
Abstract
Pulmonary arterial hypertension (PAH) is a rare disease with a complex aetiology characterized by elevated pulmonary artery resistance, which leads to right heart ventricular afterload and ultimately progressing to right ventricular failure and often death. In addition to other factors, metabolites of arachidonic acid cascade play an important role in the pulmonary vasculature, and disruption of signaling pathways of arachidonic acid plays a central role in the pathogenesis of PAH. 15-Lipoxygenase (15-LO) is upregulated in pulmonary artery endothelial cells and smooth muscle cells of PAH patients, and its metabolite 15-hydroxyeicosatetraenoic acid (15-HETE) in particular seems to play a central role in the contractile machinery, and in the initiation and propagation of cell proliferation via its effects on signal pathways, mitogens, and cell cycle components. Here, we focus on our important research into the role played by 15-LO/15-HETE, which promotes a proliferative, antiapoptotic, and vasoconstrictive physiological milieu leading to hypoxic pulmonary hypertension.
Similar content being viewed by others


Introduction
Pulmonary hypertension (PH) is a severe and frequently fatal disease characterized by elevated mean pulmonary arterial (PA) pressure greater than 25 mmHg at rest or greater than 30 mmHg with exercise [1], and which contributes to the morbidity and mortality of adult and pediatric patients with various lung and heart diseases. According to the Venice Classification of Pulmonary Hypertension in 2003, PH is currently classified into five categories as listed in Table 1. Importantly, many of these diseases or conditions are associated with persistent or intermittent hypoxia, either globally or regionally, within confined areas of the lung [2]. The acute hypoxia-induced pulmonary vasoconstriction (HPV) is an important mechanism that aids in matching ventilation with perfusion by directing blood flow from poorly ventilated regions of the lung to areas with normal or relatively high ventilation. Although acute HPV benefits gas exchange and maximizes oxygenation of venous blood in the pulmonary artery, sustained HPV or chronic exposure to hypoxia is a major cause for the elevated pulmonary vascular resistance and pulmonary arterial remodeling (PAR) in patients with pulmonary arterial hypertension (PAH) associated with hypoxic cardiopulmonary diseases [3]. Vascular remodeling is characterized largely by medial hypertrophy and hyperplasia due to enhanced vascular smooth muscle cell (VSMC) proliferation or attenuated apoptosis and endothelial cell over-proliferation [4, 5]. However, the mechanism of pulmonary vascular remodeling (PVR) and pulmonary hypertension is still unknown.
The arachidonic acid cascade plays a vital role in homeostasis of the endothelium and VSMCs, and has been observed in dysregulation of downstream pathways of arachidonic acid in patients with PAH and in animal models. More and more biological data suggest that arachidonic acid metabolites of lipoxygenases (LOs) play pivotal roles in the pathological development of PAH. LOs are a family of non-heme iron-containing enzymes which dioxygenate polyunsaturated fatty acids to hydroperoxyl metabolites. As shown in Table 2, LOs mainly include four isoforms, 5-lipoxygenase (5-LO) [6], 8-lipoxygenase (8-LO) [7, 8], 12-lipoxygenase (12-LO) [9], and 15-lipoxygenase (15-LO) [10], which correspond to the carbon position of arachidonic acid oxygenation, whereas 8-LO was not found to be expressed in human tissues. LOs are involved in biosynthesis of vasoactive mediators, growth factors, adhesion molecules, and cytokines [10–12], and hence are important targets for the atherogenesis, vasoconstriction, and vascular remodeling. Moreover, LOs-derived compounds impact the metabolic characteristics of vascular cells, particularly those of endothelial cells (EC) and smooth muscles cells (SMC) [13, 14]. In our laboratory, we have reported that 15-LO/15-HETE played an important role in hypoxic pulmonary hypertension (HPH). Therefore, this review is intended to provide a comprehensive overview of the effect of 15-LO/15-HETE on HPH, and also to propose underlying the important aspects of 5-LO and 12-LO in PAH.
Basic properties of LOs
Two distinct types of 15-LO have been identified in humans: reticulocyte type of 15-LO-1 [15] and epidermis type of 15-LO-2 [16]. 15-LO-1 was initially discovered in a rabbit reticulocytes mass at a 75-kD, single-polypeptide chain [17]; the enzyme has a two-domain structure [18] with one non-heme iron per molecule. 15-LO-2 was subsequently identified, and its expression has been reported in human prostate, skin, and cornea [17]. Both enzymes convert arachidonic acid to 15-hydroperoxyeicosatetraenoic acid (15(S)-HPETE). 15(S)-HPETE is unstable and can be reduced by peroxidases to the corresponding 15-hydroxyeicosatetraenoic acid (15-HETE), but they share only 40% amino acid homology, and there are two major differences between the isozymes. The first difference is that 15-LO-1 converts AA to 15(S)-HPETE (90%) and lesser amounts of 12(S)-HPETE (10%), whereas 15-LO-2 produces exclusively 15(S)-HPETE [17]. The second difference is that, although both arachidonic acid and linoleic acid are preferred substrates for 15-LO-1, only arachidonic acid is a substrate for 15-LO-2 [19, 20].
The human 5-LO consists of 673 amino acids and a non-heme iron, and the sequence is highly homologous with those of other mammalian LOs and soybean 15-LO [21]. 5-LO catalyzes conversion of arachidonic acid to leukotriene (LT) A4, which can be subsequently converted into the potent chemoattractant LTB4, or into cysteinyl leukotrienes (Cys-LTs: LTC4, LTD4, and LTE4) [22]. 8-LO is consisted of 677 amino acids, which displays 78% sequence identity to human 15-LO-2 considered to be its human orthologue. 8-LO expression or activity has only been detected in tissues of mice and rats [7, 8, 23], and is predominantly expressed in epithelia of mice, hair follicle, forestomach, and footsole. 8-HETE is a major arachidonic acid metabolite for 8-LO [24]. There are three isoforms of 12-LO named after the cells where they were first identified; platelet, leukocyte, and epidermis. The leukocyte-type enzyme is widely distributed among cells, but the tissue distribution varies substantially from species to species. The platelet and epidermal enzymes are present in only a relatively limited number of cell types, but murine epidermis-type 12-LOX is not a functional human gene [9]. The 12-LO pathway metabolizes AA to a variety of products with numerous biological activities, and the major products of this pathway are 12(S)-HETE, hydroxyepoxy-containing hepoxilins, and trihydroxy-containing trioxilins [25].
