
Abstract
Cardiac myocytes are terminally differentiated cells and possess extremely limited regenerative capacity; therefore, preservation of mature cardiac myocytes throughout the individual’s entire life span contributes substantially to healthy living. Autophagy, a lysosome-dependent cellular catabolic process, is essential for normal cardiac function and mitochondria maintenance. Therefore, it may be reasonable to hypothesize that if endurance exercise promotes cardiac autophagy and mitochondrial autophagy or mitophagy, exercise-induced cardiac autophagy (EICA) or exercise-induced cardiac mitophagy (EICM) may confer propitious cellular environment and thus protect the heart against detrimental stresses, such as an ischemia–reperfusion (I/R) injury. However, although the body of evidence supporting EICA and EICM is growing, the molecular mechanisms of EICA and EICM and their possible roles in cardioprotection against an I/R injury are poorly understood. Here, we introduce the general mechanisms of autophagy in an attempt to integrate potential molecular pathways of EICA and EICM and also highlight a potential insight into EICA and EICM in cardioprotection against an I/R insult.
Similar content being viewed by others


Introduction
Despite several decades of incessant pharmaceutical and clinical efforts to develop therapeutic strategies against heart diseases, the outcomes of those trials have been dismal and unsuccessful. In contrast, endurance exercise, a nonpharmacological remedy, has been shown to ameliorate morbidity and mortality caused by ischemic injuries because it improves cardiac contractile function and cellular homeostasis and to protect hearts against a wide range of heart diseases [1, 2]. Interestingly, even acute endurance exercise (3–5 days) provides cardioprotection against an ischemia–reperfusion (I/R) insult, although the molecular mechanisms of exercise-induced cardioprotection remain unclear.
In terms of the characteristics of cardiomyocytes (e.g., discontinuation of cell division and absence of regenerative capacity), maintaining and/or enhancing proper damage control over intracellular disturbance caused by oxidative stress and calcium overload is a key feature required to sustain normal cardiac function throughout an individual’s entire lifespan and confer cardiac fitness and wellness. In this regard, there is an abundance of evidence from a variety of studies which shows that cardiac autophagy—a conserved mammalian catabolic process by which unwanted cellular cargos, such as damaged proteins, lipids, DNA, and dysfunctional organelles, such as mitochondria, are discarded in a lysosome-dependent manner—is an essential element for maintaining cardiac function and providing cardioprotection against hypoxia and an I/R insult [3,4,5]. Therefore, it seems reasonable to hypothesize that if exercise increases autophagy, the exercise-induced autophagy in the heart may be a novel cardioprotective avenue. In support of this notion, recent studies have shown that both acute and chronic endurance exercise enhance autophagy in the heart post endurance exercise [6,7,8,9]. However, whether exercise-induced cardiac autophagy (EICA) is a substantial component contributing to cardioprotection against I/R injuries remain completely unknown. Moreover, the exact underlying mechanisms of EICA have not yet been clearly elucidated.
Although cardiac autophagy contributes substantially to cell viability, it is the proper regulation of autophagy which the basic underlying rationale because dysregulated autophagy (e.g., chronic upregulation of autophagy) exerts a detrimental influence. Thus, a cellular auto-recognition system that modulates ideal duration, frequency, and degree of autophagy via an automatic turn-on/turn-off switch should exist in cells to preempt any unnecessary chronic activation of autophagy. This scenario can be questioned in a situation where people participate in a long-term endurance exercise program. For example, does chronic endurance exercise continue to raise autophagy levels in response to daily exercise or does it only episodically activate it upon demand. It is tempting to speculate that exercise-induced autophagy in hearts occurs in an episodic manner rather than in a random chronic manner although this postulation remains to be demonstrated. To examine the precise pattern of exercise-induced cardiac autophagy, an understanding of acute bouts of exercise (at least 1–5 days of exercise)-induced autophagy is essential to monitor daily autophagy patterns in response to exercise, from which the effects of long-term chronic endurance exercise-induced autophagy on cardiac health can be inferred.
Not only is the proper disposal of proteins and lipids via autophagy important for cardioprotection, the timely removal of dysfunctional mitochondria via autophagy (hereafter mitophagy) is also a critical process that contributes to sustaining optimal cardiac function. Mitochondria are critical organelles that are required to provide a biological form of energy, namely, ATPs; however, once becoming dysfunctional, they can trigger apoptotic as well as necrotic cell death [10, 11]. To avoid undesired mitochondria-mediated cell death, mammalian cells are able to promptly activate and promote mitophagy to selectively eliminate impaired mitochondria, thereby preventing further collateral cell death [12]. Relating to this observation, a few studies have recently shown evidence of exercise-induced cardiac mitophagy (EICM), but the molecular signaling pathways of EICM remain undefined. Thus, to improve our current understanding of molecular mechanisms of EICM, here we first provide a general review of mitophagy and then we highlight potential signaling pathways and roles of EICM in cardioprotection against an I/R injury.
In summary, this review will advance our body of knowledge associated with EICA and EICM by accomplishing the following three objectives: (1) to summarize canonical/noncanonical mechanisms of autophagy; (2) to present current molecular mechanisms of EICA and EICM; (3) to suggest a potential, cardioprotective role of EICA and EICM against I/R injury.
Autophagy
Autophagy is a conserved lysosomal catabolic process in mammals that is typically regulated by nutrient/energy availability, hypoxia, and reactive oxygen species (ROS). Although first observed in 1963 [13], autophagy has not been actively investigated in various research fields, including neurodegenerative diseases, diabetes, heart diseases, and exercise, until recently. Studies have identified autophagy as an important mediator of maintaining normal cardiac function, as any deficiency of autophagy impairs cardiac function. For example, conditional cardiac-specific deletion of ATG5, a critical protein for autophagy induction, results in cardiac dysfunction [14].
In general, autophagy follows canonical pathways in which activation of AMP-activated kinase (AMPK) coincides with inactivation of mammalian target of rapamycin (mTOR). However, results from recent studies suggest that autophagy can also be accomplished independent of canonical routes in certain conditions. Therefore, in this section, both canonical and noncanonical pathways of autophagy will be discussed.
Canonical autophagy pathways
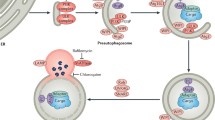
As illustrated in Fig. 1, canonical autophagy is tightly regulated by a series of protein–protein interactions and completed by lysosomes through consecutive steps of autophagy: initiation, nucleation, elongation, maturation, and fusion/degradation.

