
Abstract
In animal models, the secretion of the cardiac hormone, brain natriuretic peptide (BNP), and its closely related peptide, atrial natriuretic peptide (ANP), are stimulated by acute hypoxia. There is extensive human evidence for a rise in ANP under acute hypoxic conditions but very little evidence regarding the BNP response to acute hypoxia in humans. We therefore subjected seven healthy subjects to an acute hypobaric hypoxic stimulus to examine if BNP secretion increases rapidly. Significant hypoxaemia (mean nadir oxygen saturation 62.3%) was induced but no significant rise in BNP occurred. This suggests that either such acute hypoxaemia is well tolerated by the healthy human heart or it is not a stimulus for BNP secretion.
Similar content being viewed by others


Introduction
In humans, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) constitute the dual natriuretic peptide system of the heart. BNP is structurally related to ANP (they share a 17-amino acid internal ring) and both hormones have a role in fluid homeostasis, oppose the renin–angiotensin–aldosterone system and are secreted from cardiomyocytes [1]. ANP is known to rise acutely under hypoxic conditions in both animal and human models. Isolated rat hearts increase ANP and BNP with an acute (30 min) exposure to normobaric hypoxic [2]. Indeed, even just 10 min of normobaric hypoxia can induce ANP release in both anaesthetized rats [3] and rat atrial and ventricular tissue [4], with the steepest rise in ANP after the first 10 min of exposure [4]. In humans, ANP has been found to rise acutely after breathing a hypoxic gas mixture (10% O2) for only 10 min [5]. Another human study found no rise in ANP breathing hypoxic air (11% O2) at rest, but a significant rise after brief (5 min) exercise [6]. Other human studies have shown a rise in ANP after 30 min of breathing a normobaric nitrogen/oxygen mixture to induce SpO2 of 75–80% [7]. Following 1 h of normobaric hypoxia (12% O2) with SpO2 reduced to 68 ± 1%, ANP levels have also been found to rise by 50% [8]. The induction of more moderate SpO2 (90 ± 0.4%) with 30 min of normobaric hypoxia (16% O2) and moderate exercise has still demonstrated a rise in ANP [9]. Similarly, exhaustive exercise during hypobaric hypoxia (equivalent to 4,300 m) causes a rise in ANP [10].
Although BNP is primarily released secondary to cardiomyocyte stretch [11], it is increasingly appreciated from animal models that BNP secretion may also be stimulated by hypoxia. proBNP is stored in cardiomyocytes and following release is cleaved into the active hormone BNP and the inactive peptide NT-proBNP. Cultured adult rat cardiomyocytes have demonstrated an increase in ventricular BNP mRNA expression after the induction of hypoxia [12] as have rats exposed to hypobaric hypoxia equivalent to an altitude of 5,500 m [13] and mice exposed to normobaric hypoxia (10% O2) [14]. Even rats exposed to hypobaric hypoxia equivalent to a moderate altitude of around 2,250–2,550 m increase ventricular BNP mRNA [15]. BNP also has a pulmonary vasorelaxant action in rats [16] and BNP appears to have a modulatory role in attenuating the vasoconstriction of the pulmonary vasculature in response to hypoxia [17]. In a different animal model, utilizing anaesthetized pigs, the induction of acute myocardial hypoxia also causes a 3.5-fold rise in ventricular BNP mRNA and also plasma proBNP [18].
Data from human studies are also beginning to accumulate in support of the animal work discussed above. A very recent and elegant study has examined BNP expression under conditions of hypoxic ischaemia with and without mechanical stress. In isolated muscle strips from human atria, it was demonstrated that ischaemia per se is a potent inducer of BNP expression independent of mechanical stress. This therefore demonstrates a mechanism for BNP stimulation separate from cardiomyocyte stretch [19]. Human ventricular myocytes cultured under hypoxic conditions also increase the synthesis and secretion of BNP [20].
In this context, we therefore aimed to clarify if BNP rises in response to acute hypobaric hypoxia in healthy humans either before or after a brief exercise stimulus.
