
Abstract
Cell-free protein synthesis (CFPS) has emerged as an attractive platform for biotechnology and synthetic biology due to its numerous advantages to cell-based technologies for specific applications. CFPS can be faster, less sensitive to metabolite toxicity, and amenable to systems that are not easily genetically manipulated. Due to these advantages, a promising application of CFPS is to characterize biosynthetic gene clusters, particularly those harbored within the genomes of microorganisms that generate secondary metabolites, otherwise known as natural products. In the postgenomic era, genome sequencing has revealed an incredible wealth of metabolic diversity. However, far more of these pathways are termed “cryptic,” i.e., unable to be produced under standard laboratory conditions than have been characterized. A major barrier to characterizing these cryptic natural products using CFPS is that many of these pathways require utilization of complex cofactors, many of which to date are not recycled efficiently or in an economically viable fashion. In this perspective, we outline strategies to regenerate cofactors relevant to secondary metabolite production in CFPS. This includes adenosine 5′-triphosphate, coenzyme A, redox cofactors (iron-sulfur clusters, nicotinamide adenine dinucleotide phosphate, flavin adenine dinucleotide), all of which play a crucial role in important biosynthetic enzymes. Such advances in cofactor recycling enable continuous production of complex metabolites in CFPS and expand the utility of this emergent platform.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cell free protein synthesis (CFPS) systems have long had a role in elucidating fundamental science in biochemistry and molecular biology. The earliest iteration of cell free synthesis was reported in 1907 by Nobel laureate Edaurd Buchner (Nobel Prize in Chemistry 1907) who demonstrated bio-ethanol production in yeast cell extracts, pioneering contributing to the development of CFPS (Buchner 1897). Another example of a Nobel Prize was the groundbreaking work of Nirenberg and colleagues deducing of genetic code in Escherichia coli cell extract (Nirenberg and Matthaei 1961). In more recent years, CFPS has been applied to prototype expression and undergo biomanufacturing applications (Hodgman and Jewett 2012). Without the cellular survival objectives, CFPS significantly shortens the protein expression process and can be applied towards optimizing specific targets of expression (Carlson et al. 2012; Sword et al. 2024). By supplementing cell extracts with amino acids, nucleotides, and secondary energy substrates, protein synthesis can be initiated in vitro using the native cellular transcriptional and translational machinery. In the absence of a cell membrane, the process of protein synthesis can be directly monitored, rapidly sampled, and directly modified (Silverman et al. 2020). Despite these advances, several obstacles have prevented their engineering potential in protein production, including low production yields, high energy expense, and no viable scale-up methods existing (Swartz 2006). However, the rapid technical development of synthetic biology in the last decade provides new possibilities for cell free biocatalysis and has addressed these limitations. Swartz and colleagues have reported a novel method for cell free system scale up and reached 100 L (Voloshin and Swartz 2005). Similarly, Sutro Biopharma, Inc. demonstrated high productivity of cost-effective cell free protein synthesis at the hundred liters reaction (Zawada et al. 2011). Recent advances demonstrated that a eukaryotic CFPS platform has been developed based on Nicotiana tabacum BY-2 cell culture (BY-2 lysate; BYL), which scales the reaction volume across a 20,000 × range and is applied in a liter-scale reaction in a commercial bioreactor (Gupta et al. 2023). These milestones show that the CFPS system has the potential to become an industrially scaled platform for producing recombinant DNA proteins.
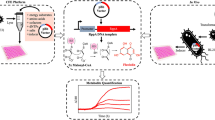
While applications of protein expression have expanded in cell free systems, one area that is an emergent application developed in recent years has been towards metabolite production. Numerous secondary metabolites gene clusters have been discovered in plant and microbial genomes and have important pharmacological applications (Fig. 1). In particular, the postgenomic era has revealed a vast wealth of untapped metabolic diversity harbored within microorganisms, most of which is called “cryptic” or “silent” inasmuch as it is not produced under standard laboratory culture conditions and thus are unable to be isolated or characterized (Ji et al. 2024; Zhao et al. 2024). A challenge in the characterization of silent is that in their natural context, secondary metabolites are produced for precise ecological reasons that are not always able to be easily recapitulated in a laboratory environment. Many metabolites are toxic when produced above physiological concentrations. For all these reasons, many metabolites that may have promising bioactivity or interesting structural diversity remain uncharacterized. A separate but related application is that the biosynthetic enzymes to generate secondary metabolites frequently harbor unusual and remarkable biocatalytic transformations, including rare domains or domains of unknown function (DUFs) which perform chemistry inaccessible via chemical synthesis (Li et al. 2024).

Schematic diagram of secondary metabolites sources, classes, and biomanufacturing applications
A CFPS system contains crude cell lysates/extracts and DNA supplemented with associated energy sources, nucleotides, amino acids, salts, and cofactors, which are essential for the reaction to generate the desired product. It is important to emphasize that there is a difference with one-pot cell free production which uses multiple purified enzymes, substrates, and cofactors but no DNA input. Most of the enzymes in current CFPS system use are cofactor-dependent such as oxidoreductases and transferases which can perform complex chemistry and catalyze many synthetically useful reactions (Zhao and van der Donk 2003). The biosynthesis and regeneration of cofactors are expensive for the cell and are typically required in stoichiometric amounts for enzymatic transformations (Bergquist et al. 2020). Therefore, cofactors must be regenerated and recycled to improve the CFPS efficiency through a redesign of metabolic pathways, considering the economic feasibility of CFPS (Ullah et al. 2023). In this review, we discuss cofactor recycling strategies particularly relevant to the generation of secondary metabolites. This includes ATP, coenzyme A, and redox cofactors (iron sulfur clusters, NADPH, and flavins). Taken together, cofactor regeneration considerations are necessary to advance the characterization of secondary metabolites with CFPS.