An overview of signaling nexus of autophagy pathways. Phosphorylated AMP-activated kinase (AMPK) initiates induction of autophagy by phosphorylating (p in orange circle) ULK1 (a protein kinase) at Ser555, which then activates a class III PI 3-kinase (PI3K) via phosphorylation in the PI3K–ATG14L (ATG refers to an autophagy-related family of proteins) protein complexes. The activated PI3K–ATG14L complexes recruit the scaffold protein Beclin 1, dissociated from Beclin 1–BCL-2 complexes upon BCL-2 (member of family of proteins which regulate apoptosis) phosphorylation, to form a PI3K–ATG14L–BECLIN 1 complex. This complex joins together with a fragment of double membrane, and the activated PI3K in the complex produces the phospholipid PI3P (phosphatidylinositol 3-phosphate) that activates WIPI1, a homolog of the yeast Atg18 protein, and begins to build a phagophore (also called the isolation membrane). To elongate the phagophore, ATG12–ATG5–ATG16–LC3-II complexes are needed. For this process, LC3, a central protein in the autophagy pathway, is cleaved by ATG4, resulting in LC-1, and the resultant LC3-I is then conjugated with phosphatidylethanolamine (PE) through ATG7 to become LC-II. The LC3-II now conjugates with the ATG12–ATG5–ATG16 complexes to form ATG12–ATG5–ATG16–LC3-II complexes. Finally, ATG12–ATG5–ATG16–LC3-II complexes are attached to an activated phagophore and begin to elongate the membrane. The elongated phagophore encloses cellular cargos, including protein aggregates and mitochondria, using the LC3-II residing in the luminal side of the phagophore, and LC3 adapter proteins, such as p62 and BNIP3 (BCL2/adenovirus E1B nineteen kDa interacting protein 3). Once cellular cargos are completely enclosed, an autophagsome (a spherical structure with double layer membrane) is formed and subsequently fused with a lysosome with the help of lysosome-associated membrane protein2 (LAMP2) and soluble N-ethylmaleimide-sensitive fusion attachment protein receptor (SNARE) proteins. Cargo materials are then processed by lysosomal proteases such as CATHEPSIN L and recycled. To enhance autophagic fluxes, lysosomal biosynthesis also occurs by upregulating a master transcription regulator of lysosomal biogenesis, transcription factor EB (TFEB), which is regulated by the modulation of the phosphorylation state. An activated CALCINEURIN, a calcium-dependent phosphatase, dephosphorylates TFEB and facilitates the translocation of TFEB to the nucleus, where TFEB promotes the transcription of lysosome-related proteins
Initiation
Adenosine monophosphate-activated kinase is an enzyme that monitors cellular energy homeostasis; thus, when it recognizes a shortage of ATP, it triggers catabolism (e.g., lipolysis) to ensure proper levels of energy [15]. Moreover, AMPK plays a critical role in initiating another catabolic process, autophagy, under ischemic and nutrient-deficient conditions [16]. Activated AMPK phosphorylates a serine/threonine protein kinase uncoordiated-51 like kinase1 (ULK1) at Ser555 that enhances autophagy induction. Next, the activated ULK1 in the ULK1–Atg13–FIP200 complex potentiates a class III phosphatidylinositol-4,5-bisphosphate 3 kinase (PI3K) and promotes the formation of complex between PI3K and ATG14L, which relays a signal for the nucleation process by engaging Beclin 1, a mammalian homolog of yeast ATG6 and a central scaffold protein, into the complex [17].
Nucleation
Nucleation (or phagophore formation) requires the formation of the Beclin 1–PI3K–ATG14L complex. For this process, BECLIN 1 needs to be released from the heterodimer complex between antiapoptotic protein BCL-2 and Beclin 1. Under a nutrient-rich condition, the dissociation of Beclin 1 from BCL-2 is hindered, resulting in the failure to construct the BECLIN 1–PI3K–ATG14L complexes, whereas under a nutrient-deficient condition, BCL-2 becomes phosphorylated, releases Beclin 1, and thus causes the formation of Beclin 1–PI3K–ATG14L complexes. The activated PI3K by ULK1 in the complex now generates phosphatidylinositol 3-phosphate (PI3P) that links WIPI-1, a protein which plays an important role in autophagy, into the complex to facilitate the transfer of proteins needed for the implementation of phagophore structure.
Elongation and maturation
Phagophores (immature or opened autophagosomes), which are created by several inductive processes of autophagy, should undertake an expansion and closure procedure for complete autophagosome conformation in conjunction with microtubule-associated protein 1A/1B light chain 3 (LC3) II and the ATG5–ATG12-ATG16 complex, both of which are combined through a series of conjugation and ligation events using ATG proteins. Briefly, once pro-LC3 is cleaved by ATG4, LC3-I is formed. Then, when the LC3-I becomes lipidated with phosphatidylethanolamine (PE) via ATG7 (ubiquitin E1-like activating enzyme) [18,19,20], LC3-II (an activated form of LC3) is generated. To form ATG5–ATG12–ATG16 complexes, ATG12 is first covalently conjugated to ATG5 by ATG7. The resultant ATG5–ATG12 is then conjugated with ATG16, resulting in the ATG5–ATG12–ATG16 complexes. This complex exerts an E3 ubiquitin ligase-like activity that tethers LC3-II with the help of ATG7 (E1-like activating enzyme) and ATG3 (E2 ubiquitin conjugating enzyme). The final ATG12–ATG5–ATG16–LC3-II complexes translocate to autophagosomal membrane and expand phagophore structures. Next, a C-shaped phagophore (open form) surrounds damaged proteins, lipids, and small organelles, such as mitochondria, designated for lysosomal degradation. In this process, LC3-II localized in the luminal side of the phagophore captures ubiquitinated protein aggregates and dysfunctional mitochondria via autophagy adaptors such as p62 and BCL2/adenovirus E1B nineteen kDa interacting protein 3 (BNIP3) respectively [21]. Finally, the elongated phagophore containing cellular cargos is completely enclosed and becomes a mature autophagsome.
Fusion
For the final autophagic degradation process, autophagosomes are fused with lysosome, forming autophagolysosomes (also referred to as autolysosomes), in which autophagic cargos are degraded by lysosomal hydrolases and recycled [22]. For this fusion process, autophagosome’s soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, such as vesicle-associated membrane proteins, syntaxins, and synaptosomal-associated proteins, interact with lysosomes to facilitate fusion events between autophagosomes and lysosomes [23,24,25]. In addition, the class C vacuolar protein sorting proteins and a GTPase family protein Rab7 play an important role in enhancing the fusion process in conjunction with lysosome-associated membrane proteins [26].
Noncanonical autophagy pathways
mTOR-independent pathways
An anabolic promoter, mTOR, has long been considered to be a potent autophagy regulator, as inhibition or activation of mTOR modulates autophagy. For example, mTOR inactivation under a nutrient-deficient condition precedes autophagy upregulation [16], whereas chronic activation of mTOR abolishes autophagy [27]. Surprisingly, however, recent studies have discovered that autophagy can be induced regardless of a mTOR state. For example, it has been shown that autophagy in the acutely exercise-trained heart occurs in the presence of mTOR activation [7] and that the heart undergoing reperfusion followed by ischemia exhibits elevated autophagy concurrent with mTOR activation [28]. Moreover, cannabinoid receptor 1 (CB1)-linked autophagy and trehalose (a natural disaccharide)-mediated autophagy occur in a mTOR-independent fashion [29, 30]. These studies indicate a strong possibility that, unlike starvation-mediated autophagy requiring mTOR inactivation, exercise-induced autophagy may have different autophagy regulatory mechanisms. Despite these novel observations, almost no attention has been given to this topic. Thus, further studies are required to establish a new mTOR-independent autophagy pathway.
BECLIN 1-independent pathways
Results from nutrition intervention studies suggest that BECLIN 1 is an important canonical autophagy protein involved in autophagy induction [31]. However, there is a growing body of evidence indicating that BECLIN 1 may not be an essential component in the autophagy process since autophagy induction remains even with knockdown of BECLIN 1 or overexpression of its antagonist BCL2 [32, 33]. In addition, a study also showed that luteolin, a member of the flavonoid family, triggers autophagy despite the loss of BECLIN 1 [34]. Consistent with these results, a recent study also found that acute endurance exercise promoted autophagy in the heart, even though BECLIN 1 was downregulated [7]. However, despite these data supporting BECLIN 1-independent autophagy, there is still a dearth of information on the exact molecular mechanisms.