Biological role of 15-LO/15-HETE
15-LOs are lipid peroxidizing enzymes that catalyze the stereoselective introduction of molecular dioxygen at carbon 15 (C-15) of arachidonic acid [26, 27], and their expression and arachidonic acid metabolites are implicated in several important inflammatory conditions, cell differentiation, carcinogenesis, atherogenesis, and other potential functions. The physiologic roles of the enzymes and arachidonic acid metabolites are dependent on the context in which (tissue- and species-specificity) they are expressed.
Tissue levels of 15-LOs and 15-HETE are often elevated during inflammation condition. Further evidence for a role of 15-HETE is that it is increased in asthma and chronic bronchitis patients and animals [28–31]. Several lines of ex vivo studies have documented that 15-LOs product 15-HETE might have anti-inflammatory properties. 15-HETE inhibits the activity of 5-LO and LT production by neutrophils [32–34]. The expression of 15-LO induced by interleukin (IL)-4 in monocytes significantly decreased the LTB4 expression [35]. 15-HETE can inhibit neutrophil migration across cytokine-activated (interleukin-1 beta or tumor necrosis factor-alpha) endothelium [36]. Transfection of rat kidney with human 15-LO can suppress inflammation [37]. According to these studies, the in vivo activity of 15-LO/15-HETE may be regarded as a protective response to limit or reverse inflammatory symptoms and to maintain basic cell function [38].
Also, it has been found that 15-LO is implicated in the maturation of rabbit reticulocytes by inhibiting mitochondria degradation [39–41]. 15-HETE improves the proliferation of Friend erythroleukemia cells, rat aortic smooth muscle cells, and calf PASMCs [42–44], and even plays an important role in differentiation and function of macrophage [45]. Moreover, 15-LO plays a role in pro- and anti-carcinogenesis [46–48], 15-HETE also triggers cell death through the release of cytochrome C, activation of caspase-3, and PARP-1 (poly (ADP) ribose polymerase-1) cleavage in the K-562 cell line [49]. Chronic inflammation plays an important role in atherogenesis. Furthermore, there is evidence for a pro-atherosclerotic effect and anti-atherosclerotic effect of 15-LO [50–52].
Although 15-LO/15-HETE participates in various physiological and pathological processes, yet the exact role and mechanism in HPH have not been explained, so here we will describe in detail the effect of 15-LO/15-HETE on hypoxic pulmonary vasoconstriction and vascular remodeling.
15-LO/15-HETE and HPH
Distribution and expression of 15-LO/15-HETE in HPH
It is reported that only 15-LO-1 is expressed in normoxic lung tissues, and that 15-LO-1 and 15-LO-2 are upregulated by hypoxia. Moreover, in hypoxic lungs, 15-LO is concentrated in the microsomes, whereas in normoxic lungs, 15-LO is localized in the cytosol, suggesting that activation of 15-LO is associated with translocation of the enzyme from the cytosol to membrane under hypoxic conditions [53]. Moreover, the 15-LO-1 mRNA and protein were localized in pulmonary artery endothelial cells (PAECs), while the 15-LO-2 mRNA and protein were localized in both PAECs and pulmonary smooth muscle cells (PASMCs) [54]. Furthermore, both 15-LO-1 and 15-LO-2 were up-regulated and localized in PASMCs and PAECs of pulmonary vessels from patients displaying severe PH [44]. Using a combination of high-pressure liquid chromatography (HPLC) and gas chromatography/mass spectrometry (GC/MS), the synthesis of 15-HETE was increased in microsomes from hypoxic lungs and this effect is dependent on the lipoxygenase pathway [53].
15-LO/15-HETE and hypoxic pulmonary vasoconstriction
15-HETE increases intracellular Ca2+ and contracts pulmonary arteries
15-HETE increases the tension of PA from hypoxic rats in a concentration-dependent manner [53]. After inhibiting the endogenous production of 15-HETE, the reaction of hypoxic pulmonary artery rings to phenylephrine (a vasoconstrictor and used to detect the viability of vessels) was markedly decreased, suggesting that endogenous 15-HETE is involved in pulmonary vasoconstriction. PA vasoconstriction induced by 15-HETE is triggered by an increase in intracellular Ca2+ concentration([Ca2+]i) in PASMCs, which is in turn caused by Ca2+ release from intracellular Ca2+ stores, or an influx of Ca2+ through ion channels such as L-type and store-operated calcium channels (SOCCs) [55].
More and more studies are showing that there is an important role for SOCCs in the chronic hypoxia-induced increase in resting [Ca2+]i which is responsible for HPV but eliminates a role for voltage-dependent calcium channels (VDCCs) during this procedure [56]. Our data also show that transient receptor potential channel 1 (TRPC1), one candidate of SOCCs, was up-regulated by 15-HETE, leading to elevation of capacitative calcium entry (CCE) via SOCCs in PASMCs [57]. It has been suggested that the mechanism of 15-HETE mobilizes [Ca2+]i signaling through Ca2+ release from intracellular Ca2+ stores via IP3 receptor- and ryanodine receptor-operated Ca2+ channels. After depletion of sarcoplasmic reticulum Ca2+ stores, the resulting Ca2+ influx, known as CCE, is mediated by SOCCs, and consists of up-regulated TRPC1. The other mechanism is directly activating Ca2+ entry from extracellular solution via L-type Ca2+ channels (VDCCs). PASMCs are required to maintain active vascular tones, then the increased [Ca2+]i can form complexes with calmodulin, which activates myosin light chain (MLC) kinase (MLCK), causing phosphorylation of MLC. Phosphorylated MLC (P-MLC) then facilitates stimulation of myosin ATPase activity by actin leading to cross-bridge cycling and contraction [58].
