
Abstract
Progestins are reported to increase the risk of invasive breast cancers in postmenopausal women receiving hormone therapy with estrogen plus progestin. We report here that estrogen and progesterone receptor positive (ER+PR+) rat mammary tumors arising in the presence of estrogen and progesterone exhibit increased invasiveness and decreased expression of E-cadherin protein compared with tumors growing in the presence of estrogen alone. A similar decrease of E-cadherin expression was observed in human ER+PR+ invasive ductal carcinoma compared with ductal carcinoma in situ. In agreement with findings in the rat, estrogen plus progestin R5020 treatment decreased E-cadherin expression in vitro in T47D human breast cancer cells. Decrease of E-cadherin protein was mediated by progesterone receptor B (PRB) and dependent on the activation of the Wnt pathway. These results suggest that progesterone signaling via PRB contributes to tumor invasiveness and can provide an important therapeutic target for treatment of invasive ER+PR+ breast cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Synthetic progestins (PS), molecules with activities similar to the natural hormone progesterone (P) [1], have been shown to increase breast cancer risk in postmenopausal women receiving estrogen plus progestin (E + PS) hormonal therapy (HT) compared with women receiving therapy with estrogen (E) alone [2]. Notably, breast cancers that develop in postmenopausal women with E + PS HT are more invasive and lead to higher mortality rate [3]. Although it has been reported that PS increases motility of human breast cancer cells in vitro [4, 5], it is not known how PS and, more importantly, endogenous P regulate invasive behavior of tumor cells in vivo.
P action in the breast is mediated by two distinct progesterone receptor (PR) isoforms, PRA and PRB. Upon activation, PRA and PRB stimulate expression of different subsets of genes leading to different biological outcomes in vitro [6, 7]. PRB is generally a more potent nuclear transcription factor than PRA [8]. PRB is critical for lobuloalveolar development of the mammary gland during pregnancy, while PRA function is essential for normal development of the uterine and ovarian epithelium [9, 10]. In the normal human breast, PRA and PRB are expressed in equimolar amounts; however, the PRA/PRB expression ratio is commonly altered in breast cancers [11]. However, the specific functional roles of the two PR isoforms in the etiology and biological behavior of mammary cancers has not been well defined.
The epithelial adhesion protein E-cadherin is a major component of tight junctions between mammary epithelial cells. The decrease or loss of E-cadherin protein is associated with poor prognosis in breast cancer patients [12]. Numerous studies have reported that E-cadherin mRNA expression is negatively regulated by transcription factors important for epithelial-to-mesenchymal transition (EMT) [13–15]. Based on in vitro studies, it has been proposed that cells composing ductal carcinoma in situ undergo EMT that is accompanied with decreased E-cadherin expression and become invasive. Alternatively, epigenetic mechanisms such as DNA hypermethylation have been shown to decrease E-cadherin mRNA and protein expression [16, 17].
The adult rat mammary gland has ductal–lobular organization that is very similar to the human breast. Furthermore, the rat is a highly relevant model to study P signaling in the normal breast and in breast cancer because PRA and PRB expression during development and PR isoform colocalization in the rat mammary gland closely resemble that in the human breast [18, 19]. In addition, the majority of mammary cancers induced by carcinogen in the rat express estrogen and progesterone receptors (ER+PR+) and are hormone-dependent [20]. By using the model of mammary cancer developing in ovariectomized rats treated with E + P versus E alone, we have identified a novel molecular mechanism by which exogenous PS or endogenous P may promote invasiveness in ER+PR+ human breast cancers. This mechanism may explain why breast cancers that develop in postmenopausal women receiving E + PS HT are more likely to metastasize into the lymph nodes than cancers that develop in postmenopausal women who have not received HT [3]. Our data strongly suggest that blocking P signaling via PRB in ER+PR+ breast cancer may curtail loss of E-cadherin, thereby preventing or restraining tumor invasiveness. Such therapies may be particularly important for women who have not benefited from conventional antiestrogen treatments.
Methods
Animals
Fifty-day-old Sprague–Dawley female rats (Charles-River Laboratory, Raleigh, NC) were ovariectomized, implanted with Silastic pellets containing E (2.5 mg/1 cm) with or without four pellets releasing P (50 mg/4 cm) (N = 16 and 17, for E and E + P treatment group, respectively) and treated with a single intragastric dose (50 mg/kg) of 7,12-dimethylbenz[α]anthracene (DMBA). Carcinogen-treated sham-operated rats received pellets containing cholesterol and served as ovary-intact control (N = 19 rats). Hormone-releasing pellets were replaced every 10 weeks. These replacement regimens produced systemic hormone levels within the physiological range (Online Resource 4). Plasma hormone levels were measured as previously described [34]. Normal mammary tissues and tumors were fixed in 10 % buffered formalin and paraffin embedded for immunohistochemical analysis or flash-frozen in liquid nitrogen and stored at −80 °C for mRNA or protein analysis. All animal handling procedures were approved by the Michigan State University committee on animal use and care.