EICA and flux
Although somewhat conflicting, results from various studies suggest that acute (1 day, 30–60 min) or long-term endurance exercise is a strong inducer of autophagy in cardiac tissues [6, 8, 9, 35, 36]. Support for the concept of EICA in the majority of studies is based on the examination of LC3-II levels or the ratio of LC3-II to LC3-I; as such, presentation of an increase in LC3-II protein level or in the ratio of LC3-II to LC3-I has been accepted as a hallmark of enhanced EICA. However, this method often leads to a misinterpretation of EICA data because LC3-II levels can rise not only due to the elevated rate of EICA (enhanced autophagic flux), but also by interruption of the autophagy maturation process between autophagosomes and lysosomes (impaired autophagy flux), resulting in the accumulation of autophagosomes without degradation. To circumvent this misinterpretation, it has been suggested that LC3-II be measured after treatment with an autophagy inhibitor, such as bafilomycin [37], chloroquine [38], NH4Cl [39], and colchicine [40], which specifically exerts inhibitory effects on lysosomes for the prevention of LC3-II degradation. Increased elevation of or no change in LC3-II level in the presence of one of these inhibitors represents enhanced autophagy flux or no change in flux, respectively. Unfortunately, very little is known on whether EICA is due to enhanced autophagy or to delayed autophagosomal degradation due to a lack of studies utilizing in vivo autophagy inhibitors.
An alternative avenue to the use of autophagy inhibitors in studies examining autophagy flux is the assessment of levels of the autophagy receptor p62 in parallel with assessment of LC3-II; p62 tethers polyubiquitinated target molecules to LC3-II in autophagosomes through its LC3 interacting region (LIR) motif and is eliminated along with autophagosomes via lysosomes [41]. Thus, reduced p62 levels implies enhanced autophagy flux. Likewise, LC3-II upregulation occurs in parallel with p62 reduction under the nutritional deficient or hypoxic state [42,43,44]. This phenomenon does not seem to be the case for EICA because, according to a recent study, levels of p62 do not decrease even though LC3-II levels are augmented after acute endurance exercise [8]. Does this mean that endurance exercise interferes in autophagy flux? The answer to this question is currently unknown as studies are not available to describe this differential response of p62. Nonetheless, it can be postulated that p62 may not be directly linked to EICA, but instead it may participate in the process of antioxidant enzyme expression since the Kelch-repeat domain of the Kelch-like ECH-associated protein 1 (KEAP1) interacting region (KIR) motif of p62 plays a critical role in activating nuclear factor erythroid 2-related factor 2 (Nrf2), a master transcription factor of numerous antioxidant enzymes. Given that two important motifs in p62 (LIR and KEAP1) are not concurrently engaged [45, 46] and that the subtle changes in the intracellular environment may alter the destiny of p62 (degradation by autophagy vs. upregulation for antioxidant enzyme expression), p62 may not be a reliable indicator that properly reflects the flux of EICA. Future studies verify an exact functional role of p62 in autophagy.
Possible mechanisms of EICA
While numerous research studies have reported underling mechanisms of autophagy in the heart under various stress conditions, very few studies have described distinct mechanisms of EICA; consequently, our current understanding of EICA is still rudimentary. In this section, as depicted in Fig. 2, we introduce both identified and possible EICA signaling pathways that may be linked to EICA.

Potential pathways of exercise-induced cardiac autophagy (EICA). Endurance exercise-induced AMPK activation initiates autophagy induction. Also, BCL2 phosphorylation (currently, responsible kinases for exercise-induced BLC2 phosphorylation have not been discovered) causes dissociation of BECLIN 1 from the BECLIN 1–BCL2 complexes and contributes to the initiation of phagophore formation and elongation. Upregulation of the rate-limiting autophagy enzyme ATG7 by endurance exercise increases autophagy flux, and studies have shown that FoxO3, a transcriptional activator which triggers apoptosis in the absence of survival factors, and hypoxia-inducible factor 1 (HIF1) upregulate LC3 and BNIP3, respectively, and thus enhance autophagy. Moreover, exercise-mediated reactive oxygen species (ROS) production has been suggested to induce autophagy elevation. Importantly, an activation of CALCINEURIN and inactivation (phosphorylation) of glycogen synthase kinase-3 (GSK3) cause the translocation of TFEB to the nucleus and gene expression associated with lysosomal biosynthesis, leading to autophagy enhancement
BCL-2 phosphorylation
Several studies have shown that BCL-2 (B cell lymphoma 2) and Beclin 1 exist as heterodimer complexes in the cytosol and that the dissociation of BECLIN 1 from BCL-2 upon cellular stress permits autophagy induction [6, 47,48,49]. A good question is “What is the potential signal for the dissociation of BECLIN 1 from BCL-2? Recent studies have reported that phosphorylation of BCL-2 is an essential step in the release of BECLIN 1 from the complex and subsequent autophagsome formation [50]. To elucidate the role of BCL-2 phosphorylation in EICA, He et al. conducted an in vivo experiment with a multiple knock-in mutation mouse model (mutation of three conserved phosphorylation sites of BCL-2) and found that endurance exercise-induced BCL-2 phosphorylation is essential for EICA, as a mutation of BLC-2 phosphorylation sites interfered in BECLIN 1 dissociation and prevented EICA [50].
This leads to the following question: Which kinases are responsible for the exercise-mediated phosphorylation of BCL-2 in the heart? While there is no ready answer at the present time, recent studies suggest that mitogen-activated protein kinase 8 (MAPK8) activation by AMPK is associated with BCL-2 phosphorylation and the dissociation of BECLIN 1 from BCL-2–BECLIN 1 complexes in cardiomyocytes [51]. Moreover, c-Jun N-terminal kinase 1 (JNK1) also induces BCL-2 phosphorylation, triggering the dissociation of BECLIN 1 from BCL-2, and is linked to autophagy induction under starvation conditions in non-cardiac cells [52]. Interestingly, He et al. reported that neither JNK nor MAPK was associated with exercise-induced BCL2- phosphorylation [50]. However, since this is the only study available showing potential kinases involved in exercise-induced BLC-2 phosphorylation and providing a possible clue for the mechanism of EICA, further studies are necessary to confirm the observation. Lastly, protein kinase C (PKC) has been suggested to phosphorylate BCL-2 and promote cell survival by suppressing apoptosis [53, 54], but it has not yet been determined whether PCK-mediated BLC2 phosphorylation is related to EICA.
ATG7
ATG7 is a ubiquitin-activating E1 enzyme (a rate-limiting enzyme) involved in facilitating autophagosomes through ATG12–ATG5–LC3-II conjugation with the help of ATG3 and ATG10, both E2 ubiquitin-conjugating enzymes [55, 56]. A direct impact of ATG7 in autophagy has recently been demonstrated in a respectable in vivo experiment in which ATG7 transgenic mice showed enhanced cardiac autophagy flux without modulation of other ATG proteins [35]. This study also showed that voluntary wheel running exercise synergizes ATG7 effects in terms of autophagy promotion and reverses crystalline αβ mutation-induced cardiac fibrosis [35]. In line with this study, Lee et al. observed that endurance exercise-induced autophagy correlates with ATG7 upregulation [7].
AMP-activated kinase
AMP-activated kinase has emerged as a potent autophagy inducer because AMPK-induced direct phosphorylation of ULK1 at Ser555 or RATOR of mTORC1 via TSC1/2 activation leads to autophagy induction [16]. As a variety of evidence has shown that endurance exercise activates AMPK in many tissues, it seems reasonable to hypothesize that exercise-induced AMPK activation may be associated with exercise-induced autophagy and that there is a plethora of literature relating to AMPK and EICA. Surprisingly, however, it remains largely unclear whether AMPK is a key modulator of acute exercise-induced autophagy in the heart. Currently, only two studies have focused on this topic, with both showing that acute endurance exercise enhances AMPK phosphorylation; however, the results do not seem to be in accordance with each other. For example, one study shows that AMPK phosphorylation levels increase immediately after exercise and return to a resting level 30 min after exercise, but that autophagy peaks at 1 h after exercise [8], whereas another study reveals that AMPK phosphorylation remains elevated up to 1 h after exercise in parallel with increased EICA levels [7]. Unfortunately, based on the results of these two studies, it cannot be inferred whether AMPK activation is necessary for EICA or should concur with EICA. Further studies are required to verify the exact role of AMPK in EICA.