15-HETE induces pulmonary vasoconstriction by inhibiting Kv channels
The resting membrane potential (E m) is primarily determined by K+ permeability and K+ concentration gradient across the plasma membrane, and therefore the activity of K+ channels in the plasma membrane is a critical determinant of E m. Inhibition of K+ channels causes membrane depolarization, opens VDCCs, promotes Ca2+ influx, increases [Ca2+]i, and triggers PASMCs contraction. Studies have suggested that K+ channels in PASMCs are inhibited by subacute hypoxia, leading to depolarization, an increase in [Ca2+]i, and constriction of pulmonary arteries [59, 60]. These K+ channels are voltage-gated and sensitive to 4-aminopyridine (4-AP) [61–63]. However, how the K+ channels are inhibited after subacute hypoxia remains elusive. Both direct and indirect effects have been proposed for the channel inhibition.
Our studies have shown that inhibiting KATP and BKCa channels could not affect 15-HETE induced vasoconstriction, but once inhibited Kv channels can completely block the effect of 15-HETE on pulmonary arteries [64], 15-HETE can inhibit the Kv currents of PASMCs [65]. Recent studies also identified that subacute hypoxia down-regulates Kv1.5, Kv2.1, and Kv3.4 channel expression and suppresses IK current through endogenous 15-HETE. 15-HETE was found to be more potent than 5-HETE and 12-HETE in mediating hypoxia-induced down-regulation of Kv3.4 channel expression. These results fill the lacunae which define how subacute hypoxia inhibits Kv channels leading to membrane depolarization and an increase in [Ca2+]i level in PASMCs, and demonstrate the link between 15-LO, 15-HETE formation, and pulmonary vasoconstriction after subacute hypoxia [66–68].
Effects of pulmonary artery endothelial cells on 15-HETE induced pulmonary vasoconstriction
PAECs can release several kinds of vascular activity factors such as PGE2, NO, and some others, which have important contributions to vasoconstriction. Hypoxia reduces the activity of eNOS to decrease the production of NO, then contracts PAs [69]. In in vitro tension studies, denuded endothelial and inhibiting the NO production of vascular rings increased the effect of 15-HETE on PAs contraction. Blockage of endogenous 15-HETE can induce the production of NO in PAECs. Moreover, 15-HETE phosphorylated eNOS at Thr495, causing reduced activity of eNOS [70]. In addition, the immunoprecipitation (IP) supported there were 15-LO, Hsp90, and Akt in an eNOS complex in PAECs, and therefore these data must be interpreted with 15-HETE overcoming the protein network of eNOS process through phosphorylating or de-phosphorylating to inactivate some sites of eNOS resulting in reduced NO, thereby contracting PAs.
15-HETE regulates pulmonary vessels rhythm by protein kinase pathways
Protein kinase pathways play an important role in HPV, such as PKC, Rho kinase, Rho-associated serine/threonine kinase (ROCK), and extracellular signal-regulated kinase 1/2 (ERK1/2). Tension measurements of responsiveness of rat PA rings have demonstrated that incubation with specific protein kinase inhibitors significantly attenuated the constriction of PA rings to 15-HETE under hypoxic conditions. 15-HETE can activate the translocation of PKC isoforms, PKC-delta and PKC-varepsilon, from the cytoplasm to the membranes of PASMCs, then down-regulate expression of Kv1.5, Kv2.1, and Kv3.4 channels to protect the effect of 15-HETE on PAs [71]. Also, 15-HETE mediated the up-regulation of ROCK expression and promoted the translocation of ROCK2 from the nucleus to the cytoplasm through G-protein and tyrosine kinase pathways under hypoxic conditions, then leading to PA vasoconstriction [72]. Furthermore, the ERK1/2 pathway was involved in 15-HETE-induced PA vasoconstriction, the ERK1/2 phosphorylation was also upregulated by 15-HETE in a dose-dependent manner, and this phosphorylation was detected in cytosol as well as in nucleus [73]. These data shown above suggested that 15-HETE mediated HPV by activating different signal transduction pathways.
15-LO/15-HETE and HPVR
Hypoxia induces pathological changes of the pulmonary vasculature mainly including: extracellular matrix components such as increase in collagen fibers and elastic fibers, smooth muscle cell hypertrophy and hyperplasia, endothelial cell swelling, hypertrophy, thereby resulting in pulmonary tube wall thickening, and luminal stenosis, reducing blood vessel flexibility in the vessel wall cavity volume level. Under conditions of prolonged hypoxia, although recovering the PO2 to normoxic conditions, the pulmonary artery pressure is still higher than normal values. It can be concluded that hypoxic pulmonary vascular remodeling (HPVR) is the main mechanism of HPH [74].
It has been reported that PA remodeling induced by hypoxia in vivo is mediated by the 15-LO/15-HETE pathway at least in part. Moreover, both 15-LO-1 and 15-LO-2 were overexpressed in the pulmonary vessels of human PH lungs and localized in PASMCs and PAECs from pulmonary vessels of patients displaying severe PH [44]. Intragastric administration of rats with 15-LO inhibitor (nordihydroguaiaretic acid, NDGA) under hypoxic conditions decreased the formation of the endogenous 15-HETE level, which also reversed all the pathological changes of PAs induced by hypoxia, including the deposition of collagen and medial thickening [44, 54].
15-HETE and PASMCs apoptosis
Especially considering the fact that maintaining the proper balance between cell apoptosis and proliferation is closely related to tissue homeostasis, when this balance is disrupted, diseases such as PAH can result [75]. The enhanced VSMC proliferation and suppressed normal VSMC apoptosis are likely the major reasons leading to medial hypertrophy, arterial remodeling, and vascular lumen narrowing [76]. Indeed, the inadequate apoptosis has been implicated in the development and maintenance of severe pulmonary hypertension. We found subtle thickening of proximal media/adventitia of the PA in rats exposed to hypoxia, which was associated with an up-regulation of the anti-apoptotic Bcl-2 expression and down-regulation of activated caspase-3 and Bax expression in PA homogenates [54].
