulocytic differentiation [99]. Kapoor and colleagues [99] reported that ATRA rescues MNT from proteasomal degradation by inhibiting E6AP. Although further studies are needed to unveil the molecular basis of E6AP inhibition by differentiating agents, the Authors suggested a potential auto-degradation mechanism for E6AP. Altogether, these studies indicate that targeting E6-AP may offer a therapeutic strategy to restore myeloid differentiation of AML blasts.
3 Conclusions
In brief, the work on the HECT E3s expands our understanding of the pathogenesis of hematological diseases, providing novel effectors, whose biological significance and therapeutic interest might be explored in the future, especially considering the development of precision oncology [100,101,102]. This field has opened several novel opportunities, thanks to new algorithms [103,104,105] and artificial intelligence [106], modernizing the field of precision oncology in general [107,108,109] as well as in specific cancers [110,111,112,113]. From this overview, it appears clear that further studies are needed to corroborate the implications of HECT-type E3s in leukemogenesis, to understand the molecular basis of their oncogenic function in hematological disorders, as well as to identify their relevant substrates and regulators. A remarkable challenge in the field is to establish whether HECT E3s can serve as targets for therapeutic intervention along with the identification of inhibitors for clinical application. Considerable effort has been made in the generation of HECT inhibitors. However, since the catalytic domain is highly conserved amongst the family members and, at least for the NEDD4-like sub-group, the E3s share an elevated degree of protein similarity, selective inhibitors have not yet been identified.
Data availability
Not applicable.
Abbreviations
- E1:
-
E1 activating enzyme,
- E2:
-
E2 conjugating enzyme,
- E3:
-
E3 ubiquitin protein ligases,
- UPS:
-
Ubiquitin proteasome system,
- HECT:
-
Homologous to E6AP C-Terminus,
- RLDs:
-
RCC [regulator of chromatin condensation 1]-like domain,
- CLL:
-
Chronic myelogenous leukemia,
- T-ALL:
-
T-acute lymphocytic leukemia,
- BCR-ABL:
-
Breakpoint cluster region-Abelson proto-oncogene
- APL:
-
Acute promyelocytic leukemia,
- ALM:
-
Acute myeloid leukemia,
- ATRA:
-
All-trans-retinoic acid,
- ATO:
-
Arsenic trioxide,
- PML:
-
Promyelocytic leukemia protein,
- RARalpha:
-
Retinoic acid receptor-alpha
- WWP1:
-
WW domain-containing E3 ubiquitin protein ligase 1,
- NEDD4:
-
Neuronal precursor cell-expressed developmentally down-regulated,
- IRF4:
-
Interferon Regulatory Factor 4,
- SMURF1:
-
SMAD-specific E3 ubiquitin protein ligase 1,
- E6AP:
-
E6-associated protein,
- ARM:
-
Armadillo repeat‐containing domain,
- AZUL:
-
Amino‐terminal Zn‐finger of Ube3a ligase domain,
- WWE:
-
WWE domain,
- BH3:
-
Bcl-2 homology 3 domain,
- ANK:
-
Ankyrin repeat‐containing domain,
- PABC:
-
Polyadenylate‐binding protein C‐terminal domain,
- UBA:
-
Ubiquitin-associated domain,
- ZnF:
-
Zinc finger domain
References
Ciechanover A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005;12:1178–90.
Ciechanover A. The unravelling of the ubiquitin system. Nat Rev Mol Cell Biol. 2015;16:322–4.
Ziv NE, Ciechanover A. A possible non-proteolytic role of ubiquitin conjugation in alleviating the pathology of Huntingtin’s aggregation. Cell Death Differ. 2021;28:814–7.
Basar MA, Beck DB, Werner A. Deubiquitylases in developmental ubiquitin signaling and congenital diseases. Cell Death Differ. 2021;28(2):538–56.
Zhang Q, Zhang ZY, Du H, Li SZ, Tu R, Jia YF, Zheng Z, Song XM, Du RL, Zhang XD. DUB3 deubiquitinates and stabilizes NRF2 in chemotherapy resistance of colorectal cancer. Cell Death Differ. 2019;26:2300–13.
Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci. 2017;42(11):873–86.