Cell Lines
Human breast cancer T47D cells co-expressing PRA and PRB were purchased from ATCC (Manassas, VA). T47D cell lines, Y (lacking PR), YA (expressing only PRA), and YB (expressing only PRB) were generously provided by Dr. KB Horwitz (University of Colorado). Cells were grown in minimal essential media and 5 % fetal bovine serum as described [35] and changed to phenol red and serum-free media for 48 h prior to treatments with synthetic PS promegestone (R5020, 20 nM), estradiol (E, 10 nM), PR inhibitor (RU486, 100 nM), Src inhibitor (PP2, 20 μM), Akt inhibitor IV (5 μM), Wnt antagonist II (IWP-2, 10 μM) that inhibits Wnt processing and secretion, Wnt antagonist III (CCT036477, 10 μM) that blocks signaling at β-catenin levels (all from Calbiochem, La Jolla, CA), PI3 kinase inhibitor (LY294002, 10 μM), MEKK1/2 inhibitor (U0126, 10 μM) (both from Cell Signaling, Beverly, MA), or the natural Wnt antagonist (Dkk1, 1 μg/ml) (R&D Systems, Minneapolis, MN) for 24 h. R5020 was used for in vitro studies because P is rapidly metabolized in vitro [36]. Kinase inhibitors were tested for their ability to block proliferation (LY and U0126) in primary culture of normal mouse mammary epithelial organoids in 3-D cultures [37], for their ability to block organogenesis and decrease phosphorylated Src levels (PP2) in 3-D cultures of normal mouse mammary epithelial organoids [37], and their ability to block activation of their respective kinases (U0126, Akt inhibitor IV) in YB breast cancer cells treated with 200 ng/ml IGF1 (Cell Sciences, Canton, MA) for 30 min (Online Resource 5).
Human Archival Breast Cancer Samples
Archival human breast cancers were collected in 2000 at Sparrow Hospital, Lansing, MI in accordance with a protocol approved by the institutional review boards of Michigan State University and Sparrow Hospital. Based on pathology reports, all samples were positive for ERα and PR and consisted of invasive ductal carcinoma component with variable proportions of intra-ductal carcinoma in situ.
Histopathological Analysis
Rat tumors were evaluated by a board-certified veterinary pathologist (I.M.L) following the criteria from the Annapolis meeting on mammary pathology of genetically engineered mice [38].
Immunoblot, Cell Cycle Analysis, and RT-PCR-Based Microarray Analysis
Analyses were performed as previously described [34, 39]. All rat and human primers for RT-PCR were from Qiagen (Frederick, MD). Protein loading in immunoblots was normalized by intensity of β-actin bands.
Immunofluorescent Staining
Cells were fixed in 10 % buffered formalin for 30 min, washed with PBS, permeabilized with 0.05 % Triton-X100, blocked in 1 % BSA, incubated overnight with primary antibody, and developed with Alexa Fluor 488-labeled goat anti-rabbit antibody (Invitrogen, Carlsbad, CA; 1:400). Immunohistochemistry was performed as previously described [19]. Antibodies used in the study are described in Online Resource 6. Alexa Fluor-labeled secondary antibodies were from Invitrogen (Eugene, OR). Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI). Quantitative analysis of immunofluorescent staining with anti-Wnt 1 antibody was performed with MetaMorph software (Molecular Devices, Sunnyvale, CA).
Quantitation and Statistical Analysis
Results were presented as mean ± SEM, and differences were considered significant at P < 0.05 by ANOVA/MANOVA with Duncan's post hoc test or by χ2-test where appropriate.
Results
E + P Treatment Promotes Basal/Myoepithelial Tumor Cell Phenotype
Control ovary-intact (OI) and ovariectomized rats treated with E alone or E + P were given carcinogen to induce mammary cancers. Mammary epithelium in humans and rodents consists of two epithelial cell types, luminal and basal/myoepithelial. Luminal cells express steroid receptors ERα, PRA, and PRB [21] and keratin 18 (K18) [22]. Basal/myoepithelial cells express α-smooth muscle actin (SMA), transcription factor p63, and keratins 5 (K5) and 14 [23]. Based on expression of luminal (K18) and basal/myoepithelial (K5 and SMA) markers, all tumors were subdivided into four groups (Fig. 1a, b). All tumors from the E-treatment group expressed K18 with less than 5 % of K5 + SMA + cells (K18+K5low, N = 13 tumors). All tumors from OI group expressed K18 with up to 32 % of K5 + SMA + cells (K18+K5high, N = 12 tumors). In contrast, the tumors from E + P-treated rats exhibited marked loss of K18 expression and variable expression of K5 and SMA. Roughly equal number of tumors contained ∼12 % K5 + SMA + cells (K18−K5low, N = 12 tumors) or ∼25 % K5 + SMA + cells (K18−K5high, N = 17 tumors). Thus, endogenous E and P (OI group) or exogenous E + P treatment promoted expansion of basal/myoepithelial tumor cells. Interestingly, levels of K5 and K18 mRNA were not significantly different among experimental groups (Fig. 1c, d), indicating a discordance between mRNA levels and protein expression in tumors.

