
Abstract
Approximately 300 kg/day of food-grade CO2 was injected through a perforated pipe placed horizontally 2–2.3 m deep during July 9–August 7, 2008 at the MSU-ZERT field test to evaluate atmospheric and near-surface monitoring and detection techniques applicable to the subsurface storage and potential leakage of CO2. As part of this multidisciplinary research project, 80 samples of water were collected from 10 shallow monitoring wells (1.5 or 3.0 m deep) installed 1–6 m from the injection pipe, at the southwestern end of the slotted section (zone VI), and from two distant monitoring wells. The samples were collected before, during, and following CO2 injection. The main objective of study was to investigate changes in the concentrations of major, minor, and trace inorganic and organic compounds during and following CO2 injection. The ultimate goals were (1) to better understand the potential of groundwater quality impacts related to CO2 leakage from deep storage operations, (2) to develop geochemical tools that could provide early detection of CO2 intrusion into underground sources of drinking water (USDW), and (3) to test the predictive capabilities of geochemical codes against field data. Field determinations showed rapid and systematic changes in pH (7.0–5.6), alkalinity (400–1,330 mg/l as HCO3), and electrical conductance (600–1,800 μS/cm) following CO2 injection in samples collected from the 1.5 m-deep wells. Laboratory results show major increases in the concentrations of Ca (90–240 mg/l), Mg (25–70 mg/l), Fe (5–1,200 ppb), and Mn (5–1,400 ppb) following CO2 injection. These chemical changes could provide early detection of CO2 leakage into shallow groundwater from deep storage operations. Dissolution of observed carbonate minerals and desorption-ion exchange resulting from lowered pH values following CO2 injection are the likely geochemical processes responsible for the observed increases in the concentrations of solutes; concentrations generally decreased temporarily following four significant precipitation events. The DOC values obtained are 5 ± 2 mg/l, and the variations do not correlate with CO2 injection. CO2 injection, however, is responsible for detection of BTEX (e.g. benzene, 0–0.8 ppb), mobilization of metals, the lowered pH values, and increases in the concentrations of other solutes in groundwater. The trace metal and BTEX concentrations are all significantly below the maximum contaminant levels (MCLs). Sequential leaching of core samples is being carried out to investigate the source of metals and other solutes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbon dioxide capture, transport, and sequestration, especially its geologic storage, is now considered one of the necessary options to stabilize atmospheric CO2 levels and global temperatures at values that are considered acceptable for society and the environment (Benson and Cook 2005; IPCC 2007). Sedimentary basins in general and deep saline aquifers in particular, are being investigated as possible repositories for large amounts of anthropogenic CO2 that must be sequestered to stabilize atmospheric CO2 concentrations (Bachu 2003; White et al. 2003). These basins are attractive for CO2 storage, because they have huge potential capacity, estimated globally at up to 11,000 Gt of CO2, and advantageous locations close to major CO2 sources (Holloway 1997; Benson and Cook 2005; Hovorka et al. 2006).
In addition to storage capacity, key environmental questions include CO2 leakage related to the storage integrity and the physical and chemical processes that are initiated by injecting CO2 underground (Emberley et al. 2005; Knauss et al. 2005; White et al. 2005; Wells et al. 2007). Leakage of CO2 to the atmosphere would negate the goal of sequestration, but an equally critical issue concerns the potential for contamination and chemical alteration of shallow groundwater (Hepple and Benson 2005; Kharaka et al. 2009). In this scenario, the buoyant CO2 would migrate from the injection reservoir via a permeable fault or an improperly sealed abandoned petroleum well, and reach then dissolve in underground sources of drinking water (USDW). The contaminated groundwater, with a lowered pH (increased acidity), could dissolve carbonate, sulfide and iron oxyhydroxide minerals present in the aquifer. Dissolution of these minerals and desorption/ion exchange could add, Fe, Pb, U, As, Cd, and other toxic metals to groundwater; some of these aqueous chemicals may exceed the Maximum Contaminant Levels (MCLs) mandated by U.S. Environmental Protection Agency (US EPA 2004) as part of their national drinking standards (Wang and Jaffe 2004; Birkholzer et al. 2008; Kharaka et al. 2009).
Organic compounds may also be mobilized by the injected CO2, as reported in laboratory experiments simulating CO2 storage in deep coal beds (Kolak and Burruss 2006) and as observed in enhanced oil recovery (EOR) operations (Shiraki and Dunn 2000). Additional investigations are required in this area, but these results suggest that mobilization of organics, including BTEX from depleted oil reservoirs and non oil-bearing aquifers, could have major implications for the environmental aspects of CO2 storage and containment. The concern here is warranted as high concentrations of toxic organic compounds, including benzene, toluene (up to 60 mg/l for BTEX), phenols (<20 mg/l), and polyaromatic hydrocarbons (PAHs up to 10 mg/l), have been reported in oil-field waters (Kharaka and Hanor 2007). Also, the enhanced solvent properties of CO2, especially in its supercritical state, could lead to leaching and transport of organics, including BTEX, phenols, PAHs, and other hazardous compounds, from organic matter and petroleum present in the reservoir or in rocks encountered in the flow path, or the target contaminated aquifer (Kolak and Burruss 2006; Kharaka et al. 2009).