Glycogen synthase kinase-3 and calcineurin
The promotion of autophagy induction and of maturation are two important steps in the autophagy process, but improved biosynthesis of lysosomal machinery for the successful degradation of autophagosomes is also critical to balance the rate of autophagosome production in order to prevent undesirable cellular stresses caused by unwanted accumulation of autophagosomes. Supporting this notion, recent studies have shown that an increase in lysosomal biosynthesis through the overexpression of a master transcription factor of lysosomal biogenesis, transcription factor EB (TFEB), enhances autophagic flux and reduces cell death [57]. Parr et al. also revealed that TFEB activation through the inhibition of glycogen synthase kinase-3 (GSK3) promotes the biosynthesis of lysosomal proteins and autophagy [58]. This latter study seems to provide a potential clue that TFEB activation via endurance exercise-induced GSK3 inactivation (phosphorylation) may be a key signaling pathway that improves autophagy by upregulating lysosomal biogenesis since endurance exercise training is known to inactivate GSK3 [59, 60]. However, whether exercise-induced GSK3 phosphorylation is linked to EICA remains unknown at the present time.
In addition to GSK3, a calcium-dependent phosphatase, calcineurin, has emerged as a potential activator of TFEB in muscle tissues. One proposed mechanism is that when TFEB become dephosphorylated by calcineurin, it translocates to a nucleus, promotes lysosomal biosynthesis, and thus enhances autophagy in muscle tissues [61]. A recent study has provided important evidence that exercise training facilitates the translocation of TFEB to the nucleus and improves lysosomal biosynthesis activities in skeletal muscle [62].
Collectively, these studies suggest that exercise-induced autophagy concurs with lysosomal biosynthesis in skeletal muscle. However, comparable research has not yet targeted the heart. Thus, studies using genetic models are warranted to confirm this potential mechanism responsible for EICA.
Oxidative stress
Reactive oxygen species are generally known to cause cellular destructive stress and cell death; however, ROS also play important roles in maintaining cellular homeostasis and transmitting protective cell signaling [63]. This implies that it is the levels of ROS generation which may determine a cell’s fate; that is, a low level of ROS is beneficial whereas excessive ROS production is detrimental. Given that acute endurance exercise increases ROS production [64] but provides cardioprotective benefits [65, 66], it appears reasonable to hypothesize that an endurance exercise-induced moderate elevation in ROS may be used to alter cell signaling required for cellular adaption and gene expression. One potential example of an association of ROS with cellular adaptation is autophagy induction [67,68,69].
Mechanisms of ROS-induced autophagy are activation of a cysteine protease ATG4 that mediates LC3 processing [69] and inactivation of mTOR [70]. Studies have also confirmed a potential role of ROS in autophagy by demonstrating that the inhibition of ROS by antioxidant administration abolishes ROS-induced autophagy [67]. Similarly, a recent study has demonstrated that mitochondrial ROS enhances autophagy but that overexpression of manganese superoxide dismutase (MnSOD), a mitochondria-specific antioxidant enzyme, countermands ROS-induced autophagy [68]. In line with these observations, elevated ROS production in response to acute endurance exercise is associated with an increase in autophagy in the heart [64]. Collectively, these results support the notion that exercise-induced ROS production may be linked to EICA.
Transcription factors
Recent studies have suggested that transcriptional regulation of autophagy proteins plays a profound role in autophagy enhancement. Transcription factors FoxO1 and -3, (members of the Forkhead box-constraining protein O subfamily) are highly expressed in the heart and induce transcription of various autophagy-related genes, such as lc3 and atg12 [71], and beclin1 and bnip3, as well as autophagy [72]. In the heart, FoxO3 overexpression itself remarkably enhances cardiac autophagy [72]. Conversely, the ablation of Foxo3 in neonatal cardiomyocytes and mice hearts resulted in a lack of autophagy and cardiac hypertrophy [73].
FoxO activities are generally regulated by phosphorylation status; for example, phosphorylated FoxO by anabolic activation loses its activity and thus remains in the cytosol, whereas dephosphorylated FoxO become activated and translocates to the nucleus for expression of its autophagy target genes [74]. Nutrient deprivation results in increased FoxO3 activities and enhances autophagy [75]. FoxO activities can be regulated by another post-translational modification, such as acetylation versus deacetylation. The Sadoshima group has provided evidence that FoxO1 deacetylation by Sirtuin 1 (SIRT1) under conditions of glucose deprivation enhances autophagy [76]. This result suggests that a FoxO-mediated upregulation of BNIP3 is a key mechanism of autophagy under nutrient deficiency [77]. However, despite this potential role of FoxO in cardiac autophagy, it is still relatively unknown whether FoxO proteins are involved in EICA, although one study has shown that FoxO3 activities are not changed by acute endurance exercise. Further studies are required to verify the exact role of FoxO proteins in EICA.
In addition to FoxO, HIF1 (hypoxia inducing factor 1) has been suggested as a possible mediator of autophagy in many cell types and tissues [78]. A possible mechanism of HIF1-induced autophagy is that HIF1 is a potent transcription factor for BNIP3 [79]. HIF1 is known to be overexpressed during hypoxia/ischemia, and thus many studies have suggested that the lack of O2 availability may be a crucial trigger for HIF1 expression. However, a recent study using rat cardiomyocytes has demonstrated that HIF1 expression can be regulated by metabolic state (e.g., protein kinase A activation via cAMP), regardless of O2 availability [80], thus raising the possibility that upregulated HIF1 concurrent with autophagy elevation may be due to a metabolic adaptive response rather than solely hypoxia. This is an important clue that help researchers explore the question of whether HIF1 upregulation in response to acute endurance exercise is mediated by a hypoxic state or a metabolic state because a recent study has shown that acute endurance exercise upregulates HIF1 expression and EICA [7]. More importantly, it seems imperative to perform future studies to determine whether HIF1 is a primary inducer of EICA.
BNIP3
As briefly introduced in the previous section, BNIP3 has emerged as a potent inducer of autophagy in various cell lines and cardiac myocytes [3, 81,82,83]. BNIP3 is upregulated in response to acute endurance exercise, and its increase correlates with autophagy [7, 64]; however, the exact molecular function of BNIP3 in EICA remains unclear. Based on recent studies demonstrating that BNIP3 curbs mTOR activity and promotes autophagy [84] and that BNIP3 downregulation by siRNA suppresses autophagy [70], it can be presumed that BNIP3-induced autophagy may be associated with mTOR inhibition. Intriguingly, in contrast to these studies, acute endurance exercise promotes cardiac autophagy in conjunction with mTOR activation and BNIP3 upregulation [7]. This result suggests that EICA may not be linked to antagonistic interplay between BNIP3 and mTOR.
Another possible mechanism is a BNIP3-induced post-translational modification (e.g., acetylation of non-histone proteins). For example, BNIP3 transgenic mice show a significantly increased acetylation levels in cardiac tissues [85] in parallel with elevated autophagy. Also, cardiomyocytes treated with histone deacetylase inhibitor enhance cardiac autophagy [4]. Although these factors were not examined in the exercise-trained heart, the results suggest that an increase in protein acetylation by acute endurance exercise-induced BNIP3 upregulation may be a key mechanism responsible for EICA.
Exercise-induced cardiac mitophagy
Mitochondria function as a power plant to produce the biological form of energy (e.g., ATP) required for sustaining normal cellular activities (e.g., muscle contraction and relaxation and ion pumps). However, they can also initiate an intrinsic cell death when damaged. For this reason, cells evolutionarily developed a crucial strategy to selectively remove damaged or dysfunctional mitochondria with the help of lysosomes to avoid unwanted cell death. This process is termed mitophagy and plays protective roles against various heart diseases [50, 86,87,88].