Combined treatment with estrogen plus progesterone alters cellular composition of rat tumors. Tumors were from ovary-intact (OI) or ovariectomized rats treated with estradiol alone (E) or estradiol plus progesterone (E + P) and treated with seven carcinogen DMBA. a Representative images of staining with anti-keratin 5 (K5, green, white arrows), anti-keratin 18 (K18, red, yellow arrows) antibodies, and 4',6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue). Scale bar, 50 μm. b Quantitation of tumor cells expressing basal/myoepithelial marker SMA (N = 8–16 samples/group). Tumors with less than 15 % of basal/myoepithelial cells are K5low; tumors with more than 15 % of basal/myoepithelial cells are K5high. *P < 0.05 for comparisons indicated by brackets. c, d Real-time RT-PCR analysis of tumor c K5 and d K18 mRNA expression (N = 5 tumors/group). Only K5high tumors were analyzed in the E + P group. The bars represent the mean ± SEM fold difference compared with mRNA expression in OI tumors that was arbitrarily taken as one
E + P Treatment Decreases E-Cadherin Protein and Increases Tumor Invasiveness
The invasiveness of human breast cancers is strongly associated with decrease or loss of the adhesion protein E-cadherin [12]. E-cadherin protein was highly expressed in normal epithelial cells adjacent to tumors in all treatment groups (Fig. 2a). Notably, E-cadherin protein was significantly decreased in tumors from E + P-treated rats, both K5low and K5high, compared to tumors from OI and E-treated rats (Fig. 2a, b). The decrease of E-cadherin protein occurred without a change of E-cadherin mRNA expression (Fig. 2c). Histopathologic analysis indicated that tumors from E + P-treated rats were also more invasive than tumors from OI or E-treated rats. The cellular phenotype of invading cells in the rat mammary cancers was similar among the treatment groups; invading cells expressed the basal/myoepithelial markers SMA, p63, and K5 (Fig. 3a, b). Furthermore, invading cells outside of the primary tumor mass frequently expressed PRB but did not express PRA, whereas cells within the tumor body expressed both PRA and PRB (Fig. 3c).

Combined treatment with estrogen plus progesterone decreases E-cadherin protein expression in vivo. a, b Mammary tissue and tumors were from carcinogen-treated ovary-intact (OI) or ovariectomized rats treated with estradiol alone (E) or estradiol plus progesterone (E + P). a Representative images of E-cadherin immunostaining (green, white arrows) in tumors and ducts and lobules surrounding the tumor from an E + P-treated rat. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. b Quantitation of the percent of tumors with normal, decreased, or absent E-cadherin expression when compared to normal mammary ducts adjacent to the tumor determined by immunostaining (N = 10–13 tumors/group). *P < 0.05, the percent of tumors with absence of E-cadherin is greater in E + P-treatment group than in OI or E treatment groups. c Real-time RT-PCR analysis of E-cadherin mRNA expression (N = 5 tumors/group). Only K5high tumors were analyzed in the E + P group. The bars represent the mean ± SEM fold difference compared with mRNA expression in OI tumors

Invasive cells in the rat mammary tumors express basal/myoepithelial markers. Tumors were from carcinogen-treated ovary-intact (OI) or ovariectomized rats treated with estradiol alone (E) or estradiol plus progesterone (E + P). a H&E staining. Low-power image of an invasive tumor (×10, left panel). Inset indicates the area of the tumor at higher power (×40, right panel) with a cord of invading cells (red arrow). b Immunolabeling of invasive cells with anti-K5 (green) and anti-SMA (red) antibodies (left panel; yellow arrow indicates a basal/myoepithelial K5 + SMA + cell) or with anti-p63 (teal) and anti-SMA (red) antibodies (right panel; white arrow indicates a basal p63 + SMA cell, yellow arrow indicates a basal/myoepithelial p63 + SMA + cell). c Immunostaining of PR isoforms in luminal cells in the tumor body and in invasive basal/myoepithelial cells. Immunolabeling with anti-K5 (red) and anti-PRA (teal) antibodies (left two panels) or anti-PRB (teal) antibodies (right two panels). White arrows indicate PRA+ or PRB+ tumor luminal cells, yellow arrows indicate basal/myoepithelial cells. b, c White dotted lines indicate the margin of the tumor. Nuclei were counterstained with DAPI (blue). Scale bars, 50 μm
Invasive ER+PR+ Human Breast Cancers Acquire Expression of Basal/Myoepithelial Marker K5 and Exhibit Decrease of E-Cadherin Expression
Next, we investigated expression of cell-type specific markers K18 and K5 and E-cadherin expression in archival samples of ER+PR+ primary human breast cancers that contained both an intra-ductal preinvasive in situ tumor component and an invasive tumor component (N = 21 patients). Ductal carcinomas in situ were composed of luminal K18+ cells circumscribed by a layer of basal/myoepithelial cells expressing K5 (Fig. 4a). Cells composing of invasive ductal carcinoma acquired expression of K5 that was ranging from only few K5 expressing cells in some tumors to the majority of cells expressing K5 (Fig. 4a). K18 was expressed in the majority of tumors, with some tumors exhibiting loss of K18 expression in some cells (Fig. 4a, a panel in the middle). Furthermore, invasive tumor cells frequently co-expressed K5 and K18 (Fig. 4a), indicating that the predominant phenotype of invasive cells in human breast cancers is a cell phenotype intermediate between luminal and basal/myoepithelial cells.