Geochemical results obtained at the Zero Emission Research and Technology (ZERT) field site, where a total of approximately 300 kg/day of food-grade CO2 was injected through a perforated pipe placed horizontally 2–2.3 m deep during July 9–August 7, 2008, are presented below. The overall goal of this multidisciplinary research project is to evaluate atmospheric and near-surface monitoring and detection techniques applicable to the subsurface storage and potential leakage of CO2 (Spangler et al. 2009, this volume; http://www.montana.edu/zert). As part of this research project, approximately 80 water samples were collected from 10 observation wells (1.5 or 3.0 m deep) located 1–6 m from the injection pipe, and from two distant monitoring wells. The samples were collected before, during, and following CO2 injection. The main objective of the study was to investigate changes in the concentrations of major, minor, and trace inorganic and organic compounds during and following CO2 injection. The observed changes are being interpreted here using the latest modified version of geochemical code SOLMINEQ (Kharaka et al. 1988); ongoing work also includes geochemical modeling by Zheng et al. (2009) using EQ3/6 (Wolery 1993) and reactive transport simulations with TOUGHREACT (Xu et al. 2006).
Regional setting and methodology
The Zero Emission Research and Technology (ZERT) field site is located on a relatively flat 12 hectare agricultural plot at the western edge of the Montana State University (MSU)-Bozeman campus in Bozeman, Montana, USA (see Spangler et al. 2009, this volume, for description of regional setting, and the various investigations completed or currently ongoing at this site). Plant coverage is about 70% grasses with the remainder being alfalfa, clover, dandelion, and thistle.
The ZERT site is located at an elevation of 1,495 m near the southeastern boundary of the Gallatin Valley, which is a north–south-trending intermontane basin of about 1350 km2 in area (Kendy and Tresch 1996). Structurally, the Gallatin Valley is an eastward-tilted graben, a broad alluvial plain filled with up to 1,800 m of poorly consolidated Cenozoic sandy gravel deposits. At the ZERT site, about a meter of silts and clays with a top soil layer of variable thickness overly the sandy gravel. Test pits and cores collected from several locations on the field consistently revealed two distinct soil zones. The topsoil of organic-rich silt and clay with some sand ranges in thickness from 0.2 to 1.2 m, and a caliche layer, high in calcite (~15%), is observed at depths of ~50–80 cm. Beneath the topsoil layer is a cohesionless deposit of coarse sandy gravel extending to 5 m, the maximum depth investigated (Spangler et al. 2009, this volume). Gravels comprise ~70% of rock volume, and andesite is the chief rock fragment among the gravels and coarse sands, but minor amounts of detrital limestone and dolostone are also observed. The sand and silt sized fraction of this sediment consists of approximately 40% quartz, 40% magnetite and magnetic rock fragments, and 20% grains of amphibole, biotite/chlorite, and feldspar.
Geology
The Gallatin Valley basement and surrounding mountains consist of complexly folded and faulted Tertiary through Archean bedrock. The eastern boundary of this valley is a series of steep normal faults along the fronts of the Bridger and Gallatin Ranges. The Gallatin Range, which borders the south margin and crests above 3,000 m elevation, provides source material to the basin-fill deposits in the portion of the Gallatin Valley which includes the ZERT field site.
The basin-fill deposits are subdivided by Kendy (2001) into three hydrogeologic units: Quaternary alluvium, Quaternary and Tertiary undifferentiated deposits, and Tertiary Bozeman group. The Tertiary Bozeman Group is considered to underlie the entire Gallatin Valley; it outcrops as pediments on the Madison Plateau and benches east of Bozeman, but it is mostly covered by a veneer of Quaternary and/or Tertiary alluvium. Quaternary and Tertiary undifferentiated deposits consist of unconsolidated Quaternary alluvial-fan deposits overlying Tertiary sedimentary pediments. Alluvial fan deposits extend into the Gallatin Valley from the Bridger and Gallatin Range fronts and consist of a heterogeneous mixture of unconsolidated poorly sorted rock fragments in a sand, silt, and clay matrix with some carbonate cement (Kendy and Tresch 1996; Lonn and English 2002). The largest of these alluvial fans extends northwards from the Gallatin Range front into the alluvial plain between Hyalite and Bozeman Creeks. The city of Bozeman and much of the surrounding land, including the ZERT field site, is located on this alluvial fan and is mapped in Kendy (2001) as Quaternary and Tertiary undifferentiated deposit.
