
Abstract
Purpose
The purpose of this article was to highlight current and future trends in radiobiology in an effort to move hadron therapy forward through the application of new knowledge in DNA damage and subsequent response to heavy ion radiotherapy, immune oncology and the interconnection between.
Methods
The subject matter begins with a description of the role of radiation in eliciting either an immunogenic or tolerogenic response to radiation exposure. The role of fragmented DNA in an immunogenic response is described, followed by the definitive role that DNA damage and subsequent repair, or not, of complex DNA damage after hadron exposure plays in the survival response of hadron irradiated cells.
Results
The process by which ionizing radiation elicits an immunogenic rather than tolerogenic response is becoming clearer. The timing of fractionated radiotherapy when combined with an immune checkpoint inhibitor is not clear and may be tumor site specific. Furthermore, whether hadron therapy is more effective at generating a durable immunogenic response is unknown.
Conclusions
Cytosolic DNA plays a significant role in eliciting an innate immune response with the likelihood that hadron therapy would generate complex DNA damage that because it is less likely to be repaired, is more likely to become cytosolic DNA, and more likely to activate an immunogenic response. Lastly, DNA repair pathway choice appears to be a credible bio-indicator for hadron therapy selection as well identify druggable targets to enhance hadron therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Traditional concepts in radiobiology include the consequences of DNA damage and DNA repair, the oxygen effect and conversely the consequences of hypoxia, radiosensitivity from genetic defects associated with identified radiosensitivity syndromes and the small differences in radiosensitivity within a population, accelerated tumor repopulation and the rare abscopal effect. The abscopal effect could only be described as a rare unpredictable immune response whose mechanism remains undefined. Furthermore, traditional radiobiology, that is, studying the effects of “sub-lethal”, in this case radiation doses that are not-ablative, were in fact stimulatory in the sense that such doses of radiation increased migration and invasion in in vitro assays but also metastasis in glioblastoma cells and tumors, respectively [Wild-Bode 2001, Cancer Res]. The in vivo response suggested at that time that if there was an immunologic effect it was one of tolerance.

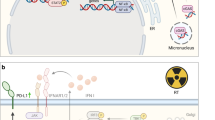
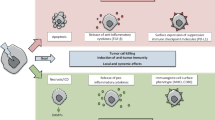
Graphical Abstract describing the interplay between radiation exposure, DNA damage and immune activation, and DNA damage itself. (A portion of this image was generated using BioRender.)
However, new approaches outside of the classic boundaries of traditional radiobiology have pointed to the potential for enhanced therapeutic options based upon the intersection of a new understanding of tumor immune responses, new ways to beneficially target that response, and how novel approaches to radiation delivery in terms of dose, temporal changes to dose fractionation and the spatial delivery of radiation can likely harness an immune response, a response that seems dependent upon the generation of cytosolic DNA as a trigger of an innate immune response if not also an adaptive response. Furthermore, as charged particle radiotherapy grows and utilizes heavier particles, DNA damage complexity and newly identified vulnerabilities to charged particles have emerged. Figure 1, is a graphical abstract that attempts to show the inter-relatedness of these concepts and describes the intent of this article.
2 Radiation-induced immunogenic response
Subsequently, Lugade et al. in 2005 [1] showed an increased tumoricidal effect with radiation generated when a large dose of radiation (15 Gy) was delivered to B16 tumors as compared to a fractionated radiation dose high dose radiation (5 Gy delivered each day for 4 days). Although these doses will not deliver the same biologically effective dose response, the single 15 Gy dose was more effective that the fractionated schema and that the single high dose was better at recruiting tumor antigen specific effector cells to the tumor that the fractionated scheme. This was followed on in 2009 by Dewan et al. [2] who using biologically equivalent dose and fractionation schemes for the primary tumor combined with an immune checkpoint inhibitor described a window of response in which the optimal abscopal immune response was determined. These results suggest radiation can cause an anti-tumor immune response when used at doses much higher than conventional radiation and with limited numbers of dose fractions. Whether an abscopal effect could be generated by 12C ions was determined in a mouse model of subcutaneous osteosarcoma tumors [3]. Here, a single dose of 5.3 Gy 12C ions was combined with a dual checkpoint antibody against PD-1 and CTLA-4. While one tumor was irradiated, the secondary tumor on the opposing leg of the syngeneic mouse was not. Tumor growth delay was measured in both irradiated and non-irradiated tumors and both tumors responded to the radiation/checkpoint inhibitor combination. Whether the effect was synergistic or not could not be determined.