The question now is just how cardiac cells identify and select dysfunctional mitochondria and undertake mitophagy. Recent studies have identified several mitophagy-related proteins and proposed potential mitophagy signaling pathways. As illustrated in Fig. 3, general mitophagy criteria are fulfilled by interplay between proteins involved in fission, fusion, and ubiquitination of mitochondria. For example, when mitochondria lose their membrane potential due to damage, fission proteins recognizing dysfunctional mitochondria converge on the mitochondrial outer membrane and subsequently recruit ubiquitin ligases that anchor the dysfunctional mitochondria with ubiquitin molecules. At this point, a ubiquitin adaptor protein connects polyuibiquitinated mitochondria to LC3-II of the autophagosome and facilitates the enclosure of targeted mitochondria into autophagosomes. The autophagosomes then fuse with the lysosome and are degraded by lysosomal digestive enzymes.

An overview of signaling nexus of mitophagy pathways. Dysfunctional mitochondria lose their membrane potential and are removed by mitophagy. A fission protein DRP1 is translocated to a dysfunctional mitochondrion and segregate it from healthy mitochondria. Then, potent mitophagy adaptor proteins BNIP3 and FUNDC1 locate to the isolated mitochondrion in which PINK1 (PTEN-induced putative kinase 1) accumulates and recruits a E3 ligase Parkin; in this process, another a E3 ligase MUL1 translocates to the mitochondrion. Parkin and MUL1 initiate ubiquitination of mitochondrial proteins, which are linked with LC3-II through a p62 ubiquitin binding motif and a LC3 interacting region and trapped inside of autophagosomes. Additionally, the LC3 interacting region of BNIP3 and FUNDC1 on the dysfunctional mitochondrial outer membrane binds to LC3 on the luminal side of an autophagosome and delivers the targeted mitochondria into the autophagosome. The delivered mitochondria are discarded through a lysosome. mPTP: Mitochondrial permeability transition pore
Given that endurance exercise induces the protective mitochondrial phenotype [65] and that mitophagy is a key event that monitors mitochondrial turnover [89, 90], it appears reasonable to hypothesize that endurance exercise may promote mitophagy, result in the collection of healthy mitochondria, and thus contribute to generating cardioprotective mitochondrial phenotypes. Unfortunately, no clear molecular mechanisms of EICM are available yet; therefore, in the following section, we first introduce general mitophagy pathways and then provide possible molecular signaling nexus of the mitophagy pathway that may link to EICM.
Fission/fusion
DRP1, MFN1/2, and OPA1
Mitochondrial turnover is regulated through the modulation of morphology dynamics (fusion and fission). Dynamin-related protein 1 (DRP1), a GTPase family protein, is associated with mitochondrial fission [91], whereas other GTPase family proteins, such as mitofusion 1 (MFN1) and mitofusin 2 (MFN2), and optic atrophy 1 (OPA1), are involved in mitochondrial outer membrane fusion and inner membrane fusion, respectively [92]. Indeed, Lee et al., using cardiac cell lines, demonstrated that DRP1-induced mitochondrial fission plays an essential role in mitophagy, as DRP1 mutants (K38E) or MFN1 overexpression abolishes mitochondrial fission [82]. Furthermore, recent in vivo conditional gene knockout (DRP1, MFN1, and MFN2) studies have provided clear evidence that improper alteration of mitochondrial dynamics leads to cardiac dysfunction [93]. For example, the deletion of DRP1 resulted in enlarged mitochondria and dilated cardiomyopathy; in contrast, MFN1 and MFN2 deletion led to mitochondrial fragmentation and induced eccentric ventricular remodeling [93].
The activity of DRP1 is also altered by post-translational modification (i.e., phosphorylation). PKA-mediated DRP1 phosphorylation at Ser637 weakens the ability of DRP1 to induce mitochondrial fission by interrupting its GTPase activity [94], whereas phosphorylation at Ser585 by cyclin-dependent kinase 1 (CDK1) promotes mitochondrial fission [95]. A recent study has also reported that dephosphorylation of DRP1 at Ser637 by a Ca2+-dependent phosphatase, calcineurin, results in mitochondrial fission [96].
In the context of mitophagy, further investigation of molecular links between the DRP1 phosphorylation state regulated by kinases and phosphatases will lead to the identification of a novel mechanism of EICM.
PINK-1, Parkin, and p62
Studies have suggested that interplay between a phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) and the E3 ligase Parkin has been reported to exert crucial influence in determining the fate of dysfunctional mitochondria via mitophagy. However, the signaling nexus of EICM pathways associated with PINK1 and Parkin is poorly understood due to the absence of mechanistic studies. Thus, in this section, we will provide some insights of potential mechanisms of EICM relating to PINK1 and Parkin.
PINK1 contains a mitochondria-targeting transmembrane domain and thus localizes to the outer membrane of mitochondria and functions as an upstream activator of Parkin. When the mitochondrial membrane potential is conserved, PINK1 is immediately degraded by a mitochondrial protease, presenilin-associated rhomboid-like protein (PARL), precluding the translocation of Parkin to normal mitochondria to prevent unintended mitophagy [97, 98]. However, when mitochondria lose their membrane potential (depolarization), PINK1 remains intact in mitochondria and recruits and activates PARKIN [99]. The activated Parkin then induces polyubiquitination on its substrates, such as voltage-dependent anion channel (VDAC) and mitofusin (MFN), and p62 subsequently binds to polymerized ubiquitin on the targeted mitochondria via its ubiquitin binding domain, tethers with LC3-II of autophagosomes through its LC3-interacting region (LIR) motif, and thus helps mitophagy to proceed [100,101,102].
Although the PNIK1–PARKIN–p62 signaling cascades described above are well-defined pathways of mitophagy in typically neuronal and cancer cell lines, it is uncertain and questionable whether this systematic mitophagy paradigm is replicated in cardiac tissues, as Gustaffson’s research team demonstrated that mitophagy is implemented in the absence of PNIK1 in the heart, providing novel evidence that PARKIN recruitment to depolarized mitochondria can be accomplished independent of PINK1 [103].
A growing number of studies has reported the potential involvement of PNIK1–PARKIN–p62 in EICM but these are all descriptive. Consequently, investigation of actual signaling pathways of EICM using isolated mitochondria and genetic models (e.g., knockout mice) are required to verify the potential association of the PINK1–PARKIN–p62 axis in EICM.
Mitochondrial E3 ubiquitin ligase 1
In addition to Parkin, recent studies have highlighted a mitochondrial E3 ubiquitin ligase 1 (MUL1) as a potential candidate involved in mitophagy because MUL1 facilitates the optimal function of DRP1 via ligase activity [104] and promotes MFN degradation [105], leading to mitochondrial fission and mitophagy. In support of this, Li et al. has demonstrated a potent role of MUL1 in mitophagy, as knockout of MUL1 failed to induce mitophagy in response to hypoxia and starvation [106]. Furthermore, knockdown of MUL1 in the Parkin knockout mouse displayed more exacerbated mitochondrial impairment compared to Parkin knockout only, indicating that MUL1 with Parkin plays a potential collateral role in mitophagy [105]. Similarly, inhibition of MUL1 degradation caused by the mitochondrial serine protease Omi/HtrA2 reduced MFN2 levels with enhanced mitophagy [107]. These studies clearly suggest that MUL1 is a critical mediator of mitophagy.
As yet no studies have shown any possible links between MUL1 and cardiac EICM; nonetheless, a study using rat neonatal cardiac myocytes did show a significant upregulation of MUL1 in response to chronic electrical stimulation (1 Hz for 48 h.), which may mimic an endurance exercise effect [108]. Obviously, this study should encourage further studies to search for a potential role of MUL1 in EICM.