The Gallatin Range provides the source material for the alluvial fan deposits in and around the ZERT site. The bedrock geology for the Gallatin Range is grouped by Kendy (2001) into four hydrogeologic units: (1) Tertiary (Eocene) volcanic rocks, which include andesite and basalt flows, breccia, agglomerate, and tuff of the Gallatin-Absaroka Volcanics; (2) Tertiary through Middle Proterozoic sedimentary bedrock, which includes a number of sandstone and limestone formations, and the Middle Proterozoic Belt Supergroup. Sandstone, conglomerate, and limestone predominate the lithologies; (3) Cretaceous through Cambrian sedimentary bedrock, in which fine-grained sandstone, shale, mudstone, siltstone, and chert predominate the lithologies; and (4) Archean metamorphic rocks, which consists of crystalline rocks, mainly biotite gneiss, with some schist, quartzite, and marble. Tertiary volcanic rocks and Archean metamorphic rocks predominate in the Gallatin Range and appear to be the major rock lithologies in poorly to moderately sorted sand and gravel at the ZERT field site.
Hydrology
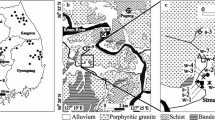
Five pairs of polyvinyl chloride (PVC) monitoring wells, 5 cm in diameter and 1.5 m (B) and 3.0 m (A) deep, were installed in the summer of 2007 at the southwestern end of the slotted section of the horizontal pipe (zone VI) to investigate changes in the chemical composition of shallow groundwater and head-space gas, following CO2 injection (Fig. 1). The wells, which are screened at the bottom 0.76 cm, are located 1–6 m from the horizontal pipe, downgradient of the groundwater flow in the area, except for wells 1A and 1B that are situated upgradient (Fig. 1). In December, 2008, three additional wells were installed, primarily to obtain core for mineral characterization and laboratory leaching experiments. Well W6 is 2.6 m deep with the bottom 0.76 m screened; wells W7 and W8 are 3 m deep with the bottom 1.5 m of the wells screened (Fig. 1).

Location of water-monitoring wells in relation to the surface trace of the slotted horizontal pipe in zone VI of the ZERT site
The depth of the water table at the ZERT field site is close to the ground surface, but varies seasonally. Spangler et al. (2009, this volume) report a variation of about 1 m in the depth of the water table over the past two and a half years. In the summers of 2007 and 2008, the depth of the water table measured in the water monitoring wells in zone (VI) averaged approximately 1.5 m below ground surface. Percolation tests made in 2006 yielded hydraulic conductivity values approximately 0.76 and 1.4 m/day in the upper soil (sandy silt) layer and in the sand-gravel aquifer, respectively. The direction of the ground water flow was estimated to be 17 degrees west of north (Fig. 1) and results of the tracer tests conducted in 2009 showed the lateral groundwater flow is high at ~2 m/day, and much higher than previously estimated (Spangler et al. 2009, this volume). Diffusion and flow rates for CO2 in the unsaturated zone are several orders of magnitude higher than the groundwater rates (Lewicki et al. 2007), and analyses of gas samples obtained from the head space of the wells yielded pCO2 values as high as 0.95 bar relatively rapidly following CO2 injection (Stratizar et al. 2008). These flow rates indicate that the rapid and systematic changes observed in the chemical composition of groundwater are driven by both the transport of groundwater with dissolved CO2, as well as by fast spreading of CO2 in the sand-gravel unsaturated zone under the upper soil layer, followed by re-dissolution into the groundwater.
Methods
Most of the water samples for this study were obtained from the shallow groundwater wells (1.5 m deep), especially after it became clear that the water from the deeper wells was not significantly impacted by the injected CO2. Lack of impact resulted because deep-well perforations are situated below the injection pipe and at some distance away. Sample collection, preservation and field and laboratory chemical analyses of water, and some relevant isotopes were carried out by methods described in Kharaka and Hanor (2007). On-site chemical analyses of groundwater samples, carried out in the laboratory truck, included measurements of pH, Eh, conductivity, alkalinity, dissolved oxygen (DO), and temperature. Sample filtration, acidification, and preservation were also carried out in the field laboratory.
Laboratory analyses for cations and metals were carried out at USGS by comprehensive ICP-MS measurements, with focus on initial concentrations of hazardous and other trace elements, including As, B, Cd, Cr, Cu, Fe, Hg, Mn, Pb, Se, U, and Zn. Sophisticated ICP-MS equipment is necessary, because these elements of interest have very low concentrations at ambient conditions, often close to the detection limits. Major and minor anions were determined in USGS laboratory by ion chromatography (IC). Benzene, toluene, ethyl-benzene, m-, p- and o-xylene (BTEX) were analyzed using an SRI-8610C gas chromatograph as described in EPA Method 5030C, a purge-and-trap procedure for the analysis of low levels of volatile organic compounds (VOCs) in aqueous samples and water miscible liquid samples. The gas chromatographic determinative steps are found in EPA methods 8015 and 8021 (US EPA 2004).
Results and discussion
Detailed chemical analyses of over 60 groundwater samples collected from the ZERT wells in July–August, 2008 have been completed. Most of the samples analyzed are from 4 of the 5 shallow 1.5 m deep “B” wells; results from well 3B are incomplete because adequate volumes of water could not be obtained for analytical requirements, as water recovery in this well was generally slow. The chemical data obtained for samples from the ZERT well 2B, located 1 m from the injection pipe and with a relatively complete data set for cations, anions, and trace metals, are listed in Table 1 as representative of the chemical results during the sampling period. The chemical composition of water obtained from the 3-m-deep “A” wells are similar to those listed for Z-109 (Table 1), and remained relatively unchanged following CO2 injection primarily because their perforations are located below the CO2 injection pipe. Results from the initial samples from the “A” wells, and from all other wells are plotted in the interpretive figures listed in this report.