The positive results described above can be interpreted to suggest that there is an optimal dose to be identified, there is a time between doses that has not been optimized and that these parameters may be different for different tumors. Not examined and which requires a better understanding is the temporal events associated with eliciting an immune response. Furthermore, immune cells are some of the most radiosensitive cells in the body and any immune cells that have entered the primary tumor are likely killed by the radiation dose targeted against the tumor. This collateral damage may be consequential to an anti-tumor immune response. While not examining directly the temporal events associated with launching an immune response Moore et al., [4] showed that radiation spaced 10 days apart rather than 1 or 4 days when combined with a checkpoint inhibitor was more tumoricidal. Furthermore, waiting until just before the second radiation to provide checkpoint inhibitor rather than providing checkpoint inhibitor with the first radiation dose was clearly more effective suggesting that for an immune checkpoint inhibitor to work requires time for the antigen to present itself. Because the application of immune checkpoint inhibitors with radiation has been based upon empirical observations, Friedrich et al., [5] developed a predictive model benchmarked across 16 data sets of radiation interaction with immune checkpoint inhibitors that took into account characteristics of the primary and secondary (distal) tumors, the kinetics of immune cell populations and subsequent signaling after radiation. The model identified a radiation dose window that maximized stimulation of the immune response while avoiding the depletion of the immune cell population due to radiation killing of immune cells within the tumor being targeted.
There is perhaps a role in the immune response for avoiding the draining lymph node given that it is a location for initiation of the immune adaptive response. It has been suggested that in tumor sites where only the involved lymph node is irradiated therapeutic responses are more durable. This supported by studies where tumor response to stereotactic radiation combined with immune checkpoint inhibitor was attenuated when the draining lymph node was irradiated [6]. Furthermore, T cell memory was also compromised when the draining lymph node was irradiated.
Avoiding depletion of the immune cell population, maximizing stimulation of the immune response and taking advantage of bystander effects is likely the foundation for the enhanced tumor response seen with spatially fractionated radiotherapy approaches. For example, given the distance between irradiated and non-irradiated tissues described for the treatment of mouse glioblastomas with minibeams there is likely a bystander effect. Furthermore, adverse normal tissue events are limited, likely given the distance between unirradiated tissue that allows for quick blood vessel repair across the irradiated regions [7]. The potential for an immune response after minibeam irradiation is analogous to PATHY (PArtial Tumour irradiation targeting the HYpoxic segment) where the peritumoral immune microenvironment is spared providing an environment for an immune response as well as a bystander effect [8, 9]. This approach has been extended to both proton and carbon radiotherapy (Particle PATHY) [8] for the treatment of recurrent large bulky tumors.
3 The role of cytosolic DNA in the immunogenic response
The bystander effect suggested for spatially fractionated radiotherapy is likely generated by signaling from irradiated tissues. Indeed, based upon Monte Carlo modelling, the bystander effect, i.e. the lethal cell signaling, besides causing the death of cells in adjacent non-irradiated tissues, was reported to have a significant role in the local killing effect that enhances cell killing [10]. The generation of this cell signaling which can cause apoptosis or DNA breaks in non-irradiated cells arises from the direct or indirect damage to the DNA by radiation. In addition, DNA can leach into the cytoplasm as condensed chromosomal fragments or as micronuclei triggering the STING pathway and an innate immune response as this DNA is considered foreign. This lack of DNA repair, whether as a result of defects in DNA repair or overwhelming damage, likely serves as a link between DNA repair processes and immune surveillance through the cGAS-cGAMP- STING pathway [11, 12]. Supporting evidence includes manipulation of the mis-match repair system which modulates immune response [13] linking radiation and immune surveillance in mismatch repair-deficient tumors. As a result, modulation of DNA repair has been suggested as a strategy in which to enhance immune surveillance and therapeutic efficacy [14]. Since then a direct link between MRE11, a member of the MRE11-RAD50-NBM (MRN) complex, which functions as DNA double-strand break repair sensor, has been shown to also have a role in the activation of the cGAS-STING pathway releasing cGAS that was sequestered in nucleosomes [15]. For normal cells, however, MRE11 may serve to promote tumorigenesis by facilitating resistance to oncogene-induced replication stress.