Mitophagy adaptors
BNIP3
In addition to BNIP3 playing a role in promoting general autophagy, the discovery that BNIP3’s transmembrane domain targets the mitochondrial outer membrane [109] and its LIR motif [110, 111] has provided a critical clue that BNIP3 may function as an important mitophagy mediator.
A commonly proposed mechanism of BNIP3-mediated mitophagy is that depolarization of the membrane potential of mitochondria by opening the mitochondrial permeability transition pore (mPTP) through BNIP3 disseminates a “discard me” signal. This signal permits LC3-II of autophagosomes to couple with the LIR of BNIP3 and to enclose mitochondria inside autophagosomes, resulting in selective removal of mitochondria via mitophagy [111]. Interestingly, a recent study using adult rat cardiomyocytes overexpressing BNIP3 has shown that BNIP3 induces mitophagy in the absence of mPTP opening and cell death [81, 83]. These studies provide the novel insight that BNIP3 may induce mitophagy without causing perturbation of mitochondrial membrane potential and that its upregulation may contribute to mitochondrial quality control and cardiac health under a normal condition, such as exercise, although it is known to cause mitochondrial dysfunction and cell death under unfavorable stress conditions. Supporting this notion, research has shown a strong correlation between endurance exercise-induced BNIP3 upregulation and autophagy promotion [7, 64], which may imply a possible connection of BNIP3 with EICM, although it remains unknown whether upregulation of BNIP3 by endurance exercise is essential for ECIM.
In addition to its role as an LC3-II adaptor, BNIP3 appears to be an essential mediator of PINK1–Parkin-mediated mitophagy. For example, BNIP3 overexpression triggers Parkin translocation to mitochondria and promotes mitophagy in adult rat cardiomyocytes [82]. Moreover, BINP3 conjugation with PINK1 promotes PINK1 accumulation in the mitochondrial outer membrane, which allows Parkin recruitment and accommodates mitophagy [112].
FUNDC1
FUNDC1, an integral mitochondrial outer-membrane protein, contains a transmembrane domain targeting the mitochondrial outer membrane [113]. Similar to other mitochondrial adaptor proteins (e.g., BNIP3), FUNDC1 directly interacts with LC3 via its LIR motif and appears to play a substantial in mitophagy, as knockdown of FUNDC1 abolished hypoxia-induced mitophagy but restores it when wild-type FUNDC1 was regained [113]. This study also discovered that a phosphorylation state of FUNDC1 modulates FUNDC1 activity and thus mitophagy. For example, phosphorylation of FUNDC1 by Src kinase inactivates FUNDC1, whereas dephosphorylation of FUNDC1 enhances FUNDC1’s LC3 binding activities [113] and leads to dissociation of OPA1 from the FUNDC1–OPA1 complexes and promotion of DRP1 recruitment and fission of mitochondria, thereby contributing to mitophagy [114]. It will be interesting to learn which phosphatases are responsible for FUNDC1 activation, but this question remains unanswered. Furthermore, the possible link between FUNDC1 and cardiac EICM needs to be determined in future studies.
Cardiolipin
Cardiolipin (CL) is a phospholipid dispersed throughout the inner membrane of mitochondria and plays a critical role in maintaining activities of mitochondrial respiratory complexes. Intriguingly, CL also mediates mitophagy. Chu et al. reported that CL contains a LC3-interacting region and that externalization of CL from the inner membrane to the outer membrane upon mitochondrial stress allows LC3-II of autophagosomes to tether the CL of stressed mitochondria and leads to mitophagy [115]. Although a study showed an increase in CL content after endurance exercise [116], whether upregulated CL by exercise is associated with EICM has not yet been investigated.
Potential role of EICA and EICM in cardiac protection against an I/R injury
Cardiac autophagy critically contributes to cardiac protection under stress conditions, such as nutrient deficiency [117, 118] and proteinopathy [35]. However, whether enhanced cardiac autophagy and mitophagy in response to acute endurance exercise are essential for cardioprotection against an I/R injury are entirely unknown. Given recent studies demonstrating that enhanced cardiac autophagy via a selective agonist of adenosine receptor 1 or an administration of a histone deacetylase inhibitor prior to I/R reduces cell death [3, 4, 119], it seems reasonable to propose that promotion of exercise-mediated cardiac autophagy may be an important protective avenue that helps mitigate myocardial cell death caused by an I/R episode. Furthermore, it would be of great interest to explore whether endurance exercise-trained hearts show enhanced autophagy during an I/R insult, since upregulation of autophagy prior to an I/R insult rescues cells. Based upon our unpublished observation that endurance exercise training increases cardiac autophagy during an ex vivo I/R insult, it can be postulated that once exercise-trained cardiomyocytes are familiarized to an autophagy induction system, a rapid autophagic response would occur during an I/R insult and provide cardioprotection against an IR injury. This hypothesis should be examined in further studies to discover a novel mechanism responsible for exercise-induced cardioprotection.
What will be the main purpose of EICM? Dysfunctional mitochondria lose their membrane potential, leading to the initiation of apoptotic cell death through the release of pro-apoptotic factors, such as cytochrome c and apoptosis inducing factor [120]. Therefore, selective removal of dysfunctional mitochondria via mitophagy is a potent process required for sustaining healthy cardiomyocytes, as evidenced by recent research showing that the accumulation of dysfunctional mitochondria due to the failure of mitophagy results in cardiac dysfunction [121]. Although a clear relationship between EICM and cardioprotection against an I/R injury and an exact molecular mechanism of EICM has not yet been elucidated, it can be inferred that the promotion of exercise-mediated cardioprotective mitochondrial phenotypes via EICM may be a primary source of cardioprotection against myocardial infarction caused by an I/R event.
Summary
Exercise training has been known to confer cardioprotection against an I/R insult; however, the mechanisms of exercise-induced cardioprotection remain unclear. Given that acute endurance exercise promotes autophagy and that enhanced autophagy prior to an I/R insult is protective against myocardial infarction, as depicted in Fig. 4, it seems plausible that EICA and EICM may play a crucial role in cardioprotection against myocardial infarction. However, since clear mechanisms regulating EICA and EICM have not been established, here we introduced molecular signaling pathways (including canonical and non-canonical pathways of general autophagy and mitophagy shown in starvation and ischemia) and reviewed published articles on EICA and EICM. Finally, we deduced a possible mechanism of molecular signaling nexus of EICA and EICM pathways and a probable association of EICA and EICM with cardioprotection against an I/R injury.

A schematic outline of exercise-induced cardiac protection against an ischemia–reperfusion (I/R) injury. Cardiac myocytes are terminally differentiated and contain insufficient regenerative capacity; therefore, excessive cellular stresses caused by damaged proteins, lipids, and organelles can lead to irreversible cell damage, resulting in cell death. This unfavorable cellular condition in cardiac myocytes will be exacerbated when an I/R event concurs. In contrast, regular endurance exercise maintains the healthy cellular environment by disposing of damaged proteins and lipids and mitochondria through autophagy and mitophagy. Thus, upon an IR insult leading to cell death, exercise-trained hearts may resist I/R injuries and thus confer cardiac protection against I/R-induced myocardial infarction. EICM Exercise-induced cardiac mitophagy
Change history
13 November 2017
The article Potential signaling pathways of acute endurance exercise-induced cardiac autophagy and mitophagy and its possible role in cardioprotection, written by Youngil Lee.