The effects of hypoxia on PASMCs apoptosis are well known; however, whether 15-HETE acts on the apoptotic responses in PASMCs remains unclear. Studies have shown that 15-HETE induced anti-apoptotic Bcl-2 expression, and down-regulated apoptotic caspase-3, Bax, FasL, Bad and caspase-9 expression to prevent PASMCs from apoptosis via ROCK, HSP90, PI3K/Akt, ERK1/2, and iNOS pathways. Some methods such as cell viability measurement, nuclear morphology determination, TUNEL assay, and mitochondrial potential analysis have also demonstrated that 15-HETE suppressed PASMC apoptosis and improved cell survival, contributing to HPVR including morphological alterations, mitochondrial depolarization, and the expression of anti-apoptotic proteins [77–80].
Activity of Kv channels also plays a major role in regulating the PASMCs population in the pulmonary vasculature, as they are involved in cell apoptosis, survival, and proliferation [3, 75, 76]. PASMCs from PAH patients demonstrate many cellular abnormalities linked to Kv channels, including decreased Kv current, down-regulated expression of various Kv channels, and inhibited apoptosis [81, 82]. It is well known that hypoxia can inhibit Kv channels, inhibiting cell apoptosis, but it remains unclear whether K+ channels participate in the 15-HETE anti-apoptotic process under hypoxic conditions. Data have also shown that 15-HETE enhanced cell survival, suppressed the expression and activity of caspase-3, up-regulated Bcl-2 and attenuated mitochondrial depolarization, prevented chromatin condensation, and partly reversed K+ channel opener-induced apoptosis in PASMCs under serum-deprived conditions, indicating that 15-HETE inhibited the apoptosis in PASMCs through, at least in part, inactivating K+ channels [83].
15-HETE and PASMCs proliferation
Studies on the exact mechanism of 15-HETE on HPVR are currently in progress. Until now, it has been found that proliferating cell nuclear antigen (PCNA) and Cyclin A were up-regulated by endogenous 15-HETE induced by hypoxia and exogenous 15-HETE in PASMCs, and that the effect was also abolished in the presence of cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC), the inhibitor of 15-LO. Moreover, hypoxia and exogenous 15-HETE significantly enhanced 5-bromodeoxyuridine (BrdU) incorporation and microtubule formation by α-tubulin, and made more PASMCs accumulation at the G2/M+S phase, respectively. CDC also suppressed the cell proliferation, α-tubulin polymerizatio,n and made more PASMCs arrest at the G0/G1 phase of the cell cycle. Our results also clarified that the ROCK pathway was responsible for 15-HETE-induced PASMCs proliferation, which contributed to media hypertrophy. These findings appeared in favor of the potential relevance of ROCK pathway and 15-HETE inhibition in the treatment of human PH [44].
15-HETE and angiogenesis
Most studies conclude that the structural changes that are thought to underlie the increased vascular resistance can be broadly classified into two processes: firstly, remodeling of the walls of the pulmonary resistance vessels, and, secondly, a reduction in the total number of blood vessels in the lung [74, 84]. The second major structural alteration caused by chronic hypoxia is loss of small blood vessels, sometimes termed rarefaction or pruning, which is said to increase vascular resistance by reducing the extent of parallel vascular pathways [85]. But some other researchers question this widely accepted paradigm, and hypoxia-induced angiogenesis in the mature pulmonary circulation, a structural adaptation that have important beneficial results for gas exchange, has recently been reported [86]. The specific vascular endothelial mitogen, the vascular endothelial growth factor (VEGF) family, plays an important role in the development of PH, and chronic hypoxia can lead to increased VEGF, PIGF, and their receptor expression in the lung [87, 88]. The ROCK pathway also mediates hypoxia-induced capillary angiogenesis [89]. However, the mechanism by which hypoxia induced the angiogenesis in PH needs to be explored further.
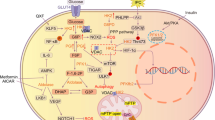
Our study also showed that hypoxia could induce angiogenesis, and that this effect can be inhibited by intragastric administration of NDGA in vivo in rats, indicating that endogenous 15-HETE was involved in hypoxia-induced angiogenesis. It has also been found that exogenous 15-HETE can induce bovine PAECs migration, tube formation in Matrigel, or vessel density in chick chorioallantoic membrane under normoxic conditions [44]. Meanwhile, hypoxia induced significant PAECs migration and tube formation was blocked by CDC. However, the inhibitory effect was partly diminished in the presence of exogenous 15-HETE [44], and also ROCK pathway mediated the effect of 15-HETE-induced PAECs migration, tube formation in vitro, and chick chorioallantoic membrane angiogenesis in vivo. These results indicated that 15-HETE was a potent mediator involved in hypoxia-induced pulmonary vascular angiogenesis. Also, we are studying whether 15-HETE could regulate other pathways to promote angiogenesis, such as VEGF and PIGF. From all the results, we can conclude 15-LO/15-HETE plays an important role in HPH as in Fig. 1.

Role of 15-LO/15-HETE in hypoxia-induced pulmonary hypertension. Hypoxia-induced 15-LO/15-HETE function and expression stimulates pulmonary arteries constriction, promotes pulmonary artery endothelial cell (PAEC) and smooth muscle cell (PASMC) proliferation and inhibits PASMC apoptosis. The increased vascular pressure, proliferation and inhibited apoptosis in PASMC may play an important role in initiation and/or progression of pulmonary vascular remodeling
5-LO/12-LO and PH
In addition to the 15-LO pathway, many other data suggest that arachidonic acid metabolism via 5-LO and 12-LO also play pivotal roles in PH, since in one of these studies, 5-LO expression was unregulated in PAECs from patients with PAH [90], in rats subjected to chronic hypoxia [91], and in mice challenged with antigen [92]. Cys-LTs was also found to be in the lung lavage fluid of neonates with persistent pulmonary hypertension [93], patients with chronic obstructive pulmonary disease [94], and animals subjected to acute hypoxia, which may increase vascular permeability and accelerate PAECs proliferation after injuring endothelial cells [95]. Targeted disruption of the 5-LO gene and protein [96] or treatment of animals with diethylcarbamazine, MK-886, an inhibitor of five-lipoxygenase-activating protein (FLAP) [91], or LT antagonists [97], reduced hypoxia and monocrotaline (MCT)-induced pulmonary hypertension, while overexpression of 5-LO accelerated the development of MCT-induced PH in rats, respectively [98]. More recently, it has been shown that adenoviral overexpression of 5-LO in BMPR2+/− exposed to MCT resulted in increased inflammation and exacerbation of PH compared with wild-type mice [99, 100].