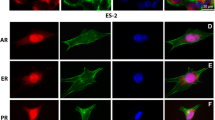
Invasiveness of human ER+PR+ breast cancers is associated with increased keratin 5 expression, decreased K18, and decreased E-cadherin expression. a Human breast cancer samples that contained in situ and invasive components were immunostained with anti-K5 (K5, green) and anti-K18 (K18, red) antibodies. Note variable expression and colocalization of K5 and K18 in invasive tumors. White arrows indicate luminal K18 + K5 cells; yellow arrows indicate basal/myoepithelial K18-K5+ cells; red arrows indicate double positive K18 + K5 cells. b, c Immunostaining with anti-E-cadherin antibody (green cytoplasmic staining) in normal duct and lobule adjacent to the tumor, carcinoma in situ, and invasive carcinoma. Carcinoma in situ exhibited normal E-cadherin expression (top panel) or decreased E-cadherin expression (bottom panel). Invasive carcinomas frequently exhibited loss of E-cadherin expression in some tumor cells (top and bottom panels). Cytoplasmic/membrane E-cadherin (white arrows), cytoplasmic E-cadherin (yellow arrow). Dotted lines indicate the edge of carcinoma in situ. c Percent of tumors with normal, decreased, or absence of E-cadherin expression. *P < 0.05, the percent of tumors with the absence of E-cadherin expression is greater in invasive carcinomas than in carcinoma in situ. d Representative images of staining with anti-PRA (teal nuclear staining) and anti-K5 (K5, red cytoplasmic staining) antibody. White arrows indicate cells co-expressing PRA and K5. Nuclei were counterstained with DAPI (blue). a, b, d Scale bars, 50 μm
E-cadherin was strongly expressed in the normal mammary epithelium adjacent to tumor (Fig. 4b). E-cadherin expression in carcinoma in situ ranged from similar to or decreased compared with E-cadherin expression in the normal tissue, whereas a more significant loss of E-cadherin was observed in IDC (Fig. 4b, c, Online Resource 1). Invasive tumor cells frequently co-expressed the luminal marker, PRA, and the basal/myoepithelial marker, K5 (Fig. 4d). Because PRA and PRB were frequently co-expressed in the same cell (Online Resource 1), we concluded that cells expressing PRA and K5 are likely to express PRB. Collectively, these data indicate that the transition from in situ to invasive tumor is associated with decreased E-cadherin expression, decreased K18 expression, and increased expression of K5.
E + PS Treatment Decreases E-Cadherin Protein in Breast Cancer Cells via Activation of the Wnt Pathway
To further investigate the effect of P/PS on E-cadherin protein, we used humanT47D breast cancer cells that co-express PRA and PRB. Indeed, treatment with E plus, the synthetic PS R5020 decreased the levels of E-cadherin precursor and mature E-cadherin protein (Fig. 5a). Accordingly, immunocytochemical analysis also showed a drastic decrease of E-cadherin staining in cells treated with E + R5020 (Fig. 5b). This effect of E + R5020 was completely abolished by the PR antagonist, RU486 (Fig. 5b). It has been shown that E-cadherin expression in breast cancer cells is regulated at the level of mRNA transcription by transcription factors important for EMT such as ZEB1, Twist, Snail, and Slug [13–15] or by epigenetic mechanisms [16, 17]; however, E-cadherin mRNA levels were not altered by E + R5020 treatment in T47D cells (Fig. 5c). We have tested tumors from ovary-intact rats and from rats treated with E or E + P for expression of Zeb1 and Snail by immunohistochemistry. We found no significant differences in expression of these transcription factors among groups. When we analyzed Twist1 mRNA expression, we found a trend for increased levels of Twist1 in tumors from E alone-treated rats compared to E + P-treated (data not shown). Overall, we found no evidence that E + P treatment induces transcription factors involved in EMT.