Adenosine 5’-triphosphate (ATP) regeneration
ATP-related functions include the synthesis of dNTP, aminoacyl-tRNA charging, and regeneration of GTP in the peptide bonding process (Jewett and Swartz 2004). In the biosynthesis of many classes of natural products, ATP is used as a phosphorylating agent. YcaO enzymes are widely studied ones to catalyze phosphorylation of peptide backbone amide bonds in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs) (Franz et al. 2017). YcaO-catalyzed reactions have been found in the formation of azolines (Burkhart et al. 2017), amidines (Travin et al. 2018), and macrolactamidines (Franz et al. 2017). In the biosynthesis of nonribosomal peptides, adenylation domains catalyze the selective incorporation of acyl building blocks. Through activating a specific acyl substrate with ATP, the domain forms an acyl adenylate intermediate and then transfers the acyl group onto the carrier protein domain (Miyanaga et al. 2022). Therefore, the ATP availability will obviously limit the enzyme-catalyzed biotransformation. Whitesides first reported a series of useful ATP-recycling reactions and methods in the 1970s and 1980s (Crans and Whitesides 1983; Pollak et al. 1977). Three enzymatic methods are the most popular: phosphoenolpyruvate (PEP) catalyzed by pyruvate kinase; acetylphosphate coupled with acetate kinase; and polyphosphate coupled with polyphosphate kinase (PPK) (Zhao and van der Donk 2003).
Acetate kinase/acetyl phosphate
ATP is synthesized and recycled from diphosphate (ADP) and/or monophosphate (AMP) through an auxiliary enzymatic reaction (acetate kinase) using acetyl phosphate as the phosphate donor (Fig. 2a). Lee and colleagues evaluated ATP regeneration with acetyl phosphate and investigated the production of sugar nucleotides in E. coli. Their initial attempts using endogenous acetate kinase-catalyzed ATP recycling showed a positive result, which is also economically attractive since acetate kinase is the most abundant enzyme in E. coli, and acetyl phosphate is a cheaper substrate (Lee et al. 2002). The subsequent findings reported that overexpression of the acetate kinase activity improved the yield (Lee et al. 2010). In another study, Easton and coworkers developed rapid regeneration of ATP from AMP with acetyl kinase and applied this to both polymerase chain reaction (PCR) to produce DNA and cell-free protein synthesis of mRNA (Alissandratos et al. 2016). They reported the conversion from dNMP to dNTP in the presence of acetyl phosphate, ATP recycling, and endogenous kinases. This work provides an alternative strategy in economics and is environmentally friendly since it overcomes the low dNTP yield and the usage of toxic chemical solvents in current chemical production (Bao and Ryu 2007).

Mechanisms of different ATP regeneration systems. a, ATP regeneration using acetyl phosphate and acetate kinase (adapted from Bartzoka et al. 2022); b, glycolysis pathway and ATP regeneration using glucose-6-phosphate, phosphoenolpyruvate, and pyruvate as the energy sources (adapted from Kim and Kim 2009; Yan et al. 2014); c, ATP regeneration from AMP and poly(P) by the PAP-ADK system (adapted from Resnick and Zehnder 2000); d, ATP regeneration from AMP by the PAP-PPK system combined with the acetyl-CoA synthesis. ADK, adenylate kinase (adapted from Zhao and van der Donk 2003); AK, acetate kinase; PAP, polyphosphate AMP phosphotransferase; PPK, polyphosphate kinase; PPase, inorganic pyrophosphatase
Pyruvate kinase/phosphoenol pyruvate and glycolytic intermediates
One of the most widely used ATP recycling systems is the PANOx system, first developed by Swartz. The PANOx system uses phosphoenolpyruvate (PEP) to drive the regeneration of ATP (Fig. 2b) (Swartz 2006). Drawbacks to this recycling method include the short reaction duration (~ 60 min) of ATP bursts, which causes the accumulation of inhibitory phosphates which is subsequently inhibitory to cell-free protein synthesis (Kim and Swartz 2001). To address these limitations, new ATP regeneration approaches focus on glycolytic intermediates such as pyruvate and glucose-6-phosphate (G6P) rather than PEP as energy sources (Fig. 2b). G6P is the first glycolysis intermediate and can be effectively used for ATP regeneration (Ullah et al. 2023). Kim and Swartz found that G6P had a much lower cost and a higher potential for ATP generation than PEP (Kim and Swartz 2001). They reported a high yield result of protein synthesis by using G6P as a secondary energy source compared to PEP by optimizing the pH of the reaction mixture. G6P can prolong the reaction period, which results in more ATP being readily available. The successful usage of G6P in cell free reactions indicates that glucose is also available to regenerate ATP (Calhoun and Swartz 2005). Pyruvate is the end-product of glycolysis and several studies have demonstrated that it can be used for ATP regeneration (Fig. 2b) (Kim and Swartz 1999, 2001; Ullah et al. 2015). Kim and colleagues introduced pyruvate oxidase into the reaction which catalyzed the condensation of pyruvate and inorganic phosphate and produced acetyl phosphate. Endogenous acetate kinase then converted the acetyl phosphate to acetate and catalyzed the ATP regeneration. This pyruvate oxidase-dependent system was able to extend the duration of protein synthesis and prevent inorganic phosphate accumulation as well (Kim and Swartz 1999). Additionally, the usage of pyruvate significantly reduces the energy cost compared to the conventional energy source (PEP). However, there are two limitations of this method to be easily scaled up. Pyruvate oxidase is an exogenous enzyme from Lactobacillus or Pediococcus sp. and is not present in E. coli cell extracts. Synthetic biology can further optimize by remove the necessity to add exogenous pyruvate oxidase by cloning the encoding gene into a plasmid vector and heterologously expressing it in E. coli. Another limitation is the requirement of oxygen utilized by pyruvate oxidase for the conversion from pyruvate into acetyl phosphate (Kim and Swartz 2001; Kim and Kim 2009). Kim and Swartz bypassed these limitations by adding cofactors, NAD, and CoA. In this process, pyruvate dehydrogenase converts pyruvate to acetyl-CoA after the addition of NAD and CoA. Acetyl-CoA is subsequently converted to acetylphosphate, thus ATP regeneration is stimulated when acetylphosphate produces acetate (Kim and Swartz 2001). The utilization of the first intermediate (G6P) and the downstream products (PEP and pyruvate) in the glycolysis pathway also inspired the examination of other intermediates. There have been several studies that reported the successful results of the glycolytic intermediates used as efficient energy sources such as fructose-1,6-bisphosphate and 3-phosphoglycerate (Kim et al. 2007; Sitaraman et al. 2004).