12-LO and 12-HETE have also contributed to PASMCs proliferation and have participated in PVR. 12-LO gene and protein expression was elevated in lung homogenates of rats exposed to chronic hypoxia. 12(S)-HETE at concentrations as low as 10−5 μM stimulated proliferation of PASMCs, most likely via the ERK1/ERK2, MAPK pathway and apparently without the involvement of p38 MAPK, while inhibition 12-LO with baicalein blocked PASMCs proliferation, suggesting a potential role for 12-LO and its metabolite 12(S)-HETE in hypoxia-induced pulmonary hypertension [101].
Summary
It is clear that PAH has a complex, multifactorial pathobiology and it is unlikely that one factor or gene mutation will explain in all forms and aspects of PAH. In addition, we have shown that hypoxic exposure promoted the expression and activity of 15-LO, catalyzed arachidonic acid to the production of 15-HETE which played a significant role in HPV and vascular remodeling. So the signal transduction pathway and mechanism of 15-HETE may be an important mechanism underlying the treatment of PAH and provide a novel therapeutic in sight for the future. Although 15-LO/15-HETE exerted an integral role in the development of pulmonary hypertension, the mechanism by which 15-LO was regulated under hypoxic conditions is still not clear. So further studies will need to evaluate the precise mechanism of hypoxia-+regulated 15-LO expression and activity.
References
Rabinovitch M (2008) Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 118(7):2372–2379
Stenmark KR, Fagan KA, Frid MG (2006) Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99(7):675–691
Mauban JR, Remillard CV, Yuan JX (2005) Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol 98(1):415–420
Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX (2004) Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res 68(2):75–103
Gurbanov E, Shiliang X (2006) The key role of apoptosis in the pathogenesis and treatment of pulmonary hypertension. Eur J Cardiothorac Surg 30(3):499–507
Radmark O (2002) Arachidonate 5-lipoxygenase. Prostaglandins Other Lipid Mediat 68–69:211–234
Jisaka M, Kim RB, Boeglin WE, Nanney LB, Brash AR (1997) Molecular cloning and functional expression of a phorbol ester-inducible 8S-lipoxygenase from mouse skin. J Biol Chem 272(39):24410–24416
Krieg P, Kinzig A, Heidt M, Marks F, Furstenberger G (1998) cDNA cloning of a 8-lipoxygenase and a novel epidermis-type lipoxygenase from phorbol ester-treated mouse skin. Biochim Biophys Acta 1391(1):7–12
Yoshimoto T, Takahashi Y (2002) Arachidonate 12-lipoxygenases. Prostaglandins Other Lipid Mediat 68–69:245–262
Kuhn H, Walther M, Kuban RJ (2002) Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat 68–69:263–290
Cuzzocrea S, Rossi A, Mazzon E, Di Paola R, Genovese T, Muia C, Caputi AP, Sautebin L (2005) 5-Lipoxygenase modulates colitis through the regulation of adhesion molecule expression and neutrophil migration. Lab Invest 85(6):808–822
Wen Y, Gu J, Chakrabarti SK, Aylor K, Marshall J, Takahashi Y, Yoshimoto T, Nadler JL (2007) The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-alpha in macrophages. Endocrinology 148(3):1313–1322
Huber J, Furnkranz A, Bochkov VN, Patricia MK, Lee H, Hedrick CC, Berliner JA, Binder BR, Leitinger N (2006) Specific monocyte adhesion to endothelial cells induced by oxidized phospholipids involves activation of cPLA2 and lipoxygenase. J Lipid Res 47(5):1054–1062
Dronadula N, Rizvi F, Blaskova E, Li Q, Rao GN (2006) Involvement of cAMP-response element binding protein-1 in arachidonic acid-induced vascular smooth muscle cell motility. J Lipid Res 47(4):767–777
Sigal E, Grunberger D, Craik CS, Caughey GH, Nadel JA (1988) Arachidonate 15-lipoxygenase (omega-6 lipoxygenase) from human leukocytes. Purification and structural homology to other mammalian lipoxygenases. J Biol Chem 263(11):5328–5332
Brash AR, Boeglin WE, Chang MS (1997) Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci USA 94(12):6148–6152
Schewe T, Halangk W, Hiebsch C, Rapoport SM (1975) A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett 60(1):149–152
Gillmor SA, Villasenor A, Fletterick R, Sigal E, Browner MF (1997) The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat Struct Biol 4(12):1003–1009
Ivanov I, Schwarz K, Holzhutter HG, Myagkova G, Kuhn H (1998) Omega-oxidation impairs oxidizability of polyenoic fatty acids by 15-lipoxygenases: consequences for substrate orientation at the active site. Biochem J 336(Pt 2):345–352
Salzmann-Reinhardt U, Kuhn H, Wiesner R, Rapoport S (1985) Metabolism of polyunsaturated fatty acids by rabbit reticulocytes. Eur J Biochem 153(1):189–194
Goetzl EJ, An S, Smith WL (1995) Specificity of expression and effects of eicosanoid mediators in normal physiology and human diseases. FASEB J 9(11):1051–1058
Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN (1987) Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237(4819):1171–1176
Yamada M, Proia AD (2000) 8(S)-hydroxyeicosatetraenoic acid is the lipoxygenase metabolite of arachidonic acid that regulates epithelial cell migration in the rat cornea. Cornea 19(3 Suppl):S13–S20