Progesterone decreases E-cadherin protein via activation of the Wnt pathway by progesterone receptor B (PRB). a T47D cells co-expressing PRA and PRB were treated with estradiol (E), R5020 (R), or E + R5020 (E + R) for 48 h and assayed by immunoblot. Numbers above bands indicate average levels of E-cadherin precursor or mature protein, respectively, compared with basal media (BM). b, c T47D cells were treated with E, R, E + R, and E + R plus progesterone receptor inhibitor RU486 for 48 h and assayed by b immunofluorescence (green cytoplasmic staining) or c processed for mRNA preparation. d, e Y, YA, and YB cells were treated with or without R5020 for 24 h and assayed by d immunoblot or e immunostaining. d Numbers above or below E-cadherin bands indicate levels compared with untreated cells for E-cadherin precursor or mature protein, respectively. f Representative images of E-cadherin immunostaining in YB cells treated with or without R5020 and with or without PR inhibitor RU486 (R + RU486) or Wnt inhibitors, Dkk1, and CCT. g-j T47D cells were treated with E, R, E + R, or E + R + RU486 for 48 h. Real-time RT-PCR analysis of g Wnt 1 mRNA or h RouU mRNA levels. The bars represent the mean ± SEM fold difference compared with mRNA levels in cells cultured in BM. i Representative images of staining with anti-Wnt 1 antibody and j quantitative analysis of Wnt 1 immunofluorescence in treated cells relative to BM. b, e, f, i Nuclei are stained with DAPI (blue). Scale bars, 50 μm
Next, we investigated which PR isoform mediated the effect of R5020 on E-cadherin protein. For these studies, we used PR-negative T47D cells (Y cells) and T47D cells expressing either only PRA (YA cells) or PRB (YB cells). Immunoblot analysis showed that treatment with R5020 significantly decreased levels of E-cadherin precursor (1.3 ± 0.08-fold, P < 0.05, N = 3 experiments) and mature E-cadherin protein (1.5 ± 0.09-fold, P < 0.05) only in cells expressing PRB (Fig. 5d). This decrease of E-cadherin protein expression was further confirmed by immunostaining (Fig. 5e). Similarly to its action in T47D cells, the PR inhibitor RU486 prevented the decrease of E-cadherin protein expression in YB cells treated with R5020 (Fig. 5f).
Several studies have shown that E-cadherin protein expression is regulated by the Wnt/β-catenin signaling pathway [17, 24]. Therefore, we investigated the ability of Wnt inhibitors to influence regulation of E-cadherin protein levels by R5020. We used Dickkopf-related protein 1, a natural antagonist of the Wnt signaling, CCT036477, an inhibitor of transcription downstream of β-catenin, and Wnt inhibitor II that blocks the release of Wnt proteins from the cells. All three inhibitors blocked the ability of R5020 to decrease E-cadherin protein expression (Fig. 5f, Online Resource 2). Consistent with these results, we found that E + R5020 treatment significantly increased mRNA levels of Wnt-1 and the Wnt-1-inducible gene RhoU [25] (Fig. 5g, h). Induction of Wnt-1 protein was further confirmed by immunocytochemistry (Fig. 5i, j). The PR inhibitor RU486 completely blocked the induction of Wnt-1, RhoU mRNAs, and Wnt-1 protein (Fig. 5g–j). Because P/PS can also activate intracellular signaling pathways such as PI3 kinase and Src, we tested the effect of inhibitors of intracellular kinases on E-cadherin protein expression. Inhibitors of Src, Akt, PI3K, and MEK did not block the R5020-induced decrease of E-cadherin protein (Online Resource 2), indicating that E-cadherin expression was not regulated via cytoplasmic signaling of PR. Altogether, these data suggest that the E + R5020 treatment decreases E-cadherin protein expression via the transcriptional activation of the Wnt pathway by PRB.
Discussion
The present study shows how P signaling contributes to more aggressive behavior of the rat mammary tumors in vivo. Specifically, E + P treatment as compared with E alone resulted in invasive mammary cancers accompanied by decreased E-cadherin levels and expansion of cells with basal/myoepithelial phenotype. Remarkably, likewise, changes were observed in invasive primary human breast cancers compared to matched carcinoma in situ. These similarities between human invasive breast cancer and E + P-treated rat mammary cancers lead us to speculate that progestogens (P, PS) play an important role in development of invasive human breast cancers. While it is well recognized that synthetic PS increases the risk of more aggressive breast cancers in postmenopausal women receiving E + PS HT [2, 3], it was considered that P does not increase the risk [26]. The current study indicates that P is highly effective in promoting invasive ER+PR+ mammary tumors in rats, and it is, therefore, likely that endogenous P promotes more invasive breast cancers in humans.
Treatment with E alone was sufficient to induce the development of luminal noninvasive tumors. The presence of P was required for the expansion of basal/myoepithelial tumor cells that frequently expressed PRB similar to normal rat basal/myoepithelial cells [19]. The expansion of basal/myoepithelial cells in tumors from rats treated with E + P is consistent with previous findings that PS treatment leads to an expansion of rare K5+ T47D breast cancer cells in vitro [27]. Moreover, it has been shown that, when combined with E, both PS and P result in accumulation of basal/myoepithelial cells in xenografts of T47D cells in immunodeficient mice [28]. Furthermore, PS directly regulates expression of K5 gene in luminal T47D cells [29]. This raises the possibility that chronic exposure to P or PS causes transdifferentiation of human luminal K18+ cells into cells co-expressing K5 and K18, the most common cellular phenotypes observed in invasive ER+PR+ breast cancers, or into cells that lose K18 and acquire K5 expression.