Le Guerroué F, Youle RJ. Ubiquitin signaling in neurodegenerative diseases: an autophagy and proteasome perspective. Cell Death Differ. 2021;28:439–54.
Weinelt N, van Wijk SJL. Ubiquitin-dependent and -independent functions of OTULIN in cell fate control and beyond. Cell Death Differ. 2021;28:493–504.
Damgaard RB, Jolin HE, Allison MED, Davies SE, Titheradge HL, McKenzie ANJ, Komander D. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. 2020;27:1457–74.
Yogesh Kulathu DK. Atypical ubiquitylation — the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–23.
Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A. 1980;77:1783–6.
Melino G, Cecconi F, Pelicci PG, Mak TW, Bernassola F. Emerging roles of HECT-type E3 ubiquitin ligases in autophagy regulation. Mol Oncol. 2019;13:2033–48.
Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021;28(2):427–38.
Ji CH, Kim HY, Heo AJ, Lee SH, Lee MJ, Kim SB, Srinivasrao G, Mun SR, Cha-Molstad H, Ciechanover A, Choi CY, Lee HG, Kim BY, Kwon YT. The N-degron pathway mediates ER-phagy. Mol Cell. 2019;75:1058-1072.e9.
Cho HI, Kim MS, Lee J, Yoo BC, Kim KH, Choe KM, Jang YK. BRPF3-HUWE1-mediated regulation of MYST2 is required for differentiation and cell-cycle progression in embryonic stem cells. Cell Death Differ. 2020;27:3273–88.
Roberts JZ, Holohan C, Sessler T, Fox J, Crawford N, Riley JS, Khawaja H, Majkut J, Evergren E, Humphreys LM, Ferris J, Higgins C, Espona-Fiedler M, Moynagh P, McDade SS, Longley DB. The SCFSkp2 ubiquitin ligase complex modulates TRAIL-R2-induced apoptosis by regulating FLIP(L). Cell Death Differ. 2020;27:2726–41.
Djajawi TM, Liu L, Gong JN, Huang AS, Luo MJ, Xu Z, Okamoto T, Call MJ, Huang DCS, van Delft MF. MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1:NOXA complex. Cell Death Differ. 2020;27:2484–99.
Nieto A, Hara MR, Quereda V, Grant W, Saunders V, Xiao K, McDonald PH, Duckett DR. βarrestin-1 regulates DNA repair by acting as an E3-ubiquitin ligase adaptor for 53BP1. Cell Death Differ. 2020;27:1200–13.
Strappazzon F, Di Rita A, Peschiaroli A, Leoncini PP, Locatelli F, Melino G, Cecconi F. HUWE1 controls MCL1 stability to unleash AMBRA1-induced mitophagy. Cell Death Differ. 2020;27:1155–68.
Jahan AS, Elbæk CR, Damgaard RB. Met1-linked ubiquitin signalling in health and disease: inflammation, immunity, cancer, and beyond. Cell Death Differ. 2021;28:473–92.
Kravtsova-Ivantsiv Y, Goldhirsh G, Ivantsiv A, Itzhak OB, Kwon YT, Pikarsky E, Ciechanover A. Excess of the NF-κB p50 subunit generated by the ubiquitin ligase KPC1 suppresses tumors via PD-L1-and chemokines-mediated mechanisms. Proc Natl Acad Sci U S A. 2020;117:29823–31.
Hakim-Eshed V, Boulos A, Cohen-Rosenzweig C, Yu-Taeger L, Ziv T, Kwon YT, Riess O, Nguyen HHP, Ziv NE, Ciechanover A. Site-specific ubiquitination of pathogenic huntingtin attenuates its deleterious effects. Proc Natl Acad Sci U S A. 2020;117:18661–9.
Mulcahy Levy JM, Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020;27:843–57.
Hübner CA, Dikic I. ER-phagy and human diseases. Cell Death Differ. 2020;27:833–42.
Li X, Guo M, Cai L, Du T, Liu Y, Ding HF, Wang H, Zhang J, Chen X, Yan C. Competitive ubiquitination activates the tumor suppressor p53. Cell Death Differ. 2020;27:1807–18.
Ding X, Jia X, Wang C, Xu J, Gao SJ, Lu C. A DHX9-lncRNA-MDM2 interaction regulates cell invasion and angiogenesis of cervical cancer. Cell Death Differ. 2019;26:1750–65.
Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differ. 2020;27:858–71.
Keller MD, Torres VJ, Cadwell K. Autophagy and microbial pathogenesis. Cell Death Differ. 2020;27:872–86.
Allen EA, Baehrecke EH. Autophagy in animal development. Cell Death Differ. 2020;27:903–18.
Xian H, Liou YC. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2021;28:827–42.
Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14:10–21.
Bernassola F, Chillemi G, Melino G. HECT-type E3 ubiquitin ligases in cancer. Trends Biochem Sci. 2019;44:1057–75.
Verdecia MA, Joazeiro CAP, Wells NJ, Ferrer JL, Bowman ME, Hunter T, Noel JP. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell. 2003;11:249–59.
Shah SS, Kumar S. Adaptors as the regulators of HECT ubiquitin ligases. Cell Death Differ. 2021;28:455–72.
Scialpi F, Malatesta M, Peschiaroli A, Rossi M, Melino G, Bernassola F. Itch self-polyubiquitylation occurs through lysine-63 linkages. Biochem Pharmacol. 2008;76:1515–21.
Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SMV, Komander D. Assembly and specific recognition of K29- and K33-linked polyubiquitin. Mol Cell. 2015;58:95–109.
Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409.
Yeh CH, Bellon M, Pancewicz-Wojtkiewicz J, Nicot C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc Natl Acad Sci U S A. 2016;113:6731–6.
Close V, Close W, Kugler SJ, Reichenzeller M, Yosifov DY, Bloehdorn J, Pan L, Tausch E, Westhoff MA, Döhner H, Stilgenbauer S, Oswald F, Mertens D. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood. 2019;133:830–9.
Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112(7):1415–23.
Zhou M, Yeager AM, Smith SD, Findley HW. Overexpression of the MDM2 gene by childhood acute lymphoblastic leukemia cells expressing the wild-type p53 gene. Blood. 1995;85:1608–14.
Sanarico AG, Ronchini C, Croce A, Memmi EM, Cammarata UA, De Antoni A, Lavorgna S, Divona M, Giacò L, Melloni GEM, Brendolan A, Simonetti G, Martinelli G, Mancuso P, Bertolini F, Coco FL, Melino G, Pelicci PG, Bernassola F. The E3 ubiquitin ligase WWP1 sustains the growth of acute myeloid leukaemia. Leukemia. 2018;32:911–9.
Bahram F, Von Der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–10.
Sanchez-Lopez E, Ghia EM, Antonucci L, Sharma N, Rassenti LZ, Xu J, Sun B, Kipps TJ, Karin M. NF-κB-p62-NRF2 survival signaling is associated with high ROR1 expression in chronic lymphocytic leukemia. Cell Death Differ. 2020;27:2206–16.
Dittmann J, Haydn T, Metzger P, Ward GA, Boerries M, Vogler M, Fulda S. Next-generation hypomethylating agent SGI-110 primes acute myeloid leukemia cells to IAP antagonist by activating extrinsic and intrinsic apoptosis pathways. Cell Death Differ. 2020;27:1878–95.
Fitzsimmons L, Cartlidge R, Chang C, Sejic N, Galbraith LCA, Suraweera CD, Croom-Carter D, Dewson G, Tierney RJ, Bell AI, Shannon-Lowe C, Herold MJ, Rickinson AB, Colman PM, Huang DCS, Strasser A, Kvansakul M, Rowe M, Kelly GL. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020;27:1554–68.
Jost PJ, Höckendorf U. Necroinflammation emerges as a key regulator of hematopoiesis in health and disease. Cell Death Differ. 2019;26:53–67.
Villalobos-Ortiz M, Ryan J, Mashaka TN, Opferman JT, Letai A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2020;27:999–1007.
Tailler M, Lindqvist LM, Gibson L, Adams JM. By reducing global mRNA translation in several ways, 2-deoxyglucose lowers MCL-1 protein and sensitizes hemopoietic tumor cells to BH3 mimetic ABT737. Cell Death Differ. 2019;26:1766–81.
Anstee NS, Bilardi RA, Ng AP, Xu Z, Robati M, Vandenberg CJ, Cory S. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 AML in mice. Cell Death Differ. 2019;26:1316–31.