Polyphosphate kinase/polyphosphate
The use of inorganic polyphosphate (poly(P)) to regenerate ATP has attracted significant attention because of its low cost, high stability, and serve as an attractive phosphoryl donor (Iwamoto et al. 2007). An E. coli recombinant producing Thermus polyphosphate kinase (PPK) has been reported to regenerate ATP when treated with exogenous polyphosphate and ADP (Fig. 2c). The E. coli was also heat-treated to allow the membrane penetration of ATP and poly(P) into the cell. This system was applied to generate fructose 1,6-diphosphate (FDP) and further tested and optimized by employing the production of glycerol 3-phosphate (G3P) from glycerol (Iwamoto et al. 2007; Restiawaty et al. 2011). In another study, Resnick and Zehnder designed an ATP regeneration system using poly(P), polyphosphate AMP phosphotransferase (PAP), and polyphosphate-independent adenylate kinase (ADK). PAP and ADK are from Acinetobacter johnsonii 210A which harbors GC content and thus poses difficultly cloning, which could be mitigated by codon replaced via gene synthesis. In this method, polyphosphate AMP phosphotransferase catalyzes the conversion from AMP to ADP and ADK regenerates ATP and AMP from ADP molecules. The system was also used to synthesize glucose-6-phosphate with hexokinase (Resnick and Zehnder 2000). Similarly, Kameda and colleagues developed a method to regenerate ATP from AMP and poly(P) catalyzed by PAP and a recombinant PPK (Fig. 2d). This method also combined with acetyl-CoA synthesis using acetyl-CoA synthase and supplemented with inorganic pyrophosphatase (PPase) which minimized the inhibitory effect of PPi and drive the regeneration system. This strategy provides a higher total turnover number for ATP and has the advantage of regenerating GTP from GMP compared with the PAP-ADK system of Resnick and Zehnder (Kameda et al. 2001; Resnick and Zehnder 2000).
Future prospects for ATP recycling: histidine kinase
Recently, a novel mechanism for ATP synthesis was uncovered by Jiang and coworkers from a bacterial histidine kinase domain HK853. The soluble domain of HK853 was shown to work effectively in vitro to generate ATP under mild conditions via recycling of ADP utilizing a substrate such as phosphorylated amino acids or phosphorylated glycerol. While it has not been explored in cell free systems, this could be a valuable tool to add to the toolkit for improved ATP recycling such that it is not such a limitation in cell free systems (Ji et al. 2024).
Coenzyme A (CoA)
Coenzyme A (CoA) is a ubiquitous essential cofactor in all domains of life and serves as an acyl group carrier and carbonyl activating group. It plays an important role in a variety of metabolic transformations, including the biosynthesis and catabolism of fatty acid, tricarboxylic acid cycle, and the biosynthesis of polyketides and nonribosomal peptides (Begley et al. 2001; Leonardi et al. 2005). As a mandatory cofactor, CoA is utilized by approximately 4% of enzymes, and it is involved in more than 100 different intermediate metabolic reactions (Jackowski 1996; Lee and Chen 1982). It is of particular importance as it serves as an important key posttranslational modification for the phosphopanetheinylation necessary for polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs) as well as the primary building blocks (e.g. acetyl-CoA, propionyl-CoA, malonyl-CoA, methylmalonyl-CoA) for polyketide synthases (Crosby and Crump 2012). CoA-dependent anabolic routes normally have three steps, including activating low energetic substrates and yielding an acyl-CoA derivative, modifying the acyl-CoA derivative through enzymatic steps, and releasing the target product by removing CoA (Ducrot et al. 2024).