Invasiveness is a characteristic of aggressive breast cancers. Tumors from rats treated with E + P exhibited increased invasiveness that was associated with the decrease or complete loss of E-cadherin and K18 proteins in the tumor; however, levels of E-cadherin or K18 mRNA were not affected by E + P treatment. Furthermore, transcription factors that are involved in the regulation of E-cadherin mRNA levels, such as Zeb1, Twist1, and Snail, were not increased in tumors from rats treated with E + P compared to tumors from OI or rats treated with E alone. There is some evidence that P may regulate the translational efficiency of several mRNAs. Indeed, P specifically shortens poly(A) tail of some mRNA [30] that may lead to less efficient translation. More recent studies also have shown that PS induces production of a number of miRNAs in breast cancer cells [31]. Some of P-dependent miRNAs are targeted to 3'UTR of genes and, thus, may regulate translation efficiency of these genes. Obviously, additional studies are required to investigate the exact molecular mechanism of posttranscriptional loss of E-cadherin and K18 in tumors developed in the presence of E + P. Data obtained in this study underscore the importance of immunohistochemical analysis that may provide information unattainable by mRNA analysis. We propose that P promotes invasiveness of the rat mammary tumors via two mechanisms. First, P may increase invasiveness of luminal tumor cells by decreasing E-cadherin protein expression via activation of the Wnt pathway. This mechanism may work in both PR-positive and PR-negative cells, since Wnt-1 may act in a paracrine manner on PR-negative cells. Second, P may promote expansion of more invasive basal/myoepithelial cells, via direct activation of PRB. Since only PRB mediates the effect of P/PS on E-cadherin, it is important to investigate the PRA/PRB ratio and understand its role in regulating E-cadherin protein and invasiveness in an individual breast cancer. However, accurate assessment of PRB levels is further complicated by the level of PRB activation, which renders PRB less stable and less detectable as shown by cytochemistry in R5020-treated YB (Online Resource 3).
Loss of E-cadherin is a hallmark of invasive behavior of luminal breast cancer cell lines cultured in vitro. E + P treatment reduced or led to a complete loss of E-cadherin protein in 10 out of 13 (77 %) tumors in the rat. Likewise, decrease or complete loss of E-cadherin was observed in 15 out of 21 (71 %) invasive human breast cancers. In agreement with the in vivo rat studies, R5020 treatment decreased E-cadherin protein in T47D cells, and this effect was mediated exclusively by PRB. Both RU486 and Wnt inhibitors blocked this effect of R5020, indicating that both the genomic action of PRB and the activation of the Wnt pathway were required to decrease the E-cadherin protein. These results are consistent with previous reports that P acting through PRB induces Wnt-1 and Wnt-4 mRNA in YB cells, and loss of Wnt1 blocks soft agar growth of YB T47D cells [32, 33]. However, other Wnt proteins may be induced by PS to regulate E-cadherin. For example, we found the induction of Wnt-2, Wnt-11, and Wnt-5b mRNA in T47D cells treated with E + P (data not shown). Collectively, obtained data indicate that P acting through PRB regulates E-cadherin protein expression or stability in human breast cancers via activation of the Wnt pathway.
In conclusion, the data obtained herein indicate that physiological levels of the natural hormone P can significantly contribute to mammary cancer development in this in vivo mammary cancer model. We propose that hormone-dependent mammary cancers in ovariectomized E + P-treated rats may serve as an appropriate preclinical model of human hormone-dependent invasive carcinoma for testing novel therapeutic interventions. Our results obtained in T47D cells and similarities between hormone-dependent human and rat mammary cancers are consistent with the hypothesis that P/PS specifically contributes to the aggressiveness of human breast cancers. Thus, assessment of the systemic P levels in breast cancer patients, analysis of PRA and PRB expression, and evaluation of biomarkers indicative of P and Wnt signaling may provide novel prognostic tools as well as targets for interventions in patients with more aggressive hormone-dependent breast cancers.
