
Abstract
Purpose
The anaerobic digestion of the organic fraction of municipal solid waste (OFMSW) is a treatment biologically optimizable. The analysis of the microbial balance within the digester allows not only to describe and improve the efficiency of the system but also to foresee potential variations after biochemical interventions. This work aims to highlight methanogenic population variations in two similar parallel digesters to identify bio-indicators for digestion performance after the inclusion of an additive.
Methods
The analysed industrial case study involved the two parallel, pilot-scale, bio-digesters of ACEA Pinerolese Industriale. They had analogous design and fed by the same OFMSW except for an additive inclusion. The analyses by qRT-PCR, on both bio-digesters, have been performed to determine an impact of the additive on the growth and metabolic activity of total methanogens, Methanosarcina spp., Methanocorpuscolum spp., and Methanospirillus spp.
Results
Collected data showed that the addition of biopolymers extracted from compost produced both total methanogens and Methanosarcina decrease (7.67 vs. 7.48 and 6.05 vs. 5.69 Log gene copies/mL sludge respectively, T-test p < 0.01) affecting slightly the methane yield (-8%). Methanosarcina was the highest expressed methanogen; however, the results suggested the involvement of a highly complex and heterogeneous methanogen community.
Conclusions
Total methanogens and active Methanosarcina modulations are able to highlight major bio-methane production even if their shift is slight. Globally only 3–4% of the present and 12–13% of the active methanogens were described. However, biological approach can supply a proxy of production performance of the digester also concerning the feeding peculiarities.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Statement of Novelty
The process of anaerobic digestion of OFMSW, as a green biotechnology, is a strategic field to focus on, because it represents a valid treatment of a risky matrix and a source of renewable energy. However, such a process is also characterized by criticalities in the digestion efficiency management. Additives with both vegetal and animal origin were proposed to improve digestion process. In this work, the determination of the variations of the methanogenic population, by biomolecular method, is exploited to identify bioindicators of digestion efficacy when a compost-derived additive was included. Significant results were produced highlighting the opportunity for biological control integration into the treatment management.
Introduction
The organic fraction of municipal solid waste (OFMSW) produced annually in the European Union is estimated approximately between 118 and 138 million tonnes (about 50% of the total amount of municipal solid waste) [1]. Therefore, owing to the content of carbohydrates, proteins, and lipids, OFMSW represents an important feedstock for the biofuel production from waste, through the anaerobic digestion (AD) process. Biogas either can be a fuel resource, for the combined heat and power units, or a starting product to upgrade into biomethane, in total accordance with the European perspective [2].
Following intensive negotiations, the EU institutions reached an agreement in June 2018 on a new Renewable Energy Directive for the next decade. The new legislation includes a legally binding EU-wide target of 32% for renewable energy by 2030, with an upward review clause in 2023, as well as sector-specific objectives, including an annual increase of 1.3% for renewable energy in the heating sector and the end target of 14% renewables in the transport sector by 2030 [2]. Biogas and biomethane must reach 65–80% greenhouse gas savings relative to the fossil fuel comparators, making the AD process increasingly appealing [3].
The AD of the OFMSW is a green biotechnology prospectively strengthening with the knowledge improvement on the biological dynamics of the process [4]. Efficient management includes a real control of the digester microbiota variation during the time, taking into account the characteristics of the feeding composition. The digester microbiota works under synergic interplay among different microbial groups, including hydrolytic, acidogenic, acetogenic, and methanogenic microorganisms [5, 6].
Various literature evidence showed a strict interaction among microbial communities following specific metabolic favourable pathways [7, 8]. For example, recently in thermophilic AD was observed that the bio-augmented M. thermophilus stimulated the growth of syntrophic acetate oxidizing bacterium Thermacetogenium phaeum, immediately after bioaugmentation [9].