3.1 The DNA damage response
DNA damage and the DNA damage response after radiation, per se, particularly by charged particles, cannot be discounted as a mechanism in and of itself in modulating the response to hadron therapy. Lesion complexity caused by hadron exposure in live cells was described by Asaithamby et al. in 2011 [16] where lesion type and lesion location were determined and then followed over time after 1 GeV 28Si (40 keV/µ) and 1 GeV 56Fe (150 keV/µ) ion irradiation. Lesion complexity, as measured by the overlap of lesions targeted by fluorescent antibody tagged DNA repair proteins that represented different DNA lesions single and double strand breaks and base damage. They described the increased time necessary for lesion resolution with increasing LET suggesting that lesion complexity impacted DNA repair capacity and cell survival. Subsequently, Bertolet et al. described and quantified lesion complexity using their Microdosimetric Gamma (MG) Model [17]. They argued that dose averaged LET may be insufficient to describe biological response given the differences in biological response seen with x-rays and protons of similar LETs, while the gamma model described a slightly increased lesion complexity determined for protons compared to X-rays.
This difference in lesion complexity determined by the MG Model can be used to explain differences in clonogenic survival for lung tumor cells deficient in DNA repair capacity, specifically homologous recombination when irradiated with X-rays vs. protons [18]. Further interrogation of this effect identified HR genes such as BRCA1, FANCA and FANCD2 as recapitulating this effect through either identification of cell lines with known genetic deficiencies or through knockdown of gene expression for these target genes. Inhibitors or ATM such as AZD0156 have been shown to exacerbate cell death in cells exposed to the Bragg peak of protons accelerated to 76.8 MeV [19], while the ATM inhibitor AZD1390, which also blocks RAD51 unloading in p53 mutant glioblastoma cells and xenografts [20]. This suggests are role for the HR pathways in the resolution of complex lesions and the potential therapeutic intervention via molecular targeting on specific genes or proteins associated with DNA repair as well as the potential to triage patients identified as having mutations in HR pathway genes to hadron therapy.
This notion of the role of the HR pathway was examined by the clonogenic response of a series of pancreatic cell lines after irradiation with 12C. Sishc et al. [21] examined clonogenic survival after γ-ray or 12C exposure combined with inhibitors of: DNA-PKcs, a member of the non-homologous end joining (NHEJ) pathway; RAD51, a member of the HR pathway; ATM; and ATR. As expected, targeting ATM and DNA-PKcs increased the radioresponse when cells were irradiated with γ-rays, however, as in Liu et al., [18] no increased radioresponse was seen with either the HR inhibitor nor the ATR inhibitor. However, when exposed to 12C ions all inhibitors caused an increase in radioresponse to varying degrees. This suggests that for hadron therapy HR inhibitors also play a role in enhancing hadron therapy with protons and other ions with higher LET like 12C. The ATR inhibitor data is somewhat confounding based upon the results of Zhou and Chen, [19, 20]. However, as outlined in Sishc et al., [21] the enhanced cell killing effect for the ATR inhibitor to 12C irradiation is likely to do with cells carrying the inherent burden of high replication stress, suggesting another pathway that may be targetable through chemistry.
Some caution should be considered when implementing the findings described here. The response of tumor and adjacent normal tissue to proton radiotherapy is likely to be similar in that while tumor response is enhanced, care should be taken in evaluating the maximally tolerated dose of the normal tissue when also treated, by default, with radiation and an HR inhibitor. That would not be the case for tumors carrying an HR defect. However, when moving to hadrons of higher LET in the SOBP, caution must be taken when choosing the DNA repair pathway to inhibit. For example, Sishc et al., also irradiated cells with 12C but in the entrance of the 12C beam where the LET was 13 keV/µ. When combined with the HR and ATR inhibitors no statistical difference was seen in clonogenic survival. However, when the NHEJ inhibitor was combined with 12C, even though cells were irradiated in the entry region of the beam, cell survival was equivalent to that seen when cells were irradiated with 12C in the SOBP. This suggests that inhibitors of NHEJ will increase the risk for serious normal tissue complications.