References
Bray JD, Jelinsky S, Ghatge R, Bray JA, Tunkey C, Saraf K, Jacobsen BM, Richer JK, Brown EL, Winneker RC, Horwitz KB, Lyttle CR (2005) Quantitative analysis of gene regulation by seven clinically relevant progestins suggests a highly similar mechanism of action through progesterone receptors in T47D breast cancer cells. J Steroid Biochem Mol Biol 97:328–341
Campagnoli C, Clavel-Chapelon F, Kaaks R, Peris C, Berrino F (2005) Progestins and progesterone in hormone replacement therapy and the risk of breast cancer. J Steroid Biochem Mol Biol 96:95–108
Chlebowski RT, Anderson GL, Gass M, Lane DS, Aragaki AK, Kuller LH, Manson JE, Stefanick ML, Ockene J, Sarto GE, Johnson KC, Wactawski-Wende J, Ravdin PM, Schenken R, Hendrix SL, Rajkovic A, Rohan TE, Yasmeen S, Prentice RL (2010) Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 304:1684–1692
Ibrahim YH, Byron SA, Cui X, Lee AV, Yee D (2008) Progesterone receptor-B regulation of insulin-like growth factor-stimulated cell migration in breast cancer cells via insulin receptor substrate-2. Mol Cancer Res 6:1491–1498
Fu XD, Goglia L, Sanchez AM, Flamini M, Giretti MS, Tosi V, Genazzani AR, Simoncini T (2012) Progesterone receptor enhances breast cancer cell motility and invasion via extranuclear activation of focal adhesion kinase. Endocr Relat Cancer 17:431–443
Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB (2002) Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem 277:5209–5218
Graham JD, Yager ML, Hill HD, Byth K, O'Neill GM, Clarke CL (2005) Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol 19:2713–2735
Giangrande PH, Kimbrel EA, Edwards DP, McDonnell DP, Tetel MJ, Leonhardt SA, Pollio G (2000) The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol Cell Biol 20:3102–3115
Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM (2003) Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA 100:9744–9749
Mulac-Jericevic B, Conneely OM (2004) Reproductive tissue selective actions of progesterone receptors. Reproduction 128:139–146
Mote PA, Bartow S, Tran N, Clarke CL (2002) Loss of coordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat 72:163–172
Siitonen SM, Kononen JT, Helin HJ, Rantala IS, Holli KA, Isola JJ (1996) Reduced E-cadherin expression is associated with invasiveness and unfavorable prognosis in breast cancer. Am J Clin Pathol 105:394–402
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G (2012) Twist2 contributes to breast cancer progression by promoting an epithelial–mesenchymal transition and cancer stem-like cell self-renewal. Oncogene 30:4707–4720
Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G (2005) SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell–cell junctions. Nucleic Acids Res 33:6566–6578
Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J (2011) Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 71:245–254
Lombaerts M, van Wezel T, Philippo K, Dierssen JW, Zimmerman RM, Oosting J, van Eijk R, Eilers PH, van de Water B, Cornelisse CJ, Cleton-Jansen AM (2006) E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer 94:661–671
Prasad CP, Mirza S, Sharma G, Prashad R, DattaGupta S, Rath G, Ralhan R (2008) Epigenetic alterations of CDH1 and APC genes: relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci 83:318–325
Russo J, Russo IH (1996) Experimentally induced mammary tumors in rats. Breast Cancer Res Treat 39:7–20
Kariagina A, Aupperlee MD, Haslam SZ (2007) Progesterone receptor isoforms and proliferation in the rat mammary gland during development. Endocrinology 148:2723–2736
Russo J, Gusterson BA, Rogers AE, Russo IH, Wellings SR, van Zwieten MJ (1990) Comparative study of human and rat mammary tumorigenesis. Lab Investig 62:244–278
Clarke RB, Howell A, Potten CS, Anderson E (1997) Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res 57:4987–4991
Bocker W, Bier B, Freytag G, Brommelkamp B, Jarasch ED, Edel G, Dockhorn-Dworniczak B, Schmid KW (1992) An immunohistochemical study of the breast using antibodies to basal and luminal keratins, alpha-smooth muscle actin, vimentin, collagen IV, and laminin. Part II: Epitheliosis and ductal carcinoma in situ. Virchows Arch 421:323–330
Heatley M, Maxwell P, Whiteside C, Toner P (1995) Cytokeratin intermediate filament expression in benign and malignant breast disease. J Clin Pathol 48:26–32
Meniel V, Clarke AR (2003) Wnt-cadherin connections in normal and neoplastic mammary epithelium. J Mammary Gland Biol Neoplasia 8:435–447
Schiavone D, Dewilde S, Vallania F, Turkson J, Di Cunto F, Poli V (2009) The RhoU/Wrch1 Rho GTPase gene is a common transcriptional target of both the gp130/STAT3 and Wnt-1 pathways. Biochem J 421:283–292
Fournier A, Berrino F, Riboli E, Avenel V, Clavel-Chapelon F (2005) Breast cancer risk in relation to different types of hormone replacement therapy in the E3N-EPIC cohort. Int J Cancer 114:448–454
Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA (2008) Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA 105:5774–5779
Sartorius CA, Harvell DM, Shen T, Horwitz KB (2005) Progestins initiate a luminal to myoepithelial switch in estrogen-dependent human breast tumors without altering growth. Cancer Res 65:9779–9788
Liu R, Zhou Z, Zhao D, Chen C (2011) The induction of KLF5 transcription factor by progesterone contributes to progesterone-induced breast cancer cell proliferation and dedifferentiation. Mol Endocrinol (Baltimore) 25:1137–1144
Wu JC, Miller WL (1991) Progesterone shortens poly(A) tails of the mRNAs for alpha and beta subunits of ovine luteinizing hormone. Biol Reprod 45:215–220