Gschwendt M, Furstenberger G, Kittstein W, Besemfelder E, Hull WE, Hagedorn H, Opferkuch HJ, Marks F (1986) Generation of the arachidonic acid metabolite 8-HETE by extracts of mouse skin treated with phorbol ester in vivo; identification by 1H-n.m.r. and GC-MS spectroscopy. Carcinogenesis 7(3):449–455
Pace-Asciak CR, Granstrom E, Samuelsson B (1983) Arachidonic acid epoxides. Isolation and structure of two hydroxy epoxide intermediates in the formation of 8,11,12- and 10,11,12-trihydroxyeicosatrienoic acids. J Biol Chem 258(11):6835–6840
Brash AR (1999) Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem 274(34):23679–23682
Kuhn H, Thiele BJ (1999) The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett 449(1):7–11
Gray PR, Derksen FJ, Broadstone RV, Robinson NE, Johnson HG, Olson NC (1992) Increased pulmonary production of immunoreactive 15-hydroxyeicosatetraenoic acid in an animal model of asthma. Am Rev Respir Dis 145(5):1092–1097
Profita M, Sala A, Riccobono L, Paterno A, Mirabella A, Bonanno A, Guerrera D, Pace E, Bonsignore G, Bousquet J, Vignola AM (2000) 15-Lipoxygenase expression and 15(S)-hydroxyeicoisatetraenoic acid release and reincorporation in induced sputum of asthmatic subjects. J Allergy Clin Immunol 105(4):711–716
Profita M, Sala A, Riccobono L, Pace E, Paterno A, Zarini S, Siena L, Mirabella A, Bonsignore G, Vignola AM (2000) 15(S)-HETE modulates LTB(4) production and neutrophil chemotaxis in chronic bronchitis. Am J Physiol Cell Physiol 279(4):C1249–C1258
Donowitz M (1985) Arachidonic acid metabolites and their role in inflammatory bowel disease. An update requiring addition of a pathway. Gastroenterology 88(2):580–587
Vanderhoek JY, Bryant RW, Bailey JM (1980) Inhibition of leukotriene biosynthesis by the leukocyte product 15-hydroxy-5,8,11,13-eicosatetraenoic acid. J Biol Chem 255(21):10064–10066
Petrich K, Ludwig P, Kuhn H, Schewe T (1996) The suppression of 5-lipoxygenation of arachidonic acid in human polymorphonuclear leucocytes by the 15-lipoxygenase product (15S)-hydroxy-(5Z,8Z,11Z,13E)-eicosatetraenoic acid: structure-activity relationship and mechanism of action. Biochem J 314(Pt 3):911–916
Vachier I, Chanez P, Bonnans C, Godard P, Bousquet J, Chavis C (2002) Endogenous anti-inflammatory mediators from arachidonate in human neutrophils. Biochem Biophys Res Commun 290(1):219–224
Profita M, Sala A, Siena L, Henson PM, Murphy RC, Paterno A, Bonanno A, Riccobono L, Mirabella A, Bonsignore G, Vignola AM (2002) Leukotriene B4 production in human mononuclear phagocytes is modulated by interleukin-4-induced 15-lipoxygenase. J Pharmacol Exp Ther 300(3):868–875
Takata S, Papayianni A, Matsubara M, Jimenez W, Pronovost PH, Brady HR (1994) 15-Hydroxyeicosatetraenoic acid inhibits neutrophil migration across cytokine-activated endothelium. Am J Pathol 145(3):541–549
Munger KA, Montero A, Fukunaga M, Uda S, Yura T, Imai E, Kaneda Y, Valdivielso JM, Badr KF (1999) Transfection of rat kidney with human 15-lipoxygenase suppresses inflammation and preserves function in experimental glomerulonephritis. Proc Natl Acad Sci USA 96(23):13375–13380
Holtzman MJ, Zhang V, Hussain H, Roswit WT, Wilson JD (1994) Prostaglandin H synthase and lipoxygenase gene families in the epithelial cell barrier. Ann N Y Acad Sci 744:58–77
Schewe T, Rapoport SM, Kuhn H (1986) Enzymology and physiology of reticulocyte lipoxygenase: comparison with other lipoxygenases. Adv Enzymol Relat Areas Mol Biol 58:191–272
Rapoport SM, Schewe T, Thiele B, Dubiel W (1982) The role of lipoxygenase and ATP-dependent proteolysis in the maturation of the reticulocyte. Prog Clin Biol Res 102(pt A):47–58
Grullich C, Duvoisin RM, Wiedmann M, van Leyen K (2001) Inhibition of 15-lipoxygenase leads to delayed organelle degradation in the reticulocyte. FEBS Lett 489(1):51–54
Postoak D, Nystuen L, King L, Ueno M, Beckman BS (1990) 15-Lipoxygenase products of arachidonate play a role in proliferation of transformed erythroid cells. Am J Physiol 259(6 Pt 1):C849–C853
Palmberg L, Lindgren JA, Thyberg J, Claesson HE (1991) On the mechanism of induction of DNA synthesis in cultured arterial smooth muscle cells by leukotrienes. Possible role of prostaglandin endoperoxide synthase products and platelet-derived growth factor. J Cell Sci 98(Pt 2):141–149
Ma C, Li Y, Ma J, Liu Y, Li Q, Niu S, Shen Z, Zhang L, Pan Z, Zhu D (2011) Key role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension 58:679–688
Miller YI, Chang MK, Funk CD, Feramisco JR, Witztum JL (2001) 12/15-lipoxygenase translocation enhances site-specific actin polymerization in macrophages phagocytosing apoptotic cells. J Biol Chem 276(22):19431–19439
Ikawa H, Kamitani H, Calvo BF, Foley JF, Eling TE (1999) Expression of 15-lipoxygenase-1 in human colorectal cancer. Cancer Res 59(2):360–366
Moody TW, Leyton J, Martinez A, Hong S, Malkinson A, Mulshine JL (1998) Lipoxygenase inhibitors prevent lung carcinogenesis and inhibit non-small cell lung cancer growth. Exp Lung Res 24(4):617–628
Shureiqi I, Chen D, Lotan R, Yang P, Newman RA, Fischer SM, Lippman SM (2000) 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res 60(24):6846–6850