Materials and methods
The study protocol was approved by the Ministry of Defence Research Ethics Committee (MODREC), UK. Seven healthy subjects from various military units gave written informed consent. All subjects passed a medical questionnaire and standard medical examination required before entry into the Royal Air Force hypobaric chamber. An indwelling venous catheter was inserted before subjects entered the hypobaric chamber. Hypobaric exposure was commenced following a 10-min equilibration period sitting at rest breathing ambient (cabin) air. Barometric pressure was then reduced at a rate equivalent to 1,219 m ascent per minute up to an altitude equivalent to 5,334 m. Subjects continued to breathe ambient air throughout the exposure. After 25 min at this altitude, a 1-min exercise step-test was performed (step height set to 25 cm, 1 complete step every 2 s). Blood samples were taken at rest before simulated ascent and immediately pre- and post-exercise. Simultaneous recordings of oxygen saturation and heart rate were performed using a Nellcor NP-20 pulse oximeter (Covidian, MA, USA). After a total exposure of 40 min at 5,334 m, the chamber was recompressed to ground level at 1,219 m per minute with subjects breathing air enriched with oxygen via a mask.
Assay measurements
Whole blood was used for BNP assays. BNP was analysed using a Biosite Triage Meter and Triage BNP test kits (Inverness Medical UK, Cheshire, UK). BNP assays were performed within 4 h at sea-level after satisfactory quality control checks. The BNP range reported by the test system is 5–5,000 pg/ml. A BNP result of ≤100 pg/ml is representative of a normal value. Coefficient of variation at a mean BNP of 71.3 pg/ml is 8.8%.
Results
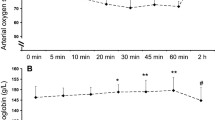
Subjects were 28 ± 2.4 years old, weighed 70.3 ± 14.8 kg, and were 172 ± 8 cm in height (mean ± SD). Mean (±SEM) SpO2 was 98.3 ± 0.3, 68.1 ± 1.1 and 62.3 ± 2 at ground level, pre-exercise and post-exercise at 5,334 m, respectively (Fig. 1). SpO2 at ground level versus 5,334 m pre- and post-exercise was highly significant (p < 0.001, paired Student’s t test), while SpO2 pre- and post exercise was also significant (p = 0.007). Mean HR (±SEM) was 70 ±3.3, 81.1 ±2.7 and 134.1 ±5.8 at ground level, pre-exercise and post-exercise at 5,334 m, respectively. HR at ground level versus 5,334 m pre-and post-exercise was significant (p = 0.013 and p < 0.001 respectively, paired Student’s t test) as was HR pre-exercise versus HR post-exercise (p < 0.001).

Oxygen saturation (mean ± SEM) at ground level, at a simulated altitude of 5,334 m and following brief exercise at 5,334 m
BNP remained <5 pg/ml in 6 of 7 subjects and showed no significant rise in the 7th subject (BNP 18.1 pg/ml pre-exercise, 18.9 post-exercise).
Discussion
We have demonstrated that BNP does not rise during an acute hypobaric hypoxic exposure that included an exercise stimulus and induced significant hypoxia and tachycardia in all subjects. The lack of an effect on BNP is in marked contrast to the effect of acute hypoxia on the related cardiac natriuretic peptide ANP in both animal and human models. This implies that a healthy human heart can withstand marked acute hypoxia without the need to release pre-stored BNP from the heart, and as such is a significant negative finding.
Other human studies regarding BNP in the context of hypoxia have usually involved co-morbidity. In cyanotic congenital heart disease patients, despite preserved ventricular function and reduced total body water, proBNP is markedly elevated [21], suggesting hypoxia is a direct stimulus for BNP secretion. In heart failure patients with sleep apnoea, the burden of hypoxemia (the time spent with oxygen saturation <90%) predicts BNP concentrations. For each 10% increase in duration of hypoxemia, BNP is increased by almost 10% [22]. Another recent study has also confirmed that in patients with sleep apnoea (but without heart failure) the duration of nocturnal oxygen desaturation correlates with pro-BNP levels [23].
Two other studies have utilized an acute hypoxic exposure to examine the effect on BNP: a very brief (1–3 min) exposure to hypobaric hypoxic (equivalent to 9,144 m) had no effect on BNP [24]; and a 60-min normobaric hypoxic exposure inducing SpO2 of 82 ± 1% caused a very slight rise in NT-proBNP from 2.2 ± 1.5 pmol/l to 2.4 ± 1.5 pmol/l [25]. This latter study involved a highly selected group of subjects with either high or low renin–angiotensin system activity.
There have also been two recent studies examining the BNP response to a more chronic hypoxic exposure: no rise in NT-proBNP occurred in healthy subjects at 5,200 m following an acclimatization period at 3,650 m [26]; and no rise in BNP occurred with gradual ascent to an altitude of 5,050 m (SpO2 84.5 ± 1.3%) [27]. The rise that occurs in ANP with acute hypoxia is not seen following acclimatization at HA [10] and this may explain these negative findings.