Finally, patients are now often screen for mutations in key cancer-associated genes and genes that might suggest a specific therapeutic intervention. It seems reasonable in the context of radiotherapy to screen for HR-associated genes. Deficiencies in HR function caused by well understood mutations could be used as a biomarker for hadron therapy and based upon clinical access that could be via either proton or heavier hadron therapy. This further suggests that inhibitors of the HR pathway should be given greater priority so that all tumors could take advantage of hadron therapy.
In summary, the role of DNA damage and repair, DNA lesion complexity, changes in the temporal and spatial delivery of radiation and how they may impact cellular and tissue responses to radiation -particularly hadrons, to elicit and immune response rather than allowing tumors to become tolerogenic, was briefly described. In addition, how the seminal role of DNA damage and repair can be manipulated to clinical advantage, with caveats, was also described. These features, specific to hadron therapy and the unique dose deposition patterns thereof, support the continued development of particle therapy using particles of higher Z and energy as a mechanism to provide personalized therapy to patients with tumors resistant to conventional therapies.
References
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG. Lord EM local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005. https://doi.org/10.4049/jimmunol.174.12.7516.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009. https://doi.org/10.1158/1078-0432.CCR-09-0265.
Takahashi Y, Yasui T, Minami K, Tamari K, Hayashi K, Otani K, Seo Y, Isohashi F, Koizumi M. Ogawa K Carbon ion irradiation enhances the antitumor efficacy of dual immune checkpoint blockade therapy both for local and distant sites in murine osteosarcoma Oncotarget 2019 https://doi.org/10.18632/oncotarget.26551.
Moore C, Hsu CC, Chen WM, Chen BPC, Han C, Story M, Aguilera T, Pop LM, Hannan R, Fu YX, Saha D, Timmerman R. Personalized Ultrafractionated Stereotactic Adaptive Radiotherapy (PULSAR) in preclinical models enhances single-Agent Immune Checkpoint Blockade. Int J Radiat Oncol Biol Phys. 2021. https://doi.org/10.1016/j.ijrobp.2021.03.047.
Friedrich T, Scholz M, Durante MA. Predictive Biophysical Model of the combined action of Radiation Therapy and Immunotherapy of Cancer. Int J Radiat Oncol Biol Phys. 2022. https://doi.org/10.1016/j.ijrobp.2022.03.030.
Marciscano AE, Ghasemzadeh A, Nirschl TR, Theodros D, Kochel CM, Francica BJ, Muroyama Y, Anders RA, Sharabi AB, Velarde E, Mao W, Chaudhary KR, Chaimowitz MG, Wong J, Selby MJ, Thudium KB, Korman AJ, Ulmert D, Thorek DLJ, Deweese TL. Drake CG Elective nodal irradiation attenuates the combinatorial efficacy of Stereotactic Radiation Therapy and Immunotherapy Clin. Cancer Res. 2018. https://doi.org/10.1158/1078-0432.CCR-17-3427.
Bertho A, Ortiz R, Juchaux M, Gilbert C, Lamirault C, Pouzoulet F, Polledo L, Liens A, Warfving N, Sebrie C, Jourdain L, Patriarca A, De Marzi L, Prezado Y. First evaluation of temporal and spatial fractionation in Proton Minibeam Radiation Therapy of glioma-bearing rats cancers (Basel) 2021 https://doi.org/10.3390/cancers13194865.
Tubin S, Fossati P, Carlino A, Martino G, Gora J, Stock M, Hug E. Novel Carbon Ion and Proton Partial Irradiation of Recurrent Unresectable Bulky Tumors (Particle-PATHY): early indication of effectiveness and safety cancers. (Basel). 2022. https://doi.org/10.3390/cancers14092232.
Tubin S, Gupta S, Grusch M, Popper HH, Brcic L, Ashdown ML, Khleif SN, Peter-Vorosmarty B, Hyden M, Negrini S, Fossati P, Hug E. Shifting the Immune-Suppressive to Predominant Immune-Stimulatory Radiation effects by SBRT-PArtial Tumor Irradiation Targeting HYpoxic Segment (SBRT-PATHY). Cancers (Basel). 2020. https://doi.org/10.3390/cancers13010050.