Jiao Y, Davis JE, Rautela J, Carrington EM, Ludford-Menting MJ, Goh W, Delconte RB, Souza-Fonseca-Guimaraes F, Koldej R, Gray D, Huang D, Kile BT, Lew AM, Ritchie DS, Huntington ND. Recipient BCL2 inhibition and NK cell ablation form part of a reduced intensity conditioning regime that improves allo-bone marrow transplantation outcomes. Cell Death Differ. 2019;26:1516–30.
Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–45.
Jiang S, Wang X, He Y, Huang H, Cao B, Zhang Z, Liu J, Wang Q, Huang Z, Mao X. Suppression of USP7 induces BCR-ABL degradation and chronic myelogenous leukemia cell apoptosis. Cell Death Dis. 2021;12:456.
Mao JH, Sun XY, Liu JX, Zhang QY, Liu P, Huang QH, Li KK, Chen Q, Chen Z, Chen SJ. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2010;107:21683–8.
Ru Y, Wang Q, Liu X, Zhang M, Zhong D, Ye M, Li Y, Han H, Yao L, Li X. The chimeric ubiquitin ligase SH2-U-box inhibits the growth of imatinib-sensitive and resistant CML by targeting the native and T315I-mutant BCR-ABL. Sci Rep. 2016;6:1–13.
Yan Z, Shanmugasundaram K, Ma D, Luo J, Luo S, Rao H. The N-terminal domain of the non-receptor tyrosine kinase ABL confers protein instability and suppresses tumorigenesis. J Biol Chem. 2020;295:9069–75.
Lo Coco F, Diverio D, Avvisati G, Petti MC, Meloni G, Pogliani EM, Biondi A, Rossi G, Carlo-Stella C, Selleri C, Martino B, Specchia G, Mandelli F. Therapy of molecular relapse in acute promyelocytic leukemia. Blood. 1999;94:2225–9.
Yoshida H, Kitamura K, Tanaka K, Omura S, Miyazaki T, Hachiya T, Ohno R, Naoe T. Accelerated degradation of PML-retinoic acid receptor α (PML-RARA) oncoprotein by all-trans-retinoic acid in acute promyelocytic leukemia: Possible role of the proteasome pathway. Cancer Res. 1996;56:2945–8.
Elzinga BM, Nyhan MJ, Crowley LC, O’Donovan TR, Cahill MR, McKenna SL. Induction of autophagy by Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am J Hematol. 2013;88:455–62.
Chen C, Zhou Z, Ross JS, Zhou W, Dong J-T. The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer. 2007;121:80–7.
Lee YR, Chen M, Lee JD, Zhang J, Lin SY, Fu TM, Chen H, Ishikawa T, Chiang SY, Katon J, Zhang Y, Shulga YV, Bester AC, Fung J, Monteleone E, Wan L, Shen C, Hsu CH, Papa A, Clohessy JG, Teruya-Feldstein J, Jain S, Wu H, Matesic L, Chen RH, Wei W, Pandolfi PP. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science (80-). 2019;364:eaau0159.
Hu X, Yu J, Lin Z, Feng R, Wang Z, Chen G. The emerging role of WWP1 in cancer development and progression. Cell Death Discov. 2021;7(1):1–8.
Hustad CM, Perry WL, Siracusa LD, Rasberry C, Cobb L, Cattanach BM, Kovatch R, Copeland NG, Jenkins NA. Molecular genetic characterization of six recessive viable alleles of the mouse agouti locus. Genetics. 1995;140:255–65.
Perry WL, Hustad CM, Swing DA, Norene O’Sullivan T, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a(18H) mice. Nat Genet. 1998;18:143–6.
Melino G, Gallagher E, Aqeilan RI, Knight R, Peschiaroli A, Rossi M, Scialpi F, Malatesta M, Zocchi L, Browne G, Ciechanover A, Bernassola F. Itch: A HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008;15:1103–12.
Fang D, Kerppola TK. Ubiquitin-mediated flourescence complementation reveals that Jun ubiquitinated by Itch/AIP4 is localized to lysosomes. Proc Natl Acad Sci U S A. 2004;101:14782–7.