The major cations and anions listed in Table 1 and plotted in the figures carry analytical uncertainties of approximately ±3%. The uncertainties are ±5% for trace metals, and these may be more than ±10% for trace metals that are close to their detection limits. For sample collection, preservation, analysis, and uncertainties applicable to these results, see Kharaka and Hanor (2007) and references therein.
Sample collection started in July 7, 2008, before the CO2 injection, which began at 15:45 on July 9, 2008. Injection was continuous, except for a few short intervals when it was stopped for various reasons. CO2 injection ended on August 7, 2008, but water sample-collection continued through August 14, 2008. During the sampling period, there were several relatively large precipitation events that impacted the chemical composition of the groundwater and raised the groundwater levels in the wells (Fig. 2). On the other hand, groundwater levels were lowered as a result of evapotranspiration and water sampling (Fig. 2).

Cumulative precipitation and groundwater levels in selected ZERT wells during the sampling period in 2008
Dissolved inorganic chemicals
The chemical data obtained for samples from shallow and deep wells prior to CO2 injection show that the groundwater in the area is a Ca–Mg–Na–HCO3 type water, with a fresh water salinity of about 600 mg/l TDS (Fig. 3, sample Z- 109, Table 1). The groundwater has a pH of approximately 7, and HCO3 is the dominant anion, but the concentrations of Cl and SO4 are relatively low. The concentrations of Fe, Mn, Zn, Pb, and other trace metals are expectedly low, at ppb levels.

Modified stiff diagrams showing concentrations (equivalent units normalized to 100%) of major cations and anions, together with salinity and pH of groundwater from well 2B before and during CO2 injection
Following CO2 injection, the pH of groundwater decreased systematically to values around 6.0 for wells 1B, 2B, and 5B, all in close proximity to the injection pipe, strongly and quickly reacting within 1 day. The largest early response occurred in well 2B, only 1 m from the injection pipe and in the direction of groundwater flow, decreasing to a pH of 5.7 (Fig. 4a). The pH of water for samples from well 4B (6 m from pipe) started decreasing only after 3 days following CO2 injection, but pH values remained above pH 6.0 (Fig. 4a). The pH in all wells started increasing towards 6.5 after CO2 injection was terminated on August 7, 2008. The measured pH values of groundwater were controlled primarily by pCO2, which was measured by Strazisar et al. (2008) in capped wells, and was computed with SOLMINEQ (Kharaka et al. 1988) using the measured temperature, pH, alkalinity, and other chemical parameters. The pCO2 values measured and computed for the ZERT samples were 0.035 bar before CO2 injection; they increased to values close to 1.0 bar following CO2 injection. It is evident from these results that pH is an excellent early indicator for detection of CO2 intrusion into groundwater.

Field-measured groundwater pH values (a), alkalinities (b), and electrical conductance (c), obtained from selected ZERT wells as a function of time of sampling. Note the systematic decrease in pH values from ~7.0 before CO2 injection to values as low as 5.6 during injection, and subsequent pH increases after CO2 injection was terminated. Alkalinities increased from about 400 mg/l before CO2 injection to values close to 1,200 mg/l as HCO3, and electrical conductance also increased from about 600 μS/cm before CO2 injection to values higher than 1,600 μS/cm during injection
The alkalinity of groundwater increased from about 400 mg/l as HCO3 to values of up to 1,330 mg/l, following CO2 injection (Fig. 4b). Alkalinity values for different wells, as with the pH values, show variable trends reflecting distance from the CO2 injection pipe, impacts from precipitation events, and possibly local variations in the mineral composition of soils and sediments. The alkalinity values for wells 2B and 5B increase first and show the highest alkalinities (up to 1,330 mg/l), and with well 4B increasing more slowly and only to about 700 mg/l. The alkalinities in all wells decrease to values approaching 400 mg/l after termination of CO2 injection (Fig. 4b) probably as a result of calcite precipitation caused by its supersaturation in groundwater that resulted from higher pH values.
Values for the electrical conductance, also measured at the site, show similar trends as alkalinity, increasing from ~600 μS/cm before CO2 injection to approximately 1,800 μS/cm following CO2 injection (Fig. 4c). The conductance values for water from wells 1B, 2B, and 5B increase first and show the highest conductance, and with well 4B increasing more slowly and only to about 1,100 μS/cm; all the conductance measurements decrease to values approaching 600 μS/cm after termination of CO2 injection on August 7, 2008 (Fig. 4c).