Moreover, to find and maintain the digester’s optimal microbial equilibrium, the literature suggested a wide range of intervention. Up today bioaugmentation—the addition in bioreactors of stress resistant or efficient biogas generating microbial [10]—seems to be the more useful strategies to improve AD process [11, 12]. Basak et al. reported a fast recovery of an overloaded-failed anaerobic digester with the addition of a selected microbial consortium that contain among others acetoclastics methanogenic species [12]. The simultaneous presence of hydrolytic microorganisms (predominant genera is generally Clostridium) with acidogens (predominant phylum Proteobacteria) favouring fast organic waste hydrolysis with a consequent higher volatile fatty acids (VFAs) production. This could be a hazard point for the final step of the methanogenesis, in fact, a high level of VFAs can lead to a marked pH decrease inhibiting methanogen metabolism and reducing the methane yield [13, 14].
Augmentation strategies include also the addiction of material derived both from animal and/or vegetal matrix (soluble bio-based substances, SBO); the augmentation effect could be achieved for the effect of a substrate composition shift and/or an induced chemical/microbial shift [10].
From a technical point of view, the cultural methods for the analysis of the microbial dynamics inside the digester are not practical due to the strict anaerobic condition. Alternative methods are being developed using biomolecular methods, including PCR and its variants, and the next-generation sequencing [15].
The biomolecular management of the AD of OFMSW shows a very interesting tool for biological intervention strategies, assessing the impact on the process of the inclusion of organic by-product coming from animal and/or vegetal waste treatment, under a circular economy perspective.
This work aims to identify methanogen bio-indicators of digestion efficacy following the inclusion of an additive, produced starting from compost, in one of two analogous parallel bio-digesters.
Materials and Methods
ACEA is a modern multi-utility company, which currently processes 60,000 t y−1 of organic fraction of municipal solid waste (OFMSW), serving roughly 1 million inhabitants. The digester section where the thermophilic reaction takes place at 55 ± 1 °C, is composed of two parallel bio-digesters (Reactor A and Reactor B). Each bioreactor has around 2500 m3 working volume, for a total capacity of 5000 m3 for the digester section. The same pre-treated OFMSW was the feeding of the two parallel reactors (A and B). During the pre-treatment process bags containing OFMSW – 92.22% of composting material and 7.78% of inorganic materials as plastics, diapers or pad - are shredded and inert materials are separated by a disc with 50 mm openings [16]. Digesters’ parameters are 34% dried total solids (TS), the 14 days of hydraulic retention time (HRT) and the organic load rate (OLR) is 6.5 kg m3/die. The Table 1 showed the main process parameters for the two parallel anaerobic digesters.
The reactor B was addicted with a SBO produced starting from compost by an alkaline hydrolysis [16]. Several tests were previously carried out to assess the effects of amount of the addicted biopolymers on the AD, both considering different origin of such SBO (for example compost and/or post-AD sludge or/and home gardening and/or park trimming residues). While no effect was observed at different SBO concentration (until to 0.20% /v) for the biogas yield, a significant impact was observed for the ammonia reduction [16].
The biopolymer addition was included only into the Reactor B in the following way:
Preliminary phase: no addition; Phase 1: 11 m3 of solution enriched in compost biopolymers was added in one day, reaching a biopolymer concentration in Reactor B of 0.042% (w/v). Phase 2: 1 m3/d of solution enriched in compost biopolymers was added for 28 days, reaching a final biopolymer concentration in Reactor B of 0.059% (w/v). Phase 3: 2 m3/d of solution enriched in compost biopolymers was added for 28 days, reaching a final biopolymer concentration in Reactor B of 0.091% (w/v).
Reactor A was the control reactor (no addition during all the phases) and it was fed with the same pre-treated OFMSW of the interventional reactor (B).
Finally, 85 samples (50 mL of sludge) were collected from both the reactors on the recirculation piping as shown in Table 2. The samples reached the laboratory within few hours (< 3 h) after sampling.
DNA and RNA Extractions and Analysis
In the environmental microbiology laboratory (DPHP), the samples were quickly homogenized and, starting from 0.250 g of each sludge sample, the DNA was extracted using the kit PowerSoil DNA Isolation Kit, Qiagen (Cat N./ID: 47016) while, starting from 0.100 g of sludge from each sample unit, the RNA was extracted using the kit GenElute Total RNA Purification, SIGMA Aldrich (RNB350), following the manufacture’s instructions.