Bellomaria A, Barbato G, Melino G, Paci M, Melino S. Recognition mechanism of p63 by the E3 ligase Itch: Novel strategy in the study and inhibition of this interaction. Cell Cycle. 2012;11:3638–48.
Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–48.
Bellomaria A, Barbato G, Melino G, Paci M, Melino S. Recognition of p63 by the E3 ligase ITCH: Effect of an ectodermal dysplasia mutant. Cell Cycle. 2010;9:3730–9.
Rossi M, Inoue S, Walewska R, Knight RA, Dyer MJS, Cohen GM, Melino G. Caspase cleavage of Itch in chronic lymphocytic leukemia cells. Biochem Biophys Res Commun. 2009;379:659–64.
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPL turnover. Cell. 2006;124:601–13.
Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation- induced conformational change. Proc Natl Acad Sci U S A. 2006;103:1717–22.
Dunn R, Klos DA, Adler AS, Hicke L. The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo. J Cell Biol. 2004;165:135–44.
Candi E, Agostini M, Melino G, Bernassola F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: regulators and effectors. Hum Mutat. 2014;35:702–14.
Dötsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol. 2010;2:a004887.
Sturm I, Bosanquet AG, Hermann S, Güner D, Dörken B, Daniel PT. Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy. Cell Death Differ. 2003;10:477–84.
Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 Induces Apoptosis via PUMA Transactivation and Bax Mitochondrial Translocation. J Biol Chem. 2004;279:8076–83.
Marton S, Garcia MR, Robello C, Persson H, Trajtenberg F, Pritsch O, Rovira C, Naya H, Dighiero G, Cayota A. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia. 2008;22:330–8.
Sampath D, Calin GA, Puduvalli VK, Gopisetty G, Taccioli C, Liu CG, Ewald B, Liu C, Keating MJ, Plunkett W. Specific activation of microRNA 106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood. 2009;113:3744–53.
Liu Q, Zhang S, Sun Z, Guo X, Zhou H. E3 ubiquitin ligase Nedd4 is a key negative regulator for non-canonical inflammasome activation. Cell Death Differ. 2019;26:2386–99.
Eide PW, Cekaite L, Danielsen SA, Eilertsen IA, Kjenseth A, Fykerud TA, Ågesen TH, Bruun J, Rivedal E, Lothe RA, Leithe E. NEDD4 is overexpressed in colorectal cancer and promotes colonic cell growth independently of the PI3K/PTEN/AKT pathway. Cell Signal. 2013;25:12–8.
Sun A, Yu G, Dou X, Yan X, Yang W, Lin Q. Nedd4-1 is an exceptional prognostic biomarker for gastric cardia adenocarcinoma and functionally associated with metastasis. Mol Cancer. 2014;14:248.
Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. NEDD4-1 Is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–39.
Fukushima T, Yoshihara H, Furuta H, Kamei H, Hakuno F, Luan J, Duan C, Saeki Y, Tanaka K, Iemura SI, Natsume T, Chida K, Nakatsu Y, Kamata H, Asano T, Takahashi SI. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat Commun. 2015;16:6780.
Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56.
Shukla V, Shukla A, Joshi SS, Lu R. Interferon regulatory factor 4 attenuates Notch signaling to suppress the development of chronic lymphocytic leukemia. Oncotarget. 2016;7:41081–94.
Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, Grunn A, Fangazio M, Capello D, Monti S, Cresta S, Gargiulo E, Forconi F, Guarini A, Arcaini L, Paulli M, Laurenti L, Larocca LM, Marasca R, Gattei V, Oscier D, Bertoni F, Mullighan CG, Foà R, Pasqualucci L, Rabadan R, Dalla-Favera R, Gaidano G. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J Exp Med. 2011;208:389–401.
Koncarevic A, Jackman RW, Kandarian SC. The ubiquitin-protein ligase Nedd4 targets Notch1 in skeletal muscle and distinguishes the subset of atrophies caused by reduced muscle tension. FASEB J. 2007;21:427–37.
Matsumoto Y, LaRose J, Kent OA, Wagner MJ, Narimatsu M, Levy AD, Omar MH, Tong J, Krieger JR, Riggs E, Storozhuk Y, Pasquale J, Ventura M, Yeganeh B, Post M, Moran MF, Grynpas MD, Wrana JL, Superti-Furga G, Koleske AJ, Pendergast AM, Rottapel R. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J Clin Invest. 2016;126:4482–96.