References
Ascensao A, Ferreira R, Magalhaes J (2007) Exercise-induced cardioprotection—biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int J Cardiol 117(1):16–30
Powers SK, Quindry JC, Kavazis AN (2008) Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic Biol Med 44(2):193–201
Hamacher-Brady A, Brady NR, Gottlieb RA (2006) Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 281(40):29776–29787
Ma XC, Godar RJ, Liu HY (2014) Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129(10):1139–1151
Zhang Y, Ren J (2014) Targeting autophagy for the therapeutic application of histone deacetylase inhibitors in ischemia/reperfusion heart injury. Circulation 129(10):1088–1091
He CC, Bassik MC, Moresi V et al (2012) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481(7382):511–515
Lee Y, Kang EB, Kwon I et al (2016) Cardiac kinetophagy coincides with activation of anabolic signaling. Med Sci Sports Exerc 48(2):219–226
Ogura Y, Iemitsu M, Naito H et al (2011) Single bout of running exercise changes LC3-II expression in rat cardiac muscle. Biochem Biophys Res Commun 414(4):756–760
Fiuza-Luces C, Delmiro A, Soares-Miranda L et al (2014) Exercise training can induce cardiac autophagy at end-stage chronic conditions: insights from a graft-versus-host-disease mouse model. Brain Behav Immunol 39:56–60
Karch J, Kwong JQ, Burr AR et al (2013) Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2:e00772
Green DR, Galluzzi L, Kroemer G (2014) Cell biology. Metabolic control of cell death. Science 345(6203):1250256
Zhang J, Nadtochiy SM, Urciuoli WR et al (2016) The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am J Physiol Heart Circ Physiol 310(1):H29–H38
Duve CD (1963) The lysosome. Sci Am 208(5):64–73
Nakai A, Yamaguchi O, Takeda T et al (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13(5):619–624
Dobrzyn P, Pyrkowska A, Duda MK et al (2013) Expression of lipogenic genes is upregulated in the heart with exercise training-induced but not pressure overload-induced left ventricular hypertrophy. Am J Physiol Endocrinol Metab 304(12):E1348–E1358
Kim J. Kundu M, Viollet B et al (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141
Russell RC, Tian Y, Yuan H et al (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15(7):741–750
Yousefi S, Perozzo R, Schmid I et al (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8(10):1124–1132
Tanida I, Tanida-Miyake E, Ueno T et al (2001) The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem 276(3):1701–1706
Tanida I, Tanida-Miyake E, Komatsu M et al (2002) Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem 277(16):13739–13744
Hanna RA, Quinsay MN, Orogo AM et al (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with BNIP3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287(23):19094–19104
Yoshimori T (2004) Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun 313(2):453–458
Guo B, Liang Q, Li L et al (2014) O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol 16(12):1215–1226
Lu Y, Zhang Z, Sun D et al (2013) Syntaxin 13, a genetic modifier of mutant CHMP2B in frontotemporal dementia, is required for autophagosome maturation. Mol Cell 52(2):264–271
Meng J, Wang J (2015) Role of SNARE proteins in tumourigenesis and their potential as targets for novel anti-cancer therapeutics. Biochim Biophys Acta 1856(1):1–12
Jager S, Bucci C, Tanida I et al (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117(Pt 20):4837–4848
Shang L, Wang X (2011) AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 7(8):924–926
Matsui Y, Takagi H, Qu X et al (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100(6):914–922
Hiebel C, Kromm T, Stark M et al (2014) Cannabinoid receptor 1 modulates the autophagic flux independent of mTOR- and BECLIN1-complex. J Neurochem 131(4):484–497
Kang YL, Saleem MA, Chan KW et al (2014) Trehalose, an mTOR independent autophagy inducer, alleviates human podocyte injury after puromycin aminonucleoside treatment. PLoS One 9(11):e113520
Wu CA, Huang DY, Lin WW (2014) Beclin-1-independent autophagy positively regulates internal ribosomal entry site-dependent translation of hypoxia-inducible factor 1alpha under nutrient deprivation. Oncotarget 5(17):7525–7539
Scarlatti F, Maffei R, Beau I et al (2008) Non-canonical autophagy: an exception or an underestimated form of autophagy? Autophagy 4(8):1083–1085
Hasima N, Ozpolat B (2014) Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis 5:e1509
Park SH, Park HS, Lee JH et al (2013) Induction of endoplasmic reticulum stress-mediated apoptosis and non-canonical autophagy by luteolin in NCI-H460 lung carcinoma cells. Food Chem Toxicol 56:100–109
Bhuiyan MS, Pattison JS, Osinska H et al (2013) Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123(12):5284–5297
Xu T, Zhou Q, Zhou J et al (2011) Carboxyl terminus of Hsp70-interacting protein (CHIP) contributes to human glioma oncogenesis. Cancer Sci 102(5):959–966
Carreira RS, Lee Y, Chochani M et al (2010) Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 6(4):462–472
Teng AC, Miyake T, Yokoe S et al (2015) Metformin increases degradation of phospholamban via autophagy in cardiomyocytes. Proc Natl Acad Sci USA 112(23):7165–7170
D’Eletto M, Farrace MG, Falasca L et al (2009) Transglutaminase 2 is involved in autophagosome maturation. Autophagy 5(8):1145–1154
Ju JS, Jeon SI, Park JY et al (2016) Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J Physiol Sci 66(5):417–430
Pankiv S, Clausen TH, Lamark T et al (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282(33):24131–24145
Tan Q, Wang M, Yu M et al (2016) Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia 18(6):347–355
Chen D, Jin Z, Zhang J et al (2016) HO-1 protects against hypoxia/reoxygenation-induced mitochondrial dysfunction in H9c2 cardiomyocytes. PLoS One 11(5):e0153587
Hariharan N, Maejima Y, Nakae J et al (2010) Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 107(12):1470–1482
Jain A, Lamark T, Sjotttem E et al (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 285(29):22576–22591
Komatsu M, Kurokawa H, Waguri S et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–223
Dai JP, Zhao XF, Zeng J et al (2013) Drug screening for autophagy inhibitors based on the dissociation of Beclin1-Bcl2 complex using BiFC technique and mechanism of eugenol on anti-influenza A virus activity. PLoS One 8(4):e61026
Zalckvar E, Berissi H, Eisenstein M et al (2009) Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5(5):720–722
Zalckvar E, Berissi H, Mizrachy L et al (2009) DAP-kinase-mediated phosphorylation on the BH3 domain of Beclin 1 promotes dissociation of Beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 10(3):285–292
He C, Bassik MC, Moresi V et al (2012) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481(7382):511–515
Zou MH, Xie Z (2013) Regulation of interplay between autophagy and apoptosis in the diabetic heart: new role of AMPK. Autophagy 9(4):624–625
Wei Y, Pattingre S, Sinha S et al (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30(6):678–688
May WS, Tyler PG, Ito T et al (1994) Interleukin-3 and bryostatin-1 mediate hyperphosphorylation of Bcl2-alpha in association with suppression of apoptosis. J Biol Chem 269(43):26865–26870
Ito T, Deng X, Carr B et al (1997) Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem 272(18):11671–11673
Kaiser SE, Qiu Y, Coats JE et al (2013) Structures of Atg7-Atg3 and Atg7-Atg10 reveal noncanonical mechanisms of E2 recruitment by the autophagy E1. Autophagy 9(5):778–780
Tanida I, Yamasaki M, Komatsu M et al (2012) The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy 8(1):88–97
Ma XC, Godar RJ, Liu HY et al (2012) Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 8(3):297–309
Parr C, Carzaniga R, Gentleman SM et al (2012) Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Mol Cell Biol 32(21):4410–4418
Lajoie C, Calderone A, Trudeau F et al (2004) Exercise training attenuated the PKB and GSK-3 dephosphorylation in the myocardium of ZDF rats. J Appl Physiol (1985) 96(5):1606–1612
Sakamoto K, Arnolds DE, Ekberg I et al (2004) Exercise regulates Akt and glycogen synthase kinase-3 activities in human skeletal muscle. Biochem Biophys Res Commun 319(2):419–425
Medina DL, Di Paola S, Peluso I et al (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17(3):288–299
Mansueto G, Armani A, Viscomi C et al (2017) Transcription factor EB controls metabolic flexibility during exercise. Cell Metab 25(1):182–196