CoA recycling in the butanol pathway
As a key example of the utility of CoA recycling, Krutsakorn and co-workers constructed the butanol pathway using 16 enzymes (Fig. 3a). The pathway first produced acetaldehyde from glucose through glycolysis and pyruvate decarboxylation, followed by CoA-acylating aldehyde dehydrogenase converting acetaldehyde into acetyl-CoA. Acetyl-CoA acts as the first intermediate for butanol synthesis, then two molecules of acetyl-CoA produce acetoacetyl-CoA by condensation. Subsequent reduction and dehydration were catalyzed by specific enzymes, which converted acetoacetyl-CoA into crotonyl-CoA and CoA was finally removed by CoA-acylating aldehyde dehydrogenase to yield crotonaldehyde, which is further reduced to the terminal alcohol (Krutsakorn et al. 2013). In another case, Karim and Jewett constructed a similar butanol biosynthetic pathway involving CoA intermediates and achieved rapid prototyping by accelerating design-build-test cycles (Fig. 3b). However, they produced precursor acetyl-CoA by using endogenous glycolytic enzymes and pyruvate dehydrogenase in the E. coli cell extracts; thus, the activation module is catalyzed by the pyruvate carboxylase rather than CoA-acylating aldehyde dehydrogenase reported by Krutsakorn et al. Crotonyl-CoA is also further reduced to butyryl-CoA, and CoA-deacetylating aldehyde reductase is subsequently removed CoA to generate butyraldehyde that is finally reduced by an alcohol dehydrogenase (Karim and Jewett 2016). These two pathways showed similar total turnover number of CoA (TTNCoA) with a value of 38 for Karim and Jewett and 35 for Krutsakorn, but almost six-fold titer of Karim and Jewett (20 mM) than 3.5 mM of Krutsakorn (Karim and Jewett 2016; Krutsakorn et al. 2013).

Schematic of CoA recycling in different pathways. a, the cell free pathway of n-butanol from glucose (adapted from Krutsakorn et al. 2013); b, the constructed biosynthetic pathway of n-butanol. Acetyl-CoA is produced from glycolysis pathway in E. coli, then utilized by CoA-dependent pathway in C. acetobutylicum to produce n-butanol (adapted from Karim and Jewett 2016); c, rosmarinic acid biosynthesis pathway with CoA and ATP regeneration (adapted from Yan et al. 2019); d, biosynthetic pathway for conversion of glucose to monoterpenes. PK, pyruvate kinase; PDC, pyruvate decarboxylase (adapted from Korman et al. 2017); ADDH, CoA-acylating aldehyde dehydrogenase; ACC, acetyl-CoA acetyltransferase; HBD, hydroxybutyryl-CoA dehydrogenase; HPD, 3-hydroxypropionyl-CoA dehydratase; NFO, NADH-dependent flavinoxidoreductase; HAD, 3-hydroxyacyl-CoA dehydrogenase; PDH, pyruvate dehydrogenase; AtoB, thiolase; CRT, crotonase; TER, butyryl-CoA dehydrogenase; ADHE, bifunctional acetaldehyde/alcohol dehydrogenase; 4CL, 4-coumarate: coenzyme A ligase; RAS, rosmarinic acid synthase; PPK, polyphosphate kinase; HMGS, hydroxymethylglutaryl-CoA synthase; HMGR, hydroxymethylglutaryl-CoA reductase; MVK, mevalonate-5-kinase; PMVK, phosphomevalonate kinase; MDC, mevalonate pyrophosphate decarboxylase; IDI, isopentenyl pyrophosphate isomerase; FPPS-S82F, farnesyl-pyrophosphate synthase-S82F mutant
CoA recycling in the production of rosmarinic acid
Rosmarinic acid is a hydroxycinnamic acid ester that is within an important class of phenolic natural products and has promising anti-carcinogenic and anti-tumorigenic properties (Bloch and Schmidt-Dannert 2014). The biosynthetic pathway of rosmarinic acid was established and engineered in E. coli, however, the yield was very low (Bloch and Schmidt-Dannert 2014; Jiang et al. 2016; Petersen et al. 2009). Yan and colleagues designed a CoA and ATP regeneration system for rosmarinic acid production through a four-enzyme cascade with high efficiency (Fig. 3c). The activation module is driven by 4-coumarate: coenzyme A ligase which catalyzed the conversion from caffeic acid to caffeoyl-CoA. 3,4-Dihydroxyphenyllactic acid was then introduced to condense with caffeoyl-CoA by rosmarinic acid synthase, producing rosmarinic acid and releasing CoA for recycling. ATP regeneration coupled with the formation of caffeoyl-CoA by adding polyphosphate kinases. The reaction conditions were also optimized to achieve a significantly high TTN for CoA and ATP, which were 444.5 and 820.6, respectively (Yan et al. 2019).
CoA recycling in terpene biosynthesis
Korman and colleagues designed a platform to produce monoterpenes from glucose using up to 27 enzymes (Fig. 3d). These enzymes were purified individually and mixed in this system, supplementing substrate and cofactors for cell free production. Glucose was converted into pyruvate through the glycolysis pathway, followed by pyruvate dehydrogenase mediating the pyruvate activation and producing acetyl-CoA. The acetyl-CoA was modified through the mevalonate pathway to yield the target monoterpenes. The pathway started from the condensation of two molecules of acetyl-CoA and produced acetoacetyl-CoA. HMG-CoA synthases then catalyzed the generation of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA). The CoA was removed by HMG-CoA reductase and produced mevalonate. Following the phosphorylation and rearranging of mevalonate, the generated isoprenoid precursors are ultimately converted to terpenes by condensation and cyclization. The platform produces various target monoterpenes by using the corresponding terpene synthase enzyme. The yield of monoterpene was in the range of 88–117 mM and TTNCoA reached 357 (Korman et al. 2017).