The alkalinity increases following CO2 injection are balanced primarily by increases in the concentrations of Ca and Mg, whereas the concentrations of Na (10 ± 2 mg/l) are relatively constant (Fig. 5). The molar and atomic concentrations of Ca increase the most, with atomic values for Ca increasing from about 80 mg/l to up to 240 mg/l, and those for Mg increasing from about 25 mg/l to up to 70 mg/l (Fig. 5). Atomic Ca/Mg ratios obtained initially and following CO2 injection (Fig. 6) cannot be explained by dissolution of dolomite alone; the values, increasing from 3.2 to 6.1 for well 2B, indicate that dissolution of calcite is dominant, but that dissolutions of both calcite and dolomite (or Mg-rich calcite) are required to explain the changes in alkalinity and concentrations of Ca and Mg (Fig. 6; Table 1). Dissolutions of calcite (reaction 1) and dolomite (reaction 2) can be represented by reaction such as

Concentrations of major cations in groundwater from the ZERT wells plotted as a function of water alkalinities. Note the relatively constant concentrations of Na and K, but the general increases in the concentrations of divalent cations with water alkalinities, possibly indicating dissolution of carbonate minerals

Ca/Mg weight ratios plotted as a function of time of sampling. Note that the ratios are higher than the value expected from dissolution of dolomite (1.6), and are relatively constant for well 4B, but continue to increase for well 2B until CO2 injection was terminated, possibly indicating dissolution of calcite
These conclusions are supported by the initial characterization of minerals in core samples that show that calcite is abundant in a caliche layer observed at depths of ~50–80 cm. Small traces of carbonates were also observed in fines above 2.5 m, and minor amounts of detrital limestone and dolostone were observed in the gravel section. Results of geochemical modeling with an updated SOLMINEQ (Kharaka et al. 1988) also support dissolution of calcite and disordered dolomite as possible reactions at all pH values; also, groundwater is undersaturated with respect to dolomite at the lowered pH values obtained following CO2 injection, allowing for its dissolution.
Desorption-ion exchange reactions on clay minerals with H+ have been suggested as an alternative explanation (Zheng et al. 2009) for the increases in the concentrations of Ca, Mg (see also for Fe and Mn below). These and other reactions will be investigated after the completion of mineral characterizations and leaching experiments.
The concentrations of Fe and Mn, the two most abundant trace metals in groundwater, also increase following CO2 injection (Fig. 7). Increased concentrations of Fe could reflect dissolution of several Fe(II) and Fe(III) minerals, including siderite and ferrihydrite, depicted in Eqs. 3 and 4, respectively. The concentration of Fe in groundwater is a strong function of Eh, which could only be measured in water from the deep wells, because shallow wells did not yield enough water volume. The Eh values obtained in water from the deeper wells that was not impacted by CO2 injection (150–200 mV) indicate oxidizing conditions that account for the low concentration of dissolved Fe; much higher Fe values are possible under reducing conditions because of the higher solubility of Fe(II) minerals (Hem 1985). The Fe concentrations (Fig. 7a) increase from ~5 to 1,200 ppb, but show very low values during July 20 to July 26, following significant precipitation events (Fig. 2), even when pH values were low. Dilution alone cannot explain the low Fe concentrations during July 20–July 26, but the low values can be attributed to the oxidizing conditions possibly caused by increased dissolved O2 content in groundwater transported with percolating water from precipitation events. Ion exchange reactions on clays with H+ and major dissolved cations, such as Ca, Mg, and Na, are other possible controls on Fe concentrations (Birkholzer et al. 2008; Zheng et al. 2009), and these will be investigated after the completion of leaching experiments. Geochemical modeling indicates that the large increases observed in concentrations of Fe (from ~5 to 1,200 ppb) could result from dissolution of siderite, but are most likely caused by dissolution of iron oxyhydroxides, depicted in redox-sensitive reactions (5 and 6).

Concentrations of Fe (a) and Mn (b) in groundwater from selected ZERT wells plotted as a function of time of sampling. Note the low Fe and Mn concentrations during July 20–July 26, which we are attributing to the oxidizing conditions possibly caused by increased dissolved O2 content in groundwater transported with percolating water from precipitation events (see Fig. 2)
The concentrations of Mn show similar trends to those of Fe, increasing from ~5 to 1,400 ppb following CO2 injection, but also show low values during July 20–July 26 (Fig. 7b). Dissolution, redox, and disproportionation reactions can be written for Mn as for Fe above, but it should be noted that Mn concentrations are higher for well 4B than for 2B and 5B, and this may be controlled by local mineral compositions.
The concentrations of Pb, As, Zn, and other trace metals (Table 1; Fig. 8) generally show an increase with increasing alkalinity (lower pH value) following CO2 injection. The values reported, however, carry high uncertainties as they are, in some cases, close to the analytical detection limits, especially with the required dilutions (up to fivefold) prior to analyses. The concentration increases are likely caused by desorption-ion exchange reactions with H+, Ca, and Mg resulting from lowered pH values. Because measured H2S values were below the detection limit (0.2 mg/l) geochemical modeling could not be used to check for solubility of sulfide minerals as controls on metal concentrations. The concentrations, it should be noted, are all significantly below the maximum contaminant levels (MCLs) for the respective trace metals (e.g., 15 ppb for Pb, 6 ppb for As). The initial values and the increases in concentrations of these trace metals, although small, are readily measured by the sampling and analytical methods used in this study. These results highlight the role of geochemical tools for early detection of CO2 leakage into groundwater.