Schneider T, Martinez-Martinez A, Cubillos-Rojas M, Bartrons R, Ventura F, Rosa JL. Large HERCS function as tumor suppressors. Front Oncol. 2019;9:524.
Opatz S, Bamopoulos SA, Metzeler KH, Herold T, Ksienzyk B, Bräundl K, Tschuri S, Vosberg S, Konstandin NP, Wang C, Hartmann L, Graf A, Krebs S, Blum H, Schneider S, Thiede C, Middeke JM, Stölzel F, Röllig C, Schetelig J, Ehninger G, Krämer A, Braess J, Görlich D, Sauerland MC, Berdel WE, Wörmann BJ, Hiddemann W, Spiekermann K, Bohlander SK, Greif PA. The clinical mutatome of core binding factor leukemia. Leukemia. 2020;34:1553–62.
Diouf B, Cheng Q, Krynetskaia NF, Yang W, Cheok M, Pei D, Fan Y, Cheng C, Krynetskiy EY, Geng H, Chen S, Thierfelder WE, Mullighan CG, Downing JR, Hsieh P, Pui CH, Relling MV, Evans WE. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298–303.
Walz C, Grimwade D, Saussele S, Lengfelder E, Haferlach C, Schnittger S, Lafage-Pochitaloff M, Hochhaus A, Cross NCP, Reiter A. Atypical mRNA fusions in PML-RARA positive, RARA-PML negative acute promyelocytic leukemia. Genes Chromosom Cancer. 2010;49:471–9.
Ali MS, Panuzzo C, Calabrese C, Maglione A, Piazza R, Cilloni D, Saglio G, Pergolizzi B, Bracco E. The giant hect e3 ubiquitin ligase herc1 is aberrantly expressed in myeloid related disorders and it is a novel bcr-abl1 binding partner. Cancers (Basel). 2021;13:341.
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505.
Louria-Hayon I, Alsheich-Bartok O, Levav-Cohen Y, Silberman I, Berger M, Grossman T, Matentzoglu K, Jiang YH, Muller S, Scheffner M, Haupt S, Haupt Y. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ. 2009;16:1156–66.
Wolyniec K, Shortt J, De Stanchina E, Levav-Cohen Y, Alsheich-Bartok O, Louria-Hayon I, Corneille V, Kumar B, Woods SJ, Opat S, Johnstone RW, Scott CL, Segal D, Pandolfi PP, Fox S, Strasser A, Jiang YH, Lowe SW, Haupt S, Haupt Y. E6AP ubiquitin ligase regulates PML-induced senescence in Myc-driven lymphomagenesis. Blood. 2012;120:822–32.
Pal P, Lochab S, Kanaujiya JK, Kapoor I, Sanyal S, Behre G, Trivedi AK. E6AP, an E3 ubiquitin ligase negatively regulates granulopoiesis by targeting transcription factor C/EBPα for ubiquitin-mediated proteasome degradation. Cell Death Dis. 2013;4:e590.
Kapoor I, Kanaujiya J, Kumar Y, Thota JR, Bhatt MLB, Chattopadhyay N, Sanyal S, Trivedi AK. Proteomic discovery of MNT as a novel interacting partner of E3 ubiquitin ligase E6AP and a key mediator of myeloid differentiation. Oncotarget. 2016;7:7640–56.
Han Y, Ye X, Wang C, Liu Y, Zhang S, Feng W, Huang K, Zhang J. Integration of molecular features with clinical information for predicting outcomes for neuroblastoma patients. Biol Direct. 2019;14:1–16.
Kim SY, Jeong HH, Kim J, Moon JH, Sohn KA. Robust pathway-based multi-omics data integration using directed random walks for survival prediction in multiple cancer studies. Biol Direct. 2019;14:1–13.
Harris ZN, Dhungel E, Mosior M, Ahn TH. Massive metagenomic data analysis using abundance-based machine learning. Biol Direct. 2019;14:1–13.
Ryan FJ. Application of machine learning techniques for creating urban microbial fingerprints. Biol Direct. 2019;14:1–13.