Mahipal SV, Subhashini J, Reddy MC, Reddy MM, Anilkumar K, Roy KR, Reddy GV, Reddanna P (2007) Effect of 15-lipoxygenase metabolites, 15-(S)-HPETE and 15-(S)-HETE on chronic myelogenous leukemia cell line K-562: reactive oxygen species (ROS) mediate caspase-dependent apoptosis. Biochem Pharmacol 74(2):202–214
Wittwer J, Hersberger M (2007) The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot Essent Fatty Acids 77(2):67–77
Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E (2000) Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol 20(9):2100–2105
Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV, Chan L (1996) Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest 98(10):2201–2208
Zhu D, Medhora M, Campbell WB, Spitzbarth N, Baker JE, Jacobs ER (2003) Chronic hypoxia activates lung 15-lipoxygenase, which catalyzes production of 15-HETE and enhances constriction in neonatal rabbit pulmonary arteries. Circ Res 92(9):992–1000
Ma J, Liang S, Wang Z, Zhang L, Jiang J, Zheng J, Yu L, Zheng X, Wang R, Zhu D (2010) ROCK pathway participates in the processes that 15-hydroxyeicosatetraenoic acid (15-HETE) mediated the pulmonary vascular remodeling induced by hypoxia in rat. J Cell Physiol 222(1):82–94
Zheng X, Li Q, Tang X, Liang S, Chen L, Zhang S, Wang Z, Guo L, Zhang R, Zhu D (2008) Source of the elevation Ca2+ evoked by 15-HETE in pulmonary arterial myocytes. Eur J Pharmacol 601(1–3):16–22
Ward JP, Robertson TP, Aaronson PI (2005) Capacitative calcium entry: a central role in hypoxic pulmonary vasoconstriction? Am J Physiol Lung Cell Mol Physiol 289(1):L2–L4
Li S, Ran Y, Zheng X, Pang X, Wang Z, Zhang R, Zhu D (2010) 15-HETE mediates sub-acute hypoxia-induced TRPC1 expression and enhanced capacitative calcium entry in rat distal pulmonary arterial myocytes. Prostaglandins Other Lipid Mediat 93(1–2):60–74
Verin AD, Cooke C, Herenyiova M, Patterson CE, Garcia JG (1998) Role of Ca2 +/calmodulin-dependent phosphatase 2B in thrombin-induced endothelial cell contractile responses. Am J Physiol 275(4 Pt 1):L788–L799
Post JM, Hume JR, Archer SL, Weir EK (1992) Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 262(4 Pt 1):C882–C890
Weir EK, Archer SL (1995) The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J 9(2):183–189
Yuan XJ (1995) Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 77(2):370–378
Post JM, Gelband CH, Hume JR (1995) [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 77(1):131–139
Archer SL, Huang JM, Reeve HL, Hampl V, Tolarova S, Michelakis E, Weir EK (1996) Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res 78(3):431–442
Han WN, Li XH, Jiang ZY, Ji HY, Huang LJ, Wang ZM, Zhu DL (2004) Effect of 15-HETE on potassium channels of rabbit pulmonary arterial smooth muscles during hypoxia. Sheng Li Xue Bao 56(6):717–722
Guo L, Tang X, Tian H, Liu Y, Wang Z, Wu H, Wang J, Guo S, Zhu D (2008) Subacute hypoxia suppresses Kv3.4 channel expression and whole-cell K+ currents through endogenous 15-hydroxyeicosatetraenoic acid in pulmonary arterial smooth muscle cells. Eur J Pharmacol 587(1–3):187–195
Chu X, Tang X, Guo L, Bao H, Zhang S, Zhang J, Zhu D (2009) Hypoxia suppresses KV1.5 channel expression through endogenous 15-HETE in rat pulmonary artery. Prostaglandins Other Lipid Mediat 88(1–2):42–50
Guo L, Qiu Z, Zhang L, Chen S, Zhu D (2010) Hypoxia suppresses Kv 2.1 channel expression through endogenous 15-hydroxyeicosatetraenoic acid in rat pulmonary artery. J Physiol Sci 60(5):373–381
Fike CD, Kaplowitz MR, Thomas CJ, Nelin LD (1998) Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am J Physiol 274(4 Pt 1):L517–L526
Guo L, Tang X, Chu X, Sun L, Zhang L, Qiu Z, Chen S, Li Y, Zheng X, Zhu D (2009) Role of protein kinase C in 15-HETE-induced hypoxic pulmonary vasoconstriction. Prostaglandins Leukot Essent Fatty Acids 80(2–3):115–123
Ye H, Bi HR, Lu CL, Tang XB, Zhu DL (2005) 15-hydroxyeicosatetraenoic acid depressed endothelial nitric oxide synthase activity in pulmonary artery. Sheng Li Xue Bao 57(5):612–618
Li X, Ma C, Zhu D, Meng L, Guo L, Wang Y, Zhang L, Li Z, Li E (2010) Increased expression and altered subcellular distribution of PKC-delta and PKC-varepsilon in pulmonary arteries exposed to hypoxia and 15-HETE. Prostaglandins Other Lipid Mediat 93(3–4):84–92
Wang Y, Liang D, Wang S, Qiu Z, Chu X, Chen S, Li L, Nie X, Zhang R, Wang Z, Zhu D (2010) Role of the G-protein and tyrosine kinase–Rho/ROK pathways in 15-hydroxyeicosatetraenoic acid induced pulmonary vasoconstriction in hypoxic rats. J Biochem 147(5):751–764
Lu C, Liu Y, Tang X, Ye H, Zhu D (2006) Role of 15-hydroxyeicosatetraenoic acid in phosphorylation of ERK1/2 and caldesmon in pulmonary arterial smooth muscle cells. Can J Physiol Pharmacol 84(10):1061–1069
Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L (1979) Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol 236(6):H818–H827
Durmowicz AG, Stenmark KR (1999) Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr Rev 20(11):e91–e102
Yu WC, Guo CH (2007) Apoptosis versus proliferation activities of pulmonary artery smooth muscle cells in pulmonary arterial hypertension associated with chronic obstructive pulmonary disease. Zhonghua Jie He He Hu Xi Za Zhi 30(9):657–661