The concentration of the extracted DNA and RNA was quantified by a spectrophotometric evaluation using a microplate NanoQuant Plate (TECAN Trading AG, Switzerland) and a multimode reader TECAN Infinite 200 PRO integrated with the software i-Control (version 1.11.10). The quality of the extracts was tested by gel-electrophoresis.
The DNA extracts were stored at − 20 °C until quantitative Real Time Polymerase Chain Reaction (qRT-PCR) analysis, while RNA extracts were stored at − 80 °C until the retro-transcription into cDNA. The concentration into the extracts was on average 45.55 ± 16.27 µg/µL for the DNA and 30.22 ± 10.37 µg/µL for the RNA. Such concentrations are suitable for the following biomolecular analysis.
The retro-transcription of the RNA was conducted using a T100 thermo-cycler (Bio-rad) starting from 2 µl of the RNA extract and using the kit iScript Reverse Transcription Supermix (Bio-rad) following the instruction and the suggested thermal protocol.
qRT-PCR was conducted on all the samples for the quantification of the total methanogens and Methanosarcina spp. both starting from DNA and RNA accounting for 170 and 165 determinations respectively; while, Methanocorpuscolum spp. and Methanospirillus spp. were evaluated on a limited number of samples (30) to assess their presence and concentration in such kind of AD system.
The Real-time qPCR was conducted using a CFX Touch Real-Time PCR Detection System (Bio-Rad-Hercules, CA) and CFX Maestro Software 1.1, following the method previously described and amplifying the mcrA gene, codifying for the α subunit of the methyl coenzyme M reductase (a methanogen functional gene) [17]. Methanosarcina spp., Methanocorpuscolum spp., and Methanospirillus spp. were determined following a similar method and including a specific probe as previously described [18].
Statistics
Statistical analyses were performed using the IBM SPSS Statistics, 27 (IBM Corp.). We applied: (1) a log transformation of non-normally distributed data, (2) the Spearman’s correlation to assess relationships between variables; (3) T-test to compare means or a paired T-test when possible, (4) an ANOVA for multivariate analysis, in which we assumed an equal variance, followed by a Tukey post hoc test for multiple comparisons. The mean differences and correlations were considered significant if p < 0.05 and highly significant if p < 0.01.
Results and Discussion
Table 3 shows an overview of the results obtained considering all the samples included in this work. The extracted DNA mean quantity is significantly higher than RNA (45.55 ± 16.27 vs. 30.22 ± 10.37 µg/µL, T-test p < 0.01). Such difference remains significant also subdividing the data for the two digesters (T-test, p > 0.05).
The DNA and RNA quantities extracted into the two different digesters are similar showing the stability and the comparability of the active biomass into the two anaerobic reactors. Moreover, there is no significant difference in the extraction efficiency among the subsequent sampling sessions, showing quite stable systems.
The estimated presence of methanogen is equal to 8.58 ± 0.54 Log gene copies/mL sludge that is a comparable data with the literature considering the same feeding and similar volatile solids concentration [19, 20].
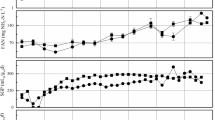
From a biological point of view, the analysis starting from different macromolecules such as DNA or RNA has a different biological meaning (Fig. 1). In literature, under different environmental conditions, abundance of mcrA transcripts changed while mcrA copy number remained almost the same, suggesting that only certain members of the methanogenic community were metabolically active and responsible for methane production [21]. Gene-expression—by the production of transcripts - is the process by which gene information is used for the synthesis of a functional gene product (typically a protein and/or an enzyme). Transcriptomic can not only determine the presence of an organism but also establish how the microorganism is adapting to the digester’s environmental changes [22]. Gene expression is common in all the cells; in prokaryotic and archaea cells, both transcription and translation occur within the cytoplasm, lacking of a defined nucleus. The RNA extraction from complex samples, as sludge, presents higher technical difficulties due to the reduced stability of such macromolecule respect the DNA. This is also confirmed by the significant difference observed for both reactors (A and B) in the total methanogen concentration evaluated comparing DNA and RNA. On the other hand, as showed on Fig. 1, a significant shift of the expressed Methanosarcina spp. can be observed during the phase 3 in both two digesters, independently by the SBO addiction. This was probably due to other influencing factors on the reactors.