Trace metal concentrations in groundwater from selected ZERT wells plotted as a function of water alkalinities. Some of the scatter shown probably results because the reported values in some cases are close to the detection limits
The chemical changes observed in the ZERT groundwater are similar in trends, though much lower in concentrations, to the changes observed in the Frio Brine Pilot test, near Houston, where 1,600 t of CO2 were injected at 1,500 m depth into a 24-m-thick “C” sandstone unit of the Frio Formation (Freifeld et al. 2005; Hovorka et al. 2006; Kharaka et al. 2006). Following CO2 breakthrough, 51 h after injection, Frio samples showed sharp drops in pH (from 6.5 to 5.7 measured at surface), pronounced increases in alkalinity (100–3,000 mg/l as HCO3) and in Fe (30–1,100 mg/l), a slug of very high DOC values, and significant shifts in the isotopic compositions of H2O, DIC, and CH4. These data, coupled with geochemical modeling, indicated rapid dissolution of minerals, especially calcite and iron oxyhydroxides, both caused by lowered pH (initially ~3.0 at subsurface conditions) of the brine in contact with supercritical CO2 (Kharaka et al. 2009). The differences between pH results from Frio and ZERT tests are related to several geochemical parameters, but an important reason relates to subsurface pCO2 value for Frio, which was approximately 150 bar (Kharaka et al. 2009), but pCO2 value measured (Stratizar et al. 2008) and computed with SOLMINEQ (Kharaka et al. 1988) for the ZERT samples ranged from 0.035 to ~1 bar. The maximum amount of CO2 dissolved in water is a strong function of fluid pressure. Thus, in a shallow aquifer such as that at the ZERT site, much less CO2 can dissolve in the groundwater compared to formation water in a deep sequestration site such as the Frio. Therefore, the decreases in pH values are much less pronounced, and fewer CO2-related chemical changes, and consequently, less contaminant mobilization are to be expected at sites such as the ZERT.
Dissolved organics
The dissolved organic carbon (DOC) values obtained in this study are generally about 4 mg/l, with a range from 2.6 to 6.9 mg/l, and these variations do not seem related to CO2 injection. The concentrations of benzene, toluene, ethyl-benzene, m-, p- and o-xylene (BTEX) were determined in a relatively large number of samples collected when it became clear that the injection of CO2 was likely responsible for the small but systematic increases in the concentrations of dissolved BTEX compounds (up to ~1.6 μg/l for m-, p-xylene) depicted in Fig. 9. It should be noted that the concentrations are all below the maximum contaminant levels (MCLs) for the respective BTEX compounds (5 ppb for benzene). There is some scatter in the results, but the concentrations are about 0.2 μg/l before CO2 injection, increasing to values greater than 1.0 μg/l for m-, p-xylene and o-xylene, and to about 1.0 and 0.8 μg/l for toluene and benzene, respectively. The measured values are higher in water from wells 1B (Fig. 9a) and 2B (Fig. 9b) relative to well 4B, which is further away from the injection pipe (Fig. 9c). All the measured BTEX values decrease to a value approaching pre-injection (~0.2 μg/l) after termination of CO2 injection on August 7, 2008 (Fig. 9d).

Concentrations of BTEX compounds detected in groundwater from selected ZERT wells plotted as a function of time of sampling. The BTEX concentrations are all significantly below the maximum contaminant levels (MCLs) (e.g., 5 ppb for benzene). The source of the low amounts of BTEX compounds detected in groundwater following CO2 injection is discussed in text below
The origin of the low amounts of BTEX compounds detected in groundwater following CO2 injection was investigated during 2008, and more recently in 2009. Three main possible sources were examined: (1) A natural source from deep organic matter or a petroleum accumulation; (2) contamination from a surface source or from the injection pipe and groundwater wells installations; or (3) contamination from trace amounts of BTEX in the injected CO2, although the gas used was of food-grade quality. Initial results obtained in 2008 were inconclusive with regard to the injected CO2 being the source of the observed BTEX. We analyzed core samples obtained from wells drilled in December 2008 for BTEX; and though progress was slow because results obtained were close to the detection limits of available analytical methods, results showed that sediments were not the source as BTEX concentrations obtained were below detection limits. Subsequently, purge-and-trap analysis of CO2 samples collected from the source tank in 2009 showed conclusively that the CO2 was the source of the trace concentrations of detected BTEX. Mobilization of organic compounds in deep subsurface by supercritical CO2, which is a very effective solvent for hydrocarbons (Kolak and Burruss 2006), is a potential concern, as these compounds may be transported to shallow potable groundwater (Kharaka et al. 2006). Results, however, indicate that BTEX compounds can be detected in groundwater early and at extremely low concentrations, below MCL levels.