Li S, Deng Y, Feng J et al (2009) Oxidative preconditioning promotes bone marrow mesenchymal stem cells migration and prevents apoptosis. Cell Biol Int 33(3):411–418
Li H, Miao W, Ma J et al (2016) Acute exercise-induced mitochondrial stress triggers an inflammatory response in the myocardium via NLRP3 inflammasome activation with mitophagy. Oxidative Med Cell Longev 2016:1987149
Lee Y, Min K, Talbert EE et al (2012) Exercise protects cardiac mitochondria against ischemia–reperfusion injury. Med Sci Sports Exerc 44(3):397–405
Quindry JC, Hamilton KL, French JP et al (2007) Exercise-induced HSP-72 elevation and cardioprotection against infarct and apoptosis. J Appl Physiol (1985) 103(3):1056–1062
Hariharan N, Zhai P, Sadoshima J (2011) Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 14(11):2179–2190
Chen Y, McMillan-Ward E, Kong J et al (2007) Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 120(Pt 23):4155–4166
Scherz-Shouval R, Shvets E, Fass E et al (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760
Byun YJ, Kim SK, Kim YM et al (2009) Hydrogen peroxide induces autophagic cell death in C6 glioma cells via BNIP3-mediated suppression of the mTOR pathway. Neurosci Lett 461(2):131–135
Sengupta A, Molkentin JD, Yutzey KE (2009) FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 284(41):28319–28331
Schips TG, Wietelmann A, Hohn K et al (2011) FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc Res 91(4):587–597
Ni YG, Berenji K, Wang N et al (2006) Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114(11):1159–1168
Zhao Y, Hu X, Liu Y et al (2017) ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer 16(1):79
Paula-Gomes S, Goncalves DAP, Baviera AM et al (2013) Insulin suppresses atrophy- and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signaling. Horm Metab Res 45(12):849–855
Hariharan N, Maejima Y, Nakae J et al (2010) Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 107(12):1470–1482
Feng CC, Lin CC, Lai YP et al (2016) Hypoxia suppresses myocardial survival pathway through HIF-1alpha-IGFBP-3-dependent signaling and enhances cardiomyocyte autophagic and apoptotic effects mainly via FoxO3a-induced BNIP3 expression. Growth Factors 34:73-86
Hu Y, Sun Q, Li Z et al (2014) High basal level of autophagy in high-altitude residents attenuates myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg 148(4):1674–1680
Guo K, Searfoss G, Krolikowski D et al (2001) Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ 8(4):367–376
Bullen JW, Tchernyshyov I, Holewinski RJ et al (2016) Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci Signal 9(430):ra56
Lee Y, Kubli DA, Hana RA et al (2015) Cellular redox status determines sensitivity to BNIP3-mediated cell death in cardiac myocytes. Am J Physiol Cell Physiol 308(12):C983–C992
Lee Y, Lee HY, Hana RA et al (2011) Mitochondrial autophagy by BNIP3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301(5):H1924–H1931
Quinsay MN, Thomas RL, Lee Y et al (2010) Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6(7):855–862
Li Y, Wang Y, Kim E et al (2007) Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem 282(49):35803–35813
Thompson JW, Wei J, Appau K et al (2015) Bnip3 binds and activates p300: possible role in cardiac transcription and myocyte morphology. PLoS One 10(8):e0136847
Zhang W, Siraj S, Zhang R et al (2017) Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 5:1–2
Yu W, Gao B, Li N et al (2016) Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta 1863(8):1973–1983
Kyrychenko V, Polakova E, Janicek R et al (2015) Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 58(2):186–195
Shirakabe A, Fritzky L, Saito T et al (2016) Evaluating mitochondrial autophagy in the mouse heart. J Mol Cell Cardiol 92:134–139
Sun N, Yun J, Liu J et al (2015) Measuring in vivo mitophagy. Mol Cell 60(4):685–696
Zhu PP, Patterson A, Stadler J et al (2004) Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem 279(34):35967–35974
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337(6098):1062–1065
Song M, Mihara K, Chen Y et al (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21(2):273–285
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282(30):21583–21587
Taguchi N, Ishihara N, Jofuku A et al (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282(15):11521–11529
Buhlman L, Damiano M, Bertolin G et al (2014) Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim Biophys Acta 1843(9):2012–2026
Jin SM, Lazarou M, Wang C et al (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191(5):933–942
Matsuda N, Sato S, Shiba K et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189(2):211–221
Kazlauskaite A, Kondapalli C, Gourlay R et al (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460(1):127–139
Narendra D, Tanaka A, Suen DF et al (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803
Narendra DP, Jin SM, Tanaka A et al (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8(1):e1000298
Geisler S, Holmstrom KM, Skujat D et al (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12(2):119–131
Kubli DA, Cortez MQ, Moyzis AG et al (2015) PINK1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS One 10(6):e0130707
Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10(7):748–754
Yun J, Puri R, Yang H et al (2014) MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 3:e01958
Li J, Qi W, Chen G et al (2015) Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 11(8):1216–1229
Cilenti L, Ambivero CT, Ward N et al (2014) Inactivation of Omi/HtrA2 protease leads to the deregulation of mitochondrial Mulan E3 ubiquitin ligase and increased mitophagy. Biochim Biophys Acta 1843(7):1295–1307
Hooper CL, Paudyal A, Dash PR et al (2013) Modulation of stretch-induced myocyte remodeling and gene expression by nitric oxide: a novel role for lipoma preferred partner in myofibrillogenesis. Am J Physiol Heart Circ Physiol 304(10):H1302–H1313
Yasuda M, Han JW, Dionne CA et al (1999) BNIP3alpha: a human homolog of mitochondrial proapoptotic protein BNIP3. Cancer Res 59(3):533–537
Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif—crucial for selective autophagy. J Cell Sci 126(Pt 15):3237–3247
Zhu YY, Massen S, Terenzio M et al (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines Pro-survival mitophagy versus apoptosis. J Biol Chem 288(2):1099–1113
Zhang T, Xue L, Li L et al (2016) BNIP3 suppresses PINK1 proteolytic cleavage to promote mitophagy. J Biol Chem 291:21616–21629
Liu L, Feng D, Cheng G et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14(2):177–185
Chen M, Chen Z, Wang Y et al (2016) Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12(4):689–702
Chu CT, Ji J, Dagda RK et al (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15(10):1197–1205
Chicco AJ, McCune SA, Emter CA et al (2008) Low-intensity exercise training delays heart failure and improves survival in female hypertensive heart failure rats. Hypertension 51(4):1096–1102
Ikeda Y, Shirakabe A, Maejima Y et al (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116(2):264–278
Sciarretta S, Volpe M, Sadoshima J (2014) NOX4 regulates autophagy during energy deprivation. Autophagy 10(4):699–701
Yitzhaki S, Huang C, Liu W et al (2009) Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol 104(2):157–167
Ljubicic V, Menzies KJ, Hood DA (2010) Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle. Mech Ageing Dev 131(2):79–88
Oka T, Hikoso S, Yamaguchi O et al (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485(7397):251–255
Acknowledgements
This project was supported by a grant from the University of West Florida though the Office of Research and Sponsored Programs to YL (#164336), UWF Florida Research Fellowship to YL (CF6672), Research Equipment Enhancement Program Award to YL (R0036).
Author information
Authors and Affiliations
Contributions
YL, IK, YJ, WS, and LMC developed a concept and collected literature and drafted the manuscript; YL, IK, and YJ prepared the figures; YL, LMC and MHR edited and revised the manuscript; YL, IK, YJ, WS, LMC and MHR approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Lee, Y., Kwon, I., Jang, Y. et al. Potential signaling pathways of acute endurance exercise-induced cardiac autophagy and mitophagy and its possible role in cardioprotection. J Physiol Sci 67, 639–654 (2017). https://doi.org/10.1007/s12576-017-0555-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-017-0555-7