Kawulok J, Kawulok M, Deorowicz S. Environmental metagenome classification for constructing a microbiome fingerprint. Biol Direct. 2019;14:1–23.
Mihaylov I, Kańduła M, Krachunov M, Vassilev D. A novel framework for horizontal and vertical data integration in cancer studies with application to survival time prediction models. Biol Direct. 2019;14:1–17.
Larmuseau M, Verbeke LPC, Marchal K. Associating expression and genomic data using co-occurrence measures. Biol Direct. 2019;14:1–14.
Amelio I, Bertolo R, Bove P, Candi E, Chiocchi M, Cipriani C, Di Daniele N, Ganini C, Juhl H, Mauriello A, Marani C, Marshall J, Montanaro M, Palmieri G, Piacentini M, Sica G, Tesauro M, Rovella V, Tisone G, Shi Y, Wang Y, Melino G. Cancer predictive studies. Biol Direct. 2020;15:1–7.
MacDonald L, Jenkins J, Purvis G, Lee J, Franco AT. The thyroid tumor microenvironment: potential targets for therapeutic intervention and prognostication. Horm Cancer. 2020;11:205–17.
Dembinski R, Prasath V, Bohnak C, Siotos C, Sebai ME, Psoter K, Gani F, Canner J, Camp MS, Azizi A, Jacobs L, Habibi M. Estrogen receptor positive and progesterone receptor negative breast cancer: the role of hormone therapy. Horm Cancer. 2020;11:148–54.
Ardolino L, Hansen A, Ackland S, Joshua A. Advanced adrenocortical carcinoma (ACC): a REVIEW WITH FOCUS ON SECOND-LINE THERAPIEs. Horm Cancer. 2020;11:155–69.
Del Moral-Morales A, González-Orozco JC, Capetillo-Velázquez JM, Piña-Medina AG, Camacho-Arroyo I. The role of mPRδ and mPRε in human glioblastoma cells: expression, hormonal regulation, and possible clinical outcome. Horm Cancer. 2020;11:117–27.
Sathyamoorthy N, Lange CA. Progesterone and breast cancer: an NCI workshop report. Horm Cancer. 2020;11:1–12.
Bolf EL, Gillis NE, Barnum MS, Beaudet CM, Yu GY, Tomczak JA, Stein JL, Lian JB, Stein GS, Frances E. HHS public access. 2021; 11: 34–41
Zhu K, Shan Z, Chen X, Cai Y, Cui L, Yao W, Wang Z, Shi P, Tian C, Lou J, Xie Y, Wen W. Allosteric auto-inhibition and activation of the Nedd4 family E3 ligase Itch. EMBO Rep. 2017;18:1618–30.
Gong W, Zhang X, Zhang W, Li J, Li Z. Structure of the HECT domain of human WWP2. Acta Crystallogr Sect Struct Biol Commun. 2015;71:1251–7.
Pandya RK, Partridge JR, Love KR, Schwartz TU, Ploegh HL. A structural element within the HUWE1 HECT domain modulates self-ubiquitination and substrate ubiquitination activities. J Biol Chem. 2010;285:5664–73.
Maspero E, Mari S, Valentini E, Musacchio A, Fish A, Pasqualato S, Polo S. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 2011;12:342–9.
Acknowledgements
We apologize for those contributions could not be cited due to space constraints.
Funding
This work was supported by PRIN_2017_2017XCXAFZ to FB, Associazione Italiana per la Ricerca contro il Cancro (AIRC) IG 23232 to FB and IG 20473 to GM, Fondazione Luigi Maria Monti IDI-IRCCS, Ministry of Health & MAECI Italy-China Science and Technology Cooperation (#PGR00961), and by Regione Lazio through LazioInnova Progetto Gruppo di Ricerca n 85–2017-14986; n 33 & 55–2021-T0002E0001.
Author information
Authors and Affiliations
Contributions
CF and SG prepared the figures. All authors contributed in writing and editing the manuscript. All the Authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Delvecchio, V.S., Fierro, C., Giovannini, S. et al. Emerging roles of the HECT-type E3 ubiquitin ligases in hematological malignancies. Discov Onc 12, 39 (2021). https://doi.org/10.1007/s12672-021-00435-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-021-00435-4