Conclusions and future plans
Results discussed in this report cover primarily the detailed changes in the concentrations of inorganic and organic chemicals in shallow groundwater at the ZERT site following CO2 injection. Rapid and systematic changes were observed in chemical parameters measured at site, including pH (7.0–5.6), alkalinity (400–1,330 mg/l as HCO3), and electrical conductance (600–1,800 μS/cm) following CO2 injection. Results obtained from laboratory analyses also show major increases in the concentrations of major and trace chemicals, including Ca (90–240 mg/l), Mg (25–70 mg/l), Fe (5–1,200 ppb), Mn (5–1,400 ppb), and BTEX (e.g. benzene 0–0.8 ppb) following CO2 injection. It should be noted that trace metal and BTEX concentrations are all significantly below the maximum contaminant levels (MCLs) (e.g., 15 ppb for Pb, 6 ppb for As, 5 ppb for benzene). The initial values and the increases in concentrations of these trace metals, though small, are readily measured by the sampling and analytical methods used in this study. These results highlight the role of geochemical tools for early detection of CO2 leakage into groundwater from deep storage operations.
In December, 2008, three additional wells (2.6 or 3 m deep) were installed, primarily to obtain core and soil samples for detailed mineral characterization and laboratory leaching experiments. Once completed, the new data will be combined with the solute concentrations and geochemical modeling to identify the minerals and processes responsible for the observed changes in water quality. Dissolution of observed calcite is clearly responsible for some of the increases in Ca and alkalinity, but the impacts of other reactive minerals will be investigated. The important role of desorption-ion exchange on clay minerals and iron oxyhydroxides resulting from lowered pH values will also be investigated. Speciation-saturation geochemical codes like SOLMINEQ (Kharaka et al. 1988) and EQ3/6 (Wolery 1993) are being used initially, while multi-dimensional reactive transport simulators such as TOUGHREACT (Xu et al. 2006, 2007) will eventually be applied to account for the temporal evolution of the system in response to relevant hydrologic, chemical and transport processes, including rain events leading to groundwater recharge and dilution, transport of CO2-charged water with the groundwater flow, and lateral spreading of CO2 in the unsaturated zone, and its re-dissolution into the groundwater (Zheng et al. 2009).
References
Bachu S (2003) Screening and ranking of sedimentary basins for sequestration of CO2 in geological media in response to climate change. Environ Geol 44:277–289
Benson SM, Cook P (2005) Underground geological storage, IPCC special report on carbon dioxide capture and storage, Intergovernmental Panel on Climate Change, Interlachen, Switzerland, Chapter 5, pp 5-1–5-134
Birkholzer J, Apps JA, Zheng L, Zhang Z, Xu T, Tsang C-F (2008) Research project on CO2 geological storage and groundwater resources: water quality effects caused by CO2 intrusion into shallow groundwater, Lawrence Berkeley National Laboratory Technical Report, LBNL-1251E, 450 pp
Emberley S, Hutcheon I, Shevalier M, Durocher K, Mayer B, Gunter WD, Perkins EH (2005) Monitoring of fluid-rock interaction and CO2 storage through produced fluid sampling at the Weyburn CO2-injection enhanced oil recovery site, Saskatchewan, Canada. Appl Geochem 20:1131–1157
Freifeld BM, Trautz RC, Kharaka YK, Phelps TJ, Myer LR, Hovorka SD, Collins DJ (2005) The U-tube: a novel system for acquiring borehole fluid samples from a deep geologic CO2 sequestration experiment. J Geophys Res 110:B10203. doi:10.1029/2005JB003735
Hem JD (1985) Study and interpretation of the chemical characteristics of natural water. US Geological Survey water-supply paper 2254, 264 pp
Hepple RP, Benson SM (2005) Geologic storage of carbon dioxide as a climate change mitigation strategy; performance requirements and the implications of surface seepage. Environ Geol 47:576–585
Holloway S (1997) An overview of the underground disposal of carbon dioxide. Energy Convers Mgmt 38:S193–S198
Hovorka SD, Benson SM, Doughty CK, Freifeld BM, Sakurai S, Daley TM, Kharaka YK, Holtz MH, Trautz RC, Nance HS, Myer LR, Knauss KG (2006) Measuring permanence of CO2 storage in saline formations—the Frio Experiment. Environ Geosci 13:105–121
Intergovernmental Panel on Climate Change (IPCC) (2007) Working Group assessment report. http://www.ipcc.ch/(working groups I, II, III and Synthesis Reports)
Kendy E (2001) Magnitude, extent, and potential sources of nitrate in ground water in the Gallatin Local Water Quality District, Southwestern Montana, 1997–1998, US Geological Survey, water-resources investigations report 01-4037, 66 pp
Kendy E, Tresch RE (1996) Geographic, geologic and hydrologic summaries of intermontane basins of the Northern Rocky Mountains, Montana, US Geological Survey water-resources investigations report 96-4025, 232 pp