Grey part: scheme of the DNA and its transcript (mRNA) biological significant in the methanogen analysis. Blank part: error bars of the methanogen determinations for the two parallel digesters (Reactor A and the addited Reactor B) during the phase 3 (last addition), subdivided in DNA and the cDNA results
As reported in the Fig. 1, starting from the RNA extract an effective involvement of the methyl coenzyme M reductase is similar into the two biodigesters. On the other hand, the ratio between the expression and presence of the same gene in the two biodigesters showed a slight inhibition into the reactor B, where the SBO additive was added (83% reactor A vs. 73% reactor B) especially in the last phase with higher additive dose (115% reactor A vs. 98% reactor B).
The results (Fig. 2) showed a methanogen functional activities 2 Log lower than their total presence (T-test, p < 0.01) while the same difference for the Methanosarcina is lower (0.23 Log), confirming the key role of such methanogen group, as observed in the literature in combination to Methanoculleus, into the AD of OFMSW [23, 24]. For Methanosarcina DNA and RNA concentration are not correlated (p > 0.05), suggesting a hyper-activation of this methanogenic species in the digesters.

Error bars of the methanogens determinations for the two parallel digesters (blank and addited) during the experimental period. Results reported starting from the extracted DNA and the cDNA
Methanogens belonging to the genera Methanocorpuscolum seem to be present but very low active while methanogens belonging to Methanospirillum seem to be absent or weakly present.
A paired T-test between the total methanogens into the biodigesters, considering the two parallel systems completely similar except the addictive treatment, shows a significant difference (p = 0.002), where the reactor B with additive is lower than the blank reactor A (7.48 vs. 7.67 Log gene copies/mL sludge). Moreover, a significantly lower functionality of Methanosarcina was observed 5.69 vs. 6.05 Log gene copies/mL sludge. Such difference is coherent with a difference in the methane production by the biodigester. The additive seems to produce both a decrement of the methane production (− 8% CH4 volume) and of the methanogens, especially at the higher addition doses. Previous studies showed as humic acids are hampering the performance of anaerobic digesters, and similar SBO (consisting of a mixture of humic compounds) was favourable for the NH3 reduction but was inhibiting for methanogen metabolisms [25].
Conclusions
The sampling methodology, including several biological and technical replicates, produces a robust data set able to descript the microbial dynamics into the two biodigesters. The difference observed was of minor entities but significant just for the methods and replicate applied. The two systems were stable and comparable in terms of extracted DNA, RNA, and methanogens dynamics. However, total methanogens and the Methanosarcina activity can be assumed as a possible indicator of major biomethane production even if their modulation was very slight between the two systems, while both Methanocorpuscolum and Methanospirullim were not so relevant in such systems, in fact no significant modulation were observed. Using the selected targets, only 3–4% of the present (target methanogen genera/total methanogens, DNA analysis) and 12–13% (target methanogen genera/total methanogens, RNA analysis) of the active methanogens are described, then this method is suitable to compare different systems but not for the exhaustive knowledge of the methanogen microbiota into the digester. In the future, the reduction of the costs for NGS methods and the reference genome’s implementation for the environmental methanogens could guarantee the application also for such kind of samples and a wider characterization of biomethane producer microbial.
Data Availability
the data set can be supply under a motivate request.