Cochrane DR, Jacobsen BM, Connaghan KD, Howe EN, Bain DL, Richer JK (2012) Progestin-regulated miRNAs that mediate progesterone receptor action in breast cancer. Mol Cell Endocrinol 355:15–24
Faivre EJ, Lange CA (2007) Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol 27:466–480
Ramamoorthy S, Dhananjayan SC, Demayo FJ, Nawaz Z (2010) Isoform-specific degradation of PR-B by E6-AP is critical for normal mammary gland development. Mol Endocrinol 24:2099–2113
Zhao Y, Tan YS, Haslam SZ, Yang C (2010) Perfluorooctanoic acid effects on steroid hormone and growth factor levels mediate stimulation of peripubertal mammary gland development in C57BL/6 mice. Toxicol Sci 115:214–224
Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, Horwitz KB (1994) New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res 54:3868–3877
Horwitz KB, Pike AW, Gonzalez-Aller C, Fennessey PV (1986) Progesterone metabolism in T47Dco human breast cancer cells–II. Intracellular metabolic path of progesterone and synthetic progestins. J Steroid Biochem 25:911–916
Meyer G, Leipprandt J, Xie J, Aupperlee MD, Haslam SZ (2012) A potential role of progestin-induced laminin-5/alpha6-integrin signaling in the formation of side branches in the mammary gland. Endocrinology 153:4990–5001
Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE (2000) The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene 19:968–988
Kariagina A, Xie J, Leipprandt JR, Haslam SZ (2010) Amphiregulin mediates estrogen, progesterone, and EGFR signaling in the normal rat mammary gland and in hormone-dependent rat mammary cancers. Horm Cancer 1:229–244
Acknowledgments
The authors thank Kristina Miller, Lyndsi Davenport, Kyle Pohl, Alicia Kramer, Kristen Bullard, Anthony Yuhas III, Sharmila Kulkarni, Bennett Cho, Howard Her, Jang Park, and Michael DeVisser for the excellent technical assistance, Dr. Valentina Factor for critical reading of the manuscript, and Dr. Horwitz for providing Y, YA, and YB cells. This work was supported by the US Army Medical Research and Materiel Command under W81XWH-07-1-0502 (to S.Z.H), W81XWH-11-1-0108 (to A.K) and by the Breast Cancer and the Environment Research Centers Grant U01 ES/CA 012800 (to S.Z.H.) from the National Institute of Environment Health Science (NIEHS) and the National Cancer Institute (NCI), National Institutes of Health, Department of Health and Human Services. The contents of the study are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NCI, NIH.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
Expression of biomarkers in ER/PR positive human breast carcinomas (PDF 29 kb)
Online Resource 2
Kinase inhibitors do not block the decrease of E-cadherin protein induced by R5020 in human T47D breast cancer cells expressing only PRB (YB cells). YB cells were precultured without serum for 48 h and treated with synthetic progestin R5020 for 24 h without or with indicated inhibitors. Representative images of E-cadherin immunostaining (green) in cells treated with R5020 without or with 10 μM of Wnt inhibitor IWP-2 (Wnt inh.2), 10 μM of Src kinases inhibitor PP2, 10 μM of PI3 kinase inhibitor LY294002 (LY), 5 μM of Akt inhibitor IV (Akt inh), and 5 μM of MEK1/2 kinase inhibitor U0126 (U126). Note that Wnt inhibitor completely blocks the effect of R5020 on E-cadherin protein. Also note that cells treated with Akt inhibitor alone exhibited massive number of dead cells (yellow arrowheads). Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm (PDF 1630 kb)
Online Resource 3
Progestin R5020 decreases nuclear expression of progesterone receptor in human T47D breast cancer cells expressing only PRB (YB cells). YB cells were precultured without serum for 48 h and treated with or without R5020 for 24 h. Representative images of PRB immunostaining (green) Nuclei were counterstained with DAPI (blue). Scale bar, 75 μm (PDF 344 kb)
Online Resource 4
Effect of hormone treatments on serum estradiol and progesterone levels ovary-intact (OI) or ovariectomized rats treated with estradiol alone (E) or estradiol plus progesterone (E+P) were treated with DMBA to induce mammary tumors. Hormone levels were measured in rats with developed mammary tumors. Serum 17-β-estradiol levels (a) (N = 5–6 animals/group) and progesterone levels (b) (N = 6–8 animals/group). *, P < 0.05, progesterone levels in E-treated rats are lower than in OI or E + P-treated rats (PDF 488 kb)
Online Resource 5
Kinase inhibitors block kinase phosphorylation in human T47D breast cancer cells expressing only PRB (YB cells) treated with growth factor. YB cells were precultured without serum for 48 h and treated with or without kinase inhibitors for 24 h. IGF1 was added for the last 30 min of incubation. a Representative images of phospho-Erk immunostaining (green) in cells treated with 5 μM of MEK1/2 kinase inhibitor U0126 (U126). b Representative images of phospho-Akt immunostaining (red) in cells treated with 5 μM of Akt kinase inhibitor IV (Akt inh.). Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm (PDF 1392 kb)
Online Resource 6
The list of primary antibodies used in the study (PDF 21 kb)
Rights and permissions
About this article
Cite this article
Kariagina, A., Xie, J., Langohr, I.M. et al. Progesterone Decreases Levels of the Adhesion Protein E-Cadherin and Promotes Invasiveness of Steroid Receptor Positive Breast Cancers. HORM CANC 4, 371–380 (2013). https://doi.org/10.1007/s12672-013-0158-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-013-0158-6