Butterworth KT, McMahon SJ, Hounsell AR, O’sullivan JM, Prise KM. Bystander signalling: exploring clinical relevance through new approaches and new models. Clin Oncol (R Coll Radiol). 2013. https://doi.org/10.1016/j.clon.2013.06.005.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu YX. Weichselbaum RR STING-Dependent cytosolic DNA sensing promotes Radiation-Induced type I Interferon-Dependent Antitumor immunity in immunogenic tumors immunity 2014 https://doi.org/10.1016/j.immuni.2014.10.019.
Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018. https://doi.org/10.1084/jem.20180139.
Ruiz-Banobre J, Goel ADNA. Mismatch Repair Deficiency and Immune checkpoint inhibitors in gastrointestinal cancers gastroenterology 2019 https://doi.org/10.1053/j.gastro.2018.11.071.
Truini A, Germano G, Bardelli A. Inactivation of DNA repair-prospects for boosting cancer immune surveillance. Genome Med. 2018. https://doi.org/10.1186/s13073-018-0603-9.
Cho MG, Kumar RJ, Lin CC, Boyer JA, Shahir JA, Fagan-Solis K, Simpson DA, Fan C, Foster CE, Goddard AM, Lerner LM, Ellington SW, Wang Q, Wang Y, Ho AY, Liu P, Perou CM, Zhang Q, Mcginty RK, Purvis JE, Gupta GP. MRE11 liberates cGAS from nucleosome sequestration during tumorigenesis. Nature. 2024. https://doi.org/10.1038/s41586-023-06889-6.
Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc Natl Acad Sci U S A. 2011. https://doi.org/10.1073/pnas.1016045108.
Bertolet A, Chamseddine I, Paganetti H, Schuemann J. The complexity of DNA damage by radiation follows a Gamma distribution: insights from the Microdosimetric Gamma. Model Front Oncol. 2023. https://doi.org/10.3389/fonc.2023.1196502.
Liu Q, Ghosh P, Magpayo N, Testa M, Tang S, Gheorghiu L, Biggs P, Paganetti H, Efstathiou JA, Lu HM, Held KD. Willers H Lung cancer cell line screen links fanconi anemia/BRCA pathway defects to increased relative biological effectiveness of proton radiation. Int J Radiat Oncol Biol Phys. 2015. https://doi.org/10.1016/j.ijrobp.2014.12.046.
Zhou Q, Howard ME, Tu X, Zhu Q, Denbeigh JM, Remmes NB, Herman MG, Beltran CJ, Yuan J, Greipp PT, Boughey JC, Wang L, Johnson N, Goetz MP, Sarkaria JN, Lou Z. Mutter RW inhibition of ATM induces hypersensitivity to Proton Irradiation by upregulating toxic end joining. Cancer Res. 2021. https://doi.org/10.1158/0008-5472.CAN-20-2960.
Chen J, Laverty DJ, Talele S, Bale A, Carlson BL, Porath KA, Bakken KK, Burgenske DM, Decker PA, Vaubel RA, Eckel-Passow JE, Bhargava R, Lou Z, Hamerlik P, Harley B, Elmquist WF, Nagel ZD, Gupta SK. Sarkaria JN aberrant ATM signaling and homology-directed DNA repair as a vulnerability of p53-mutant GBM to AZD1390-mediated radiosensitization. Sci Transl Med. 2024. https://doi.org/10.1126/scitranslmed.adj5962.
Sishc BJ, Saha J, Polsdofer E, Ding L, Lu H, Wang S-Y, Swancutt KL, Nicholson JH, Facoetti A, Pompos A, Ciocca M, Aguilera TA, Story MD. Davis AJ Defective Homologous Recombination and genomic instability predict increased responsiveness to Carbon Ion Radiotherapy in Pancreatic Cancer bioRxiv 2024 https://doi.org/10.1101/2024.04.08.588534.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
No human research was performed.
Consent to participate
No human research was performed.
Consent to publish
No human research was performed.
Competing interests
The authors have no relevant financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Story, M.D., Davis, A.J. & Sishc, B.J. Current trends and future perspectives in hadron therapy: radiobiology. Health Technol. (2024). https://doi.org/10.1007/s12553-024-00895-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12553-024-00895-y