References
Siebert, S., Gilbert, J., Ricci-Jürgensen, M.: Compost production in Europe (2019)
European Parliament and the Council of the European Union: Directives Directive (EU): 2018/2001 of the European Parliament and of the Council of 11 December 2018 on the promotion of the use of energy from renewable sources (recast) (2018). (Text with EEA relevance)
Istituto Superiore per la Protezione e la Ricerca Ambientale: :Rapporto Rifiuti Urbani 2021 ISPRA (2021)
Blair, E.M., Dickson, K.L., O’Malley, M.A.: Microbial communities and their enzymes facilitate degradation of recalcitrant polymers in anaerobic digestion. Curr. Opin. Microbiol. 64, 100–108 (2021). https://doi.org/10.1016/j.mib.2021.09.008
Amin, F.R., Khalid, H., El-Mashad, H.M., Chen, C., Liu, G., Zhang, R.: Functions of bacteria and archaea participating in the bioconversion of organic waste for methane production. Sci. Total Environ. (2021). https://doi.org/10.1016/j.scitotenv.2020.143007
Ghosh, P., Kumar, M., Kapoor, R., Kumar, S.S., Singh, L., Vijay, V., Vijay, V.K., Kumar, V., Thakur, I.S.: Enhanced biogas production from municipal solid waste via co-digestion with sewage sludge and metabolic pathway analysis. Bioresour. Technol. (2020). https://doi.org/10.1016/j.biortech.2019.122275
Mamindlapelli, T.A.S.J., Begum, N.K., Arelli, S., Juntupally, V., Ahuja, S., Dugyala, S., Anupoju, S.K.: Anaerobic mono and co-digestion of organic fraction of municipal solid waste and landfill leachate at industrial scale: Impact of volatile organic loading rate on reaction kinetics, biogas yield and microbial diversity. Sci. Total Environ. (2020). https://doi.org/10.1016/j.scitotenv.2020.142462
Tonanzi, B., Crognale, S., Gianico, A., della Sala, S., Miana, P., Zaccone, M.C., Rossetti, S.: Microbial community successional changes in a full-scale mesophilic anaerobic digester from the start-up to the steady-state conditions. Microorganisms 9, 25–81 (2021). https://doi.org/10.3390/microorganisms9122581
Tian, H., Mancini, E., Treu, L., Angelidaki, I., Fotidis, I.A.: Bioaugmentation strategy for overcoming ammonia inhibition during biomethanation of a protein-rich substrate. Chemosphere 231, 415–422 (2019). https://doi.org/10.1016/j.chemosphere.2019.05.140
Ma, H., Zhao, Y., Yang, K., Wang, Y., Zhang, C., Ji, M.: Application oriented bioaugmentation processes: Mechanism, performance improvement and scale-up. Bioresour. Technol. 344, 126192 (2022)
Westerholm, M., Liu, T., Schnürer, A.: Comparative study of industrial-scale high-solid biogas production from food waste: process operation and microbiology. Bioresour. Technol. (2020). https://doi.org/10.1016/j.biortech.2020.122981
Basak, B., Patil, S.M., Saha, S., Kurade, M.B., Ha, G.S., Govindwar, S.P., Lee, S.S., Chang, S.W., Chung, W.J., Jeon, B.H.: Rapid recovery of methane yield in organic overloaded-failed anaerobic digesters through bioaugmentation with acclimatized microbial consortium. Sci. Total Environ. (2021). https://doi.org/10.1016/j.scitotenv.2020.144219
Ao, T., Xie, Z., Zhou, P., Liu, X., Wan, L., Li, D.: Comparison of microbial community structures between mesophilic and thermophilic anaerobic digestion of vegetable waste. Bioprocess. Biosyst. Eng. 44, 1201–1214 (2021). https://doi.org/10.1007/s00449-021-02519-5
Braz, G.H.R., Fernandez-Gonzalez, N., Lema, J.M., Carballa, M.: Organic overloading affects the microbial interactions during anaerobic digestion in sewage sludge reactors. Chemosphere 222, 323–332 (2019). https://doi.org/10.1016/j.chemosphere.2019.01.124
Sanz, J.L., Köchling, T.: Next-generation sequencing and waste/wastewater treatment: a comprehensive overview. Rev. Environ. Sci. Bio Technol. 18(4), 1–46 (2019)