Our exercise stimulus was intentionally brief. Given that BNP is known to rise with prolonged exercise under normoxic conditions [28] and given the effect of acute hypoxia on ANP and considering that BNP is stored in secretory granules of atrial and ventricular myocytes [29], we wanted to assess whether, specifically, an acute hypoxic stimulus could produce a detectable rise in BNP. Our study is the only one to induce such a low SpO2 (62.3 ± 2%) and to include an exercise stimulus, albeit brief, in an unselected population. We have demonstrated that BNP does not rise acutely despite this significant hypoxaemia, suggesting that in normal healthy humans either such acute hypoxaemia is well tolerated by the heart or is not a stimulus for BNP secretion. This is in contrast to previous findings regarding the closely related cardiac natriuretic peptide ANP. It remains possible that a more prolonged period of hypobaric hypoxia with physical exercise may stimulate a rise in BNP secondary to changes in gene transcription.
References
Liang F, Kapour AM, Lam A, Damm DL, Quan D, O’Connell M, Protter AA (2007) B-type natriuretic peptide inhibited angiotensin II-stimulated cholesterol biosynthesis, cholesterol transfer, and steroidogenesis in primary human adrenocortical cells. Endocrinology 148:3722–3729
Tóth M, Vuorinen KH, Vuolteenaho O, Hassinen IE, Uusimaa PA, Leppäluoto J, Ruskoaho H (1994) Hypoxia stimulates release of ANP and BNP from perfused rat ventricular myocardium. Am J Physiol 266:H1572–H1580
Albert TS, Tucker VL, Renkin EM (1997) Acute alveolar hypoxia increases blood-to-tissue albumin transport: role of atrial natriuretic peptide. J Appl Physiol 82:111–117
Ljusegren ME, Andersson RG (1994) Hypoxia induces release of atrial natriuretic peptide in rat atrial tissue: a role for this peptide during low oxygen stress. Naunyn Schmiedebergs Arch Pharmacol 350:189–193
Kawashima A, Kubo K, Hirai K, Yoshikawa S, Matsuzawa Y, Kobayashi T (1989) Plasma levels of atrial natriuretic peptide under acute hypoxia in normal subjects. Respir Physiol 76:79–91
Lordick F, Hauck RW, Senekowitsch R, Emslander HP (1995) Atrial natriuretic peptide in acute hypoxia-exposed healthy subjects and in hypoxaemic patients. Eur Respir J 8:216–221
Cargill RI, McFarlane LC, Coutie WJ, Lipworth BJ (1996) Acute neurohormonal responses to hypoxaemia in man. Eur J Appl Physiol 72:256–260
Lawrence DL, Skatrud JB, Shenker Y (1990) Effect of hypoxia on atrial natriuretic factor and aldosterone regulation in humans. Am J Physiol 258(2 Pt 1):E243–E248
Lawrence DL, Shenker Y (1991) Effect of hypoxic exercise on atrial natriuretic factor and aldosterone regulation. Am J Hypertens 4:341–347
Rock PB, Kraemer WJ, Fulco CS, Trad LA, Malconian MK, Rose MS, Young PM, Cymerman A (1993) Effects of altitude acclimatization on fluid regulatory hormone response to submaximal exercise. J Appl Physiol 75:1208–1215
Hall C (2005) NT-ProBNP: the mechanism behind the marker. J Card Fail 11:S81–S83
Weidemann A, Klanke B, Wagner M, Volk T, Willam C, Wiesener MS, Eckardt KU, Warnecke C (2008) Hypoxia, via stabilization of the hypoxia-inducible factor HIF-1alpha, is a direct and sufficient stimulus for brain-type natriuretic peptide induction. Biochem J 409:233–242
Nakanishi K, Tajima F, Itoh H, Nakata Y, Osada H, Hama N, Nakagawa O, Nakao K, Kawai T, Takishima K, Aurues T, Ikeda T (2001) Changes in atrial natriuretic peptide and brain natriuretic peptide associated with hypobaric hypoxia-induced pulmonary hypertension in rats. Virchows Arch 439:808–817
Sun JZ, Chen SJ, Li G, Chen YF (2000) Hypoxia reduces atrial natriuretic peptide clearance receptor gene expression in ANP knockout mice. Am J Physiol Lung Cell Mol Physiol 279:L511–L519