Wang S, Wang Y, Jiang J, Wang R, Li L, Qiu Z, Wu H, Zhu D (2010) 15-HETE protects rat pulmonary arterial smooth muscle cells from apoptosis via the PI3K/Akt pathway. Prostaglandins Other Lipid Mediat 91(1–2):51–60
Zhang L, Ma J, Li Y, Guo L, Ran Y, Liu S, Jiang C, Zhu D (2010) 15-Hydroxyeicosatetraenoic acid (15-HETE) protects pulmonary artery smooth muscle cells against apoptosis via HSP90. Life Sci 87(7–8):223–231
Jiang J, Wang S, Wang Z, Ma J, Liu S, Li W, Zhu D (2011) The role of ERK1/2 in 15-HETE-inhibited apoptosis in pulmonary arterial smooth muscle cells. J Recept Signal Transduct Res 31(1):45–52
Nie X, Song S, Zhang L, Qiu Z, Shi S, Liu Y, Yao L, Zhu D (2012) 15-Hydroxyeicosatetraenoic acid (15-HETE) protects pulmonary artery smooth muscle cells from apoptosis via inducible nitric oxide synthase (iNOS) pathway. Prostaglandins Other Lipid Mediat 97:50–59
Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX (2000) Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279(5):C1540–C1549
Burg ED, Remillard CV, Yuan JX (2008) Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br J Pharmacol 153(Suppl 1):S99–S111
Li Y, Li Q, Wang Z, Liang D, Liang S, Tang X, Guo L, Zhang R, Zhu D (2009) 15-HETE suppresses K(+) channel activity and inhibits apoptosis in pulmonary artery smooth muscle cells. Apoptosis 14(1):42–51
Hislop A, Reid L (1976) New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol 57(5):542–554
Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P, Eddahibi S (2000) Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol 23(6):762–771
Howell K, Preston RJ, McLoughlin P (2003) Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547(Pt 1):133–145
Christou H, Yoshida A, Arthur V, Morita T, Kourembanas S (1998) Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 18(6):768–776
Sands M, Howell K, Costello CM, McLoughlin P (2011) Placenta growth factor and vascular endothelial growth factor B expression in the hypoxic lung. Respir Res 12:17
Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P (2005) Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 97(2):185–191
Wright L, Tuder RM, Wang J, Cool CD, Lepley RA, Voelkel NF (1998) 5-Lipoxygenase and 5-lipoxygenase activating protein (FLAP) immunoreactivity in lungs from patients with primary pulmonary hypertension. Am J Respir Crit Care Med 157(1):219–229
Voelkel NF, Tuder RM, Wade K, Hoper M, Lepley RA, Goulet JL, Koller BH, Fitzpatrick F (1996) Inhibition of 5-lipoxygenase-activating protein (FLAP) reduces pulmonary vascular reactivity and pulmonary hypertension in hypoxic rats. J Clin Invest 97(11):2491–2498
Chu SJ, Tang LO, Watney E, Chi EY, Henderson WR Jr (2000) In situ amplification of 5-lipoxygenase and 5-lipoxygenase-activating protein in allergic airway inflammation and inhibition by leukotriene blockade. J Immunol 165(8):4640–4648
Stenmark KR, James SL, Voelkel NF, Toews WH, Reeves JT, Murphy RC (1983) Leukotriene C4 and D4 in neonates with hypoxemia and pulmonary hypertension. N Engl J Med 309(2):77–80
Piperno D, Pacheco Y, Hosni R, Moliere P, Gharib C, Lagarde M, Perrin-Fayolle M (1993) Increased plasma levels of atrial natriuretic factor, renin activity, and leukotriene C4 in chronic obstructive pulmonary disease. Chest 104(2):454–459
Walker JL, Loscalzo J, Zhang YY (2002) 5-Lipoxygenase and human pulmonary artery endothelial cell proliferation. Am J Physiol Heart Circ Physiol 282(2):H585–H593
Morganroth ML, Stenmark KR, Zirrolli JA, Mauldin R, Mathias M, Reeves JT, Murphy RC, Voelkel NF (1984) Leukotriene C4 production during hypoxic pulmonary vasoconstriction in isolated rat lungs. Prostaglandins 28(6):867–875
Morganroth ML, Stenmark KR, Morris KG, Murphy RC, Mathias M, Reeves JT, Voelkel NF (1985) Diethylcarbamazine inhibits acute and chronic hypoxic pulmonary hypertension in awake rats. Am Rev Respir Dis 131(4):488–492
Jones JE, Walker JL, Song Y, Weiss N, Cardoso WV, Tuder RM, Loscalzo J, Zhang YY (2004) Effect of 5-lipoxygenase on the development of pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol 286(5):H1775–H1784
Song Y, Jones JE, Beppu H, Keaney JF Jr, Loscalzo J, Zhang YY (2005) Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 112(4):553–562
Song Y, Coleman L, Shi J, Beppu H, Sato K, Walsh K, Loscalzo J, Zhang YY (2008) Inflammation, endothelial injury, and persistent pulmonary hypertension in heterozygous BMPR2-mutant mice. Am J Physiol Heart Circ Physiol 295(2):H677–H690
Preston IR, Hill NS, Warburton RR, Fanburg BL (2006) Role of 12-lipoxygenase in hypoxia-induced rat pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 290(2):L367–L374
Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos. 30370578, 30470752 and 31071007), Science Foundation of Education Department of Heilongjiang Provence (Nos. 11531181 and 11551275) and Science Foundation of Health Department of Heilongjiang Provence (Nos. 2009-250 and 2009-251). We also thank Anuradha Dhanasekaran from Anna University for reviewing the draft.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Zhu, D., Ran, Y. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J Physiol Sci 62, 163–172 (2012). https://doi.org/10.1007/s12576-012-0196-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-012-0196-9