Francavilla, M., Beneduce, L., Gatta, G., Montoneri, E., Monteleone, M., Mainero, D.: Biochemical and chemical technology for a virtuous bio-waste cycle to produce biogas without ammonia and speciality bio-based chemicals with reduced entrepreneurial risk. J. Chem. Technol. Biotechnol. 91, 2679–2687 (2016). https://doi.org/10.1002/jctb.4875
Traversi, D., Villa, S., Lorenzi, E., Degan, R., Gilli, G.: Application of a real-time qPCR method to measure the methanogen concentration during anaerobic digestion as an indicator of biogas production capacity. J. Environ. Manag. 111, 173–177 (2012). https://doi.org/10.1016/j.jenvman.2012.07.021
Traversi, D., Romanazzi, V., Degan, R., Lorenzi, E., Carraro, E., Gilli, G.: Microbial-chemical indicator for anaerobic digester performance assessment in full-scale wastewater treatment plants for biogas production. Bioresour. Technol. 186, 179–191 (2015). https://doi.org/10.1016/j.biortech.2015.03.042
Steinberg, L.M., Regan, J.M.: mcrA-targeted real-time quantitative PCR method to examine methanogen communities. Appl. Environ. Microbiol. 75, 4435–4442 (2009). https://doi.org/10.1128/AEM.02858-08
Ho, D.P., Jensen, P.D., Batstone, D.J.: Methanosarcinaceae and acetate-oxidizing pathways dominate in high-rate thermophilic anaerobic digestion of waste-activated sludge. Appl. Environ. Microbiol. 79, 6491–6500 (2013). https://doi.org/10.1128/AEM.01730-13
Alvarado, A., Montañez-Hernández, L.E., Palacio-Molina, S.L., Oropeza-Navarro, R., Luévanos-Escareño, M.P., Balagurusamy, N.: Microbial trophic interactions and mcrA gene expression in monitoring of anaerobic digesters. Front. Microbiol. 5, 597 (2014)
Vanwonterghem, I., Jensen, P.D., Ho, D.P., Batstone, D.J., Tyson, G.W.: Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr. Opin. Biotechnol. 27, 55–64 (2014)
Zhang, Y., Li, J., Liu, F., Yan, H., Li, J., Zhang, X., Jha, A.K.: Specific quorum sensing signal molecules inducing the social behaviors of microbial populations in anaerobic digestion. Bioresour. Technol. 273, 185–195 (2019). https://doi.org/10.1016/j.biortech.2018.11.022
Tyagi, V.K., Fdez-Güelfo, L.A., Zhou, Y., Álvarez-Gallego, C.J., Garcia, L.I.R., Ng, W.J.: Anaerobic co-digestion of organic fraction of municipal solid waste (OFMSW). Progress and challenges. Renew. Sustain. Energy Rev. 93, 380–399 (2018)
Yap, S.D., Astals, S., Lu, Y., Peces, M., Jensen, P.D., Batstone, D.J., Tait, S.: Humic acid inhibition of hydrolysis and methanogenesis with different anaerobic inocula. Waste Manag. 80, 130–136 (2018). https://doi.org/10.1016/j.wasman.2018.09.001
Funding
Open access funding provided by Università degli Studi di Torino within the CRUI-CARE Agreement. The research activities was funded by ACEA Pinerolese S.p.A. and University of Turin.
Author information
Authors and Affiliations
Contributions
Conceptualization, DT and DM; methodology, DT; software, DT; validation, DT; digester manage VN: and DM, sampling, VN, EA, CC; extraction EA; biomolecular analysis DT, EA, EF, CC; resources, DT and MD; data curation, DT; writing—original draft preparation, VN, CC; writing—review and editing, DT, DM. All authors have read and agreed to the submitted version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no conflict of interest.
Ethical Approval
No human or animal experimentation is included.
Consent for Publication
The involved private and public organizations consent to the publication of the produced results.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Negro, V., Mainero, D., Caredda, C. et al. Analyses of Methanogenic Population as Key Indicator for Anaerobic Digestion: A Case-Study in the Organic Fraction of Municipal Solid Waste Treatment. Waste Biomass Valor 14, 2461–2468 (2023). https://doi.org/10.1007/s12649-022-01875-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12649-022-01875-x