Redox cofactors
Oxidoreductases are widespread groups of biocatalysts, providing high activity and exquisite stereoselectivity in the synthetic strategy of pharmaceuticals, agrochemicals and food ingredients (Zheng and Xu 2011). They require redox cofactors such as NADPH, flavin, and iron-sulfur clusters to transfer energy in the form of reducing equivalents and keep redox homeostasis; thus, the regeneration of redox cofactors in CFPS is crucial (Chen et al. 2014). These cofactors’ recycling has already been further applied in the biotechnological production of valuable chemicals and synthetic biology (Shi et al. 2018; Wang et al. 2017; Weckbecker et al. 2010). A variety of strategies have been designed to optimize the yield of metabolites as well as support dynamic homeostasis between different redox states of cofactors (Partipilo et al. 2021).
Iron-sulfur clusters
Iron-sulfur clusters (Fe-S) serve as important prosthetic groups in many redox-active proteins and have been found in all types of organisms (Evans et al. 1966). One example that requires iron-sulfur cluster is radical S-adenosyl-l-methionine (SAM) enzymes, in which the iron-sulfur cluster coordinates their three cysteine residues, showing oxygen sensitivity in CFPS (Benjdia et al. 2017a, b). The radical SAM enzymes mediate key transformations in a variety of RiPPs pathways including thiopeptides (Kelly et al. 2009), epipeptides (Benjdia et al. 2017a, b), and bottromycins (Huo et al. 2012). Iron-sulfur clusters play a role in transporting electrons and are involved in numerous enzymatic reactions in metabolic and respiratory networks, radical chain reactions, DNA synthesis, protein translation, RNA modifications, and regulatory sensing (Lill and Freibert 2020). There are multiple Fe-S stoichiometries and coordination in which [2Fe-2S] and [4Fe-4S] types are the most common examples, and the [3Fe-4S] type is rarer (Beinert et al. 1997). Three helper protein systems have been developed for Fe/S protein assembly. The NIF (nitrogen fixation) system involved in Fe-S cluster assembly required nitrogenase to assimilate N2 (Jacobson et al. 1989). The ISC (iron-sulfur cluster) and SUF (mobilization of sulfur) systems are the more general ones for a variety of Fe-S protein assembly (Braymer et al. 2021; Zheng et al. 1998), while the SUF system is more O2-tolerant and resistant to iron starvation and oxidative stress (Boyd et al. 2014; Dai and Outten 2012). Swartz group first reported high-yield production of mature [2Fe-2S] Fd in a cell-free system. They utilized ISC helper proteins for production, but mature [2Fe-2S] Fd assembly was not facilitated by proteins probably because the ISC expression and activity were limited under aerobic conditions (Boyer et al. 2006). McGlynn and Fujishima developed a cell free mature [4Fe-4S] protein synthesis system using the SUF helper protein system and overcame the O2 lability of [4Fe-4S] by introducing an O2-scavenging cascade (Fig. 4). The SUF system can be dissected into three distinct steps, (i) S2− extraction from cysteine by SufS-SufE pair, (ii), the assembly of the [4Fe-4S] cluster on SufBC2D complex, (iii), SufA transfer [4Fe-4S] cluster to recipient proteins. The SUF subunits in the SUF system and enzymes in the O2-scavenging system were individually purified, then reconstituted the SUF system stepwise in vitro and test the O2-scavenging system function in the PUREfrex system. These systems were added into the PUREfrex system as a one-pot, two-step reaction which was examined by using enzyme aconitase and thermophilic ferredoxin and yielded holo-aconitase of ∼0.15 mg/mL (Wang et al. 2023).

Schematic diagram of cell free mature [4Fe-4S] protein synthesis system. In vitro translation of mRNA was conducted with formate dehydrogenase (FDH), flavin reductase (FRE), and catalase (CAT). SUF pathway incorporates with [4Fe-4S] cluster to form the matured protein as the second step. Figure was adapted from (Wang et al. 2023)
Nicotinamide adenine dinucleotide phosphate (NADP(H))
NADP+ and NADPH are generally distributed redox cofactors, playing a major role in anabolism (Fukuzumi et al. 2018; Kim and Gadd 2008) and are necessary for many reductive steps in the biosynthesis of secondary metabolites such as polyketides and terpenes (Korman et al. 2017; Metsa-Ketela et al. 2013; Ng et al. 2015; Piasecki et al. 2011). It is important to note that cell-free systems must have NADP+/NADPH balance as part of their pathway design at the start of the cycle (Taniguchi et al. 2017; Zhang 2015). Therefore, an appropriate redox cofactor regeneration system is normally coupled with the reactions for chemical manufacturing to avoid stoichiometric consumption of cofactor and keep continuous utilization of substrate (Shi et al. 2018; Suryatin Alim et al. 2023). To date, the NAD(P)H regeneration systems include chemical, electrochemical, photochemical, and enzymatic methods (Immanuel et al. 2020; Shi et al. 2006; Wu et al. 2013). Enzymatic methods are most widely used due to their efficiency, reliability, and reproducibility and provide a high TTN value (Ullah et al. 2023). The most frequently used classification of enzymatic approaches uses another substrate and its respective enzyme. Common tool enzymes include alcohol dehydrogenase (ADH) (Jia et al. 2022), formate dehydrogenase (FDH) (Bozic et al. 2010), glucose dehydrogenase (GDH) (Xu et al. 2007), and lactate dehydrogenase (LDH) (Lee and Whitesides 1985) (Fig. 5a). These four representative enzymes drive the reaction to generate acetone, carbon dioxide, gluconate and pyruvate coupled with NAD(P)H regeneration. Additionally, NAD(P)H can be regenerated through cascade enzymes. An example is the 12-enzyme system developed by Wang and co-workers to produce NAD(P)H at a high yield of 11.4 mol NADPH per cellobiose (Wang et al. 2011).

a, typical substrate-coupled approaches to regenerate NAD(P)H, ADH, alcohol dehydrogenase; FDH, formate dehydrogenase; GDH, glucose dehydrogenase; LDH, lactate dehydrogenase (adapted from Jia et al. 2022; Shi et al. 2018); b, chemoenzymatic approach with [Cp*Rh(bpy)(H2O)]2+ to regenerate FADH2 (adapted from Hollmann et al. 2003); c, electrochemical approach with artificial electrode to regenerate FADH2 (adapted from Hollmann et al. 2005); d, enzymatic approach using formate dehydrogenase (FDH) and flavin reductase to regenerate FADH2 (adapted from (Hofstetter et al. 2004)
Flavin adenine dinucleotide (FAD)
Flavin adenine dinucleotide and dihydroflavine adenine dinucleotide (FAD/FADH2) serve as electron shuttles and are involved in many biocatalytic reactions including D-Amino acid oxidase (DAAO) catalyzed the oxidative deamination of D-amino acids (Pollegioni et al. 2008), succinate dehydrogenase (SDH) catalyzed succinate oxidation (Kim and Winge 2013), and glucose dehydrogenase (GDH) catalyzed glucose oxidation (Tsujimura 2019). Hollmann’s group first reported a direct regeneration method of flavin-dependent monooxygenases using the artificial organometallic complex [Cp*Rh(bpy)(H2O)]2+ (Fig. 5b). The FADH2-dependent monooxygenase StyA can be regenerated directly through redox catalysts [Cp*Rh(bpy)(H2O)]2+ which converts FAD into FADH2. By utilizing this cell-free chemoenzymatic approach, enantiopure epoxides can be synthesized asymmetrically (Hollmann et al. 2003). Complex [Cp*Rh(bpy)(H2O)]2+ has also been applied in FADH2-dependent halogenation (Unversucht et al. 2005). An electrochemical FAD/FADH2 regeneration system has been developed to investigate the biocatalytic asymmetric epoxidation by means of an artificial electrode (Fig. 5c). Other examples include the production of fumarate and xylose (Jin et al. 2005) and d-amino acid oxidase (d-AAO) catalyzed resolution of a methionine racemate (Kohlmann et al. 2009). However, both chemical and electrochemical lead to the inactivation of FADH2-dependent enzymes, thus the regeneration rate and TTN are low (Poizat et al. 2010). For enzymatic regeneration, additional NAD(P)H is required to serve as indirect reducing agent and formate dehydrogenase (FDH) is introduced to regenerate NAD(P)H to minimize the cost (Fig. 5d). Hofstetter et al. demonstrated an enzymatic regeneration system for asymmetric cell-free epoxidation with the FADH2-dependent monooxygenase StyA and flavin reductase StyB. Their results showed a high TTN value and average reaction rates of StyA in the range of 1800–2800 and 3–4.3 min−1, respectively (Hofstetter et al. 2004). In another case, Frese and Sewald developed an enzymatic halogenation system to produce 7-bromo-tryptophan utilizing FADH2-dependent L-tryptophan-7-halogenase RebH, flavin reductase PrnF and ADH (Frese and Sewald 2015). For FAD-dependent oxidative deamination, Hou and colleagues constructed a FAD/FADH2 regeneration system including FADH2-dependent halogenase and FAD-dependent L-amino acid deaminase (L-AAD) to coproduce 7-chloro-tryptophan and indole pyruvic acid (Hou et al. 2023). McGlynn and Fujishima used an O2-scavenging cascade which has a similar mechanism to remove O2 and produce [4Fe-4S] proteins. Formate dehydrogenase (FDH) was introduced to oxidize formate and was responsible for NADH regeneration. Flavin reductase transferred reducing equivalents and converted FAD to FADH2 by consuming NADH. Subsequently, catalase coupled with bifunctional flavin reductase removed O2 from the reaction and provided FADH2 for [4Fe-4S] protein maturation (Wang et al. 2023).
Conclusions
CFPS system has advantages to accelerate rapid prototyping and overcoming cellular toxicity. It has been widely applied in the high-throughput production of high-value products (Liu et al. 2019), biosensing (Silverman et al. 2020), integration of orthogonal genetic codes (Chemla et al. 2015), and the assembly of bacteriophages (Shin et al. 2012). With the substantial development of cell free production, low-cost and stable enzymes and cofactor balance and regeneration systems are still the main concern. In this review, we focus on biotechnologically relevant cofactor regeneration systems in CFPS, specifically cofactors found in complex metabolites. The strategies discussed, including ATP regeneration, coenzyme A recycling, and redox cofactor management, represent significant advancements in constructing of cofactor recycling systems and enhancing the production of secondary metabolites. However, there are still challenges for cofactor regeneration systems to be applied in practical production, such as precise dynamic regulation of cofactors, the maintenance of stable cellular redox state, etc. Therefore, the cofactor regeneration technology is expected to be further improved, expanding its application in the field of biocatalysis and biomanufacturing applications. A few examples of success in CFPS systems, where attention to cofactor optimization has led to notable improvements in productivity, would be useful here. Recent advances showed that optimizing the concentration of energy source PEP, and magnesium in CFPS improves the expression of nonribosomal peptide synthetases (Dinglasan et al. 2023). In a developed Streptomyces cell-free system, Moore and coworkers investigated most cofactors to identify any nonessential components and optimized the secondary energy source 3-phosphoglyceric acid (3-PGA) and NTP level to have a 52% increase of the protein production (Moore et al. 2021). Many of the pathways described in this review could be integrated into lysates via strategies such as CRISPR/Cas9 so that the enzyme does not need to be added after the fact, sometimes with purification. DNA synthesis and codon optimization also make some of these genes from rare or high GC organisms more accessible. Taken together, new cell free systems, including cofactor balance and lysate composition can be further optimized for utility for secondary metabolite generation which would expand the utility of using these systems for discovery and biosynthetic design prototyping.
Data availability
No datasets were generated or analyzed during the current study.
References
Alissandratos A, Caron K, Loan TD, Hennessy JE, Easton CJ (2016) ATP recycling with cell lysate for enzyme-catalyzed chemical synthesis, protein expression and PCR. ACS Chem Biol 11(12):3289–3293. https://doi.org/10.1021/acschembio.6b00838
Bartzoka N, Loan TD, Onagi H, Alissandratos A (2022) A simple recombinant E. coli cell lysate-based biocatalyst for ATP-dependent multi-step reactions. Methods Mol Biol 2487:297–315. https://doi.org/10.1007/978-1-0716-2269-8_18
Bao J, Ryu DD (2007) Total biosynthesis of deoxynucleoside triphosphates using deoxynucleoside monophosphate kinases for PCR application. Biotechnol Bioeng 98(1):1–11. https://doi.org/10.1002/bit.21498
Begley TP, Kinsland C, Strauss E (2001) The biosynthesis of coenzyme A in bacteria. Vitam Horm 61:157–171. https://doi.org/10.1016/s0083-6729(01)61005-7
Beinert H, Holm RH, Münck E (1997) Iron-sulfur clusters: nature’s modular multipurpose structures. Science 277(5326):653–659. https://doi.org/10.1126/science.277.5326.653
Benjdia A, Balty C, Berteau O (2017a) Radical SAM enzymes in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). Front Chem 5:87. https://doi.org/10.3389/fchem.2017.00087
Benjdia A, Guillot A, Ruffie P, Leprince J, Berteau O (2017b) Post-translational modification of ribosomally synthesized peptides by a radical SAM epimerase in Bacillus subtilis. Nat Chem 9(7):698–707. https://doi.org/10.1038/nchem.2714
Bergquist PL, Siddiqui S, Sunna A (2020) Cell-free biocatalysis for the production of platform chemicals [Review]. Front Energy Res 8. https://doi.org/10.3389/fenrg.2020.00193
Bloch SE, Schmidt-Dannert C (2014) Construction of a chimeric biosynthetic pathway for the de novo biosynthesis of rosmarinic acid in Escherichia coli. ChemBioChem 15(16):2393–2401. https://doi.org/10.1002/cbic.201402275
Boyd ES, Thomas KM, Dai Y, Boyd JM, Outten FW (2014) Interplay between oxygen and Fe–S cluster biogenesis: insights from the suf pathway. Biochemistry 53(37):5834–5847. https://doi.org/10.1021/bi500488r
Boyer ME, Wang CW, Swartz JR (2006) Simultaneous expression and maturation of the iron-sulfur protein ferredoxin in a cell-free system. Biotechnol Bioeng 94(1):128–138. https://doi.org/10.1002/bit.20830
Bozic M, Pricelius S, Guebitz GM, Kokol V (2010) Enzymatic reduction of complex redox dyes using NADH-dependent reductase from Bacillus subtilis coupled with cofactor regeneration. Appl Microbiol Biotechnol 85(3):563–571. https://doi.org/10.1007/s00253-009-2164-8
Braymer JJ, Freibert SA, Rakwalska-Bange M, Lill R (2021) Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim Biophys Acta Mol Cell Res 1868(1):118863. https://doi.org/10.1016/j.bbamcr.2020.118863
Buchner E (1897) Alkoholische Gärung ohne Hefezellen. Ber Dtsch Chem Ges 30(1):1110–1113. https://doi.org/10.1002/cber.189703001215
Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA (2017) YcaO-dependent posttranslational amide activation: biosynthesis, structure, and function. Chem Rev 117(8):5389–5456. https://doi.org/10.1021/acs.chemrev.6b00623
Calhoun KA, Swartz JR (2005) Energizing cell-free protein synthesis with glucose metabolism. Biotechnol Bioeng 90(5):606–613. https://doi.org/10.1002/bit.20449
Carlson ED, Gan R, Hodgman CE, Jewett MC (2012) Cell-free protein synthesis: applications come of age. Biotechnol Adv 30(5):1185–1194. https://doi.org/10.1016/j.biotechadv.2011.09.016
Chemla Y, Ozer E, Schlesinger O, Noireaux V, Alfonta L (2015) Genetically expanded cell-free protein synthesis using endogenous pyrrolysyl orthogonal translation system. Biotechnol Bioeng 112(8):1663–1672. https://doi.org/10.1002/bit.25587
Chen X, Li S, Liu L (2014) Engineering redox balance through cofactor systems. Trends Biotechnol 32(6):337–343. https://doi.org/10.1016/j.tibtech.2014.04.003
Crans DC, Whitesides GM (1983) A convenient synthesis of disodium acetyl phosphate for use in in situ ATP cofactor regeneration. J Org Chem 48(18):3130–3132. https://doi.org/10.1021/jo00166a048
Crosby J, Crump MP (2012) The structural role of the carrier protein–active controller or passive carrier. Nat Prod Rep 29(10):1111–1137. https://doi.org/10.1039/c2np20062g
Dai Y, Outten FW (2012) The E. coli SufS–SufE sulfur transfer system is more resistant to oxidative stress than IscS–IscU. FEBS Lett 586(22):4016–4022. https://doi.org/10.1016/j.febslet.2012.10.001
Dinglasan JLN, Sword TT, Barker JW, Doktycz MJ, Bailey CB (2023) Investigating and optimizing the lysate-based expression of nonribosomal peptide synthetases using a reporter system. ACS Synth Biol 12(5):1447–1460. https://doi.org/10.1021/acssynbio.2c00658
Ducrot L, Lopez IL, Orrego AH, Lopez-Gallego F (2024) Coenzyme A thioester intermediates as platform molecules in cell-free chemical biomanufacturing. ChemBioChem 25(2):e202300673. https://doi.org/10.1002/cbic.202300673
Evans MC, Buchanan BB, Arnon DI (1966) A new ferredoxin-dependent carbon reduction cycle in a photosynthetic bacterium. Proc Natl Acad Sci U S A 55(4):928–934. https://doi.org/10.1073/pnas.55.4.928
Franz L, Adam S, Santos-Aberturas J, Truman AW, Koehnke J (2017) Macroamidine formation in bottromycins is catalyzed by a divergent YcaO enzyme. J Am Chem Soc 139(50):18158–18161. https://doi.org/10.1021/jacs.7b09898
Frese M, Sewald N (2015) Enzymatic halogenation of tryptophan on a gram scale. Angew Chem Int Ed Engl 54(1):298–301. https://doi.org/10.1002/anie.201408561
Fukuzumi S, Lee YM, Nam W (2018) Artificial photosynthesis for production of ATP, NAD (P) H, and hydrogen peroxide. ChemPhotoChem 2(3):121–135. https://doi.org/10.1002/cptc.201700146
Gupta MD, Flaskamp Y, Roentgen R, Juergens H, Armero-Gimenez J, Albrecht F, Hemmerich J, Arfi ZA, Neuser J, Spiegel H, Schillberg S, Yeliseev A, Song L, Qiu J, Williams C, Finnern R (2023) Scaling eukaryotic cell-free protein synthesis achieved with the versatile and high-yielding tobacco BY-2 cell lysate. Biotechnol Bioeng 120(10):2890–2906. https://doi.org/10.1002/bit.28461
Hodgman CE, Jewett MC (2012) Cell-free synthetic biology: thinking outside the cell. Metab Eng 14(3):261–269. https://doi.org/10.1016/j.ymben.2011.09.002
Hofstetter K, Lutz J, Lang I, Witholt B, Schmid A (2004) Coupling of biocatalytic asymmetric epoxidation with NADH regeneration in organic-aqueous emulsions. Angew Chem Int Ed Engl 43(16):2163–2166. https://doi.org/10.1002/anie.200353338
Hollmann F, Lin PC, Witholt B, Schmid A (2003) Stereospecific biocatalytic epoxidation: the first example of direct regeneration of a FAD-dependent monooxygenase for catalysis. J Am Chem Soc 125(27):8209–8217. https://doi.org/10.1021/ja034119u
Hollmann F, Hofstetter K, Habicher T, Hauer B, Schmid A (2005) Direct electrochemical regeneration of monooxygenase subunits for biocatalytic asymmetric epoxidation. J Am Chem Soc 127(18):6540–6541. https://doi.org/10.1021/ja050997b
Hou Y, Zhao W, Ding X, Zhang X, Li Z, Tan Z, Zhou J, Wang H, Jia S (2023) Co-production of 7-chloro-tryptophan and indole pyruvic acid based on an efficient FAD/FADH(2) regeneration system. Appl Microbiol Biotechnol 107(15):4873–4885. https://doi.org/10.1007/s00253-023-12619-9
Huo L, Rachid S, Stadler M, Wenzel SC, Muller R (2012) Synthetic biotechnology to study and engineer ribosomal bottromycin biosynthesis. Chem Biol 19(10):1278–1287. https://doi.org/10.1016/j.chembiol.2012.08.013
Immanuel S, Sivasubramanian R, Gul R, Dar MA (2020) Recent progress and perspectives on electrochemical regeneration of reduced nicotinamide adenine dinucleotide (NADH). Chem Asian J 15(24):4256–4270. https://doi.org/10.1002/asia.202001035
Iwamoto S, Motomura K, Shinoda Y, Urata M, Kato J, Takiguchi N, Ohtake H, Hirota R, Kuroda A (2007) Use of an Escherichia coli recombinant producing thermostable polyphosphate kinase as an ATP regenerator to produce fructose 1,6-diphosphate. Appl Environ Microbiol 73(17):5676–5678. https://doi.org/10.1128/AEM.00278-07
Jackowski S (1996) Biosynthesis of pantothenic acid and coenzyme A. In: Neidhardt FC, Curtiss R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (eds) Escherichia coli and salmonella typhimurium: cellular and molecular biology, 2nd edn. American Society for Microbiology, Washington, DC, pp 687–694. https://cir.nii.ac.jp/crid/1573387449106804