Kharaka YK, Hanor JS (2007) Deep fluids in the continents: sedimentary basins. In: Drever JI (ed) Surface and ground water, weathering and soils, vol 5. Treatise on Geochemistry (Holland HD and Turekian KK (eds)), Elsevier, Oxford, pp 1–48
Kharaka YK, Gunter WD, Aggarwal PK, Perkins EH, DeBraal JD (1988) SOLMINEQ.88: a computer program for geochemical modeling of water-rock interactions. U.S. Geological Survey water resources investigations report 88-4227
Kharaka YK, Cole DR, Hovorka SD, Gunter WD, Knauss KG, Freifeld BM (2006) Gas–Water–Rock interactions in Frio formation following CO2 injection: implications to the storage of greenhouse gases in sedimentary basins. Geology 34:577–580
Kharaka YK, Thordsen JJ, Hovorka SD, Nance HS, Cole DR, Phelps TJ, Knauss KG (2009) Potential environmental issues of CO2 storage in deep saline aquifers: geochemical results from the Frio-I Brine Pilot test, Texas, USA. Appl Geochem 24:1106–1112
Knauss KG, Johnson JW, Steefel CI (2005) Evaluation of the impact of CO2, co-contaminant gas, aqueous fluid and reservoir–rock interactions on the geologic sequestration of CO2. Chem Geol 217:339–350
Kolak JJ, Burruss RC (2006) Geochemical investigation of the potential for mobilizing non-methane hydrocarbons during carbon dioxide storage in deep coal beds. Energy Fuels 20(2):566–574
Lewicki J, Oldenburg C et al (2007) Surface CO2 leakage during the first shallow subsurface CO2 release experiment. Geophys Res Lett 34:L24402
Lonn JD, English AR (2002) Preliminary geologic map of the eastern part of the Gallatin Valley, Montana, Montana Bureau of Mines and Geology open file report 457, 20 pp
Shiraki R, Dunn TL (2000) Experimental study on water–rock interactions during CO2 flooding in the Tensleep Formation, Wyoming, USA. Appl Geochem 15:265–279
Spangler LH, Dobeck LM et al. (2009) A controlled field pilot in Bozeman, Montana, USA, for testing near surface CO2 detection techniques and transport models. Environ Geol (this volume)
Strazisar BR, Wells AW et al. (2008) Soil gas monitoring for the ZERT shallow CO2 injection project. Prepr Pap Am Chem Soc Div Fuel Chem 52(2) (in press)
US Environmental Protection Agency (2003) Purge-and-trap for aqueous samples, method 5030C at: http://www.epa.gov/osw/hazard/testmethods/sw846/new_meth.htm#5030C
US Environmental Protection Agency (2004) 2004 edition of the drinking water standards and health advisories, EPA 822-R-04-005, January
Wang S, Jaffe PR (2004) Dissolution of trace metals in potable aquifers due to CO2 releases from deep formations. Energy Convers Manag 45:2833–2848
Wells AW, Diehl JR, Bromhal G, Strazisar BR, Wilson TH, White CM (2007) The use of tracers to assess leakage from the sequestration of CO2 in a depleted oil reservoir, New Mexico. USA Appl Geochem 22:996–1016
White CM, Strazisar BR, Granite EJ, Hoffman JS, Pennline HW (2003) Separation and capture of CO2 from large stationary sources and sequestration in geological formations—coalbeds and deep saline aquifers. J Air Waste Manag Assoc 53:645–715
White SP, Allis RG, Moore J, Chidsey T, Morgan C, Gwynn W, Adams M (2005) Simulation of reactive transport of injected CO2 on the Colorado Plateau, Utah, USA. Chem Geol 217:387–405
Wolery TJ (1996) EQ3/6, A software package for geochemical modelling of aqueous systems (Version 7.2): Lawrence Livermore Nat. Lab. UCRL-MA 110662
Wood RH (1958) The second ionization constant of hydrogen selenide. J Am Chem Soc 80:1559–1562
Xu T, Sonnenthal E, Spycher N, Pruess K (2006) TOUGHREACT: a simulation program for non-isothermal multiphase reactive geochemical transport in variably saturated geologic media. Comput Geosci 32:145–165
Xu T, Apps JA, Pruess K, Yamamoto H (2007) Numerical modeling of injection and mineral trapping of CO2 with H2S and SO2 in a sandstone formation. Chem Geol 242:319–346
Zheng L, Apps J, Spycher N, Birkholzer J, Kharaka YK, Thordsen J, Kakouros E, Trautz R, Rauch H, Gullickson K (2009) Changes in shallow groundwater chemistry at the 2008 ZERT CO2 injection experiment: II-modeling analysis. Abstract, eight carbon capture and sequestration conference, Pittsburgh, PA, May 4–7
Acknowledgments
This research was conducted within the ZERT project directed by Lee Spangler and managed by Laura Dobeck, MSU, Bozeman, MT. http://www.montana.edu/zert. We thank the entire ZERT team and participating organizations for creating a supportive and exciting research environment. This research was funded primarily by the Electric Power Research Institute, EPRI, but funds were also obtained from EPA, DOE, LBNL, and USGS.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kharaka, Y.K., Thordsen, J.J., Kakouros, E. et al. Changes in the chemistry of shallow groundwater related to the 2008 injection of CO2 at the ZERT field site, Bozeman, Montana. Environ Earth Sci 60, 273–284 (2010). https://doi.org/10.1007/s12665-009-0401-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-009-0401-1