Perhonen M, Takala TE, Vuolteenaho O, Mäntymaa P, Leppäluoto J, Ruskoaho H (1997) Induction of cardiac natriuretic peptide gene expression in rats trained in hypobaric hypoxic conditions. Am J Physiol 273:R344–R352
Hill NS, Klinger JR, Warburton RR, Pietras L, Wrenn DS (1994) Brain natriuretic peptide: possible role in the modulation of hypoxic pulmonary hypertension. Am J Physiol 266:L308–L315
Klinger JR, Warburton RR, Pietras L, Hill NS (1998) Brain natriuretic peptide inhibits hypoxic pulmonary hypertension in rats. J Appl Physiol 84:1646–1652
Goetze JP, Gore A, Moller CH, Steinbruchel DA, Rehfeld JF, Nielsen LB (2004) Acute myocardial hypoxia increases BNP gene expression. FASEB J 17:1105–1107
Möllmann H, Nef HM, Kostin S, Dragu A, Maack C, Weber M, Troidl C, Rolf A, Elsässer A, Böhm M, Brantner R, Hamm CW, Holubarsch CJ (2010) Ischemia triggers BNP expression in the human myocardium independent from mechanical stress. Int J Cardiol 143:289–297
Casals G, Ros J, Sionis A, Davidson MM, Morales-Ruiz M, Jiménez W (2009) Hypoxia induces B-type natriuretic peptide release in cell lines derived from human cardiomyocytes. Am J Physiol Heart Circ Physiol 297:H550–H555
Hopkins WE, Chen Z, Fukagawa NK, Hall C, Knot HJ, LeWinter MM (2004) Increased atrial and brain natriuretic peptides in adults with cyanotic congenital heart disease: enhanced understanding of the relationship between hypoxia and natriuretic peptide secretion. Circulation 109:2872–2877
Gottlieb JD, Schwartz AR, Marshall J, Ouyang P, Kern L, Shetty V, Trois M, Punjabi NM, Brown C, Najjar SS, Gottlieb SS (2009) Hypoxia, not the frequency of sleep apnea, induces acute hemodynamic stress in patients with chronic heart failure. J Am Coll Cardiol 54:1706–1712
Sarıman N, Levent E, Cubuk R, Yurtlu S, Benli Aksungar F (2010) Bronchial hyperreactivity and airway wall thickening in obstructive sleep apnea patients. Sleep Breath (Epub ahead of print)
Karadag R, Sen A, Yildirim N, Basmak H, Golemez H, Cakir E, Akin A (2010) The relation between intraocular pressure change and plasma natriuretic peptide under simulated hypobaric conditions. Indian J Ophthalmol 58:195–198
Due-Andersen R, Pedersen-Bjergaard U, Høi-Hansen T, Olsen NV, Kistorp C, Faber J, Boomsma F, Thorsteinsson B (2008) NT-pro-BNP during hypoglycemia and hypoxemia in normal subjects: impact of renin–angiotensin system activity. J Appl Physiol 104:1080–1085
Toshner MR, Thompson AAR, Irving JB, Baillie JK, Morton JJ, Peacock AJ (2008) NT-proBNP does not rise on acute ascent to high altitude. High Alt Med Biol 9:1–4
Feddersen B, Ausserer H, Haditsch B, Frisch H, Noachtar S, Straube A (2009) Brain natriuretic peptide at altitude: relationship to diuresis, natriuresis, and mountain sickness. Aviat Space Environ Med 80:108–111
Hew-Butler T, Noakes TD, Soldin SJ, Verbalis JG (2008) Acute changes in endocrine and fluid balance markers during high-intensity, steady-state, and prolonged endurance running: unexpected increases in oxytocin and brain natriuretic peptide during exercise. Eur J Endocrinol 159:729–737
Levin ER, Gardner DG, Samson WK (1998) Natriuretic peptides. N Engl J Med 339:321–328
Acknowledgments
The authors thank the Drummond Foundation for their charitable funding, the volunteers and their military units, and Inverness Medical UK, Cheshire, for the unconditional loan of the Triage meter.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Woods, D., Hooper, T., Mellor, A. et al. Brain natriuretic peptide and acute hypobaric hypoxia in humans. J Physiol Sci 61, 217–220 (2011). https://doi.org/10.1007/s12576-011-0141-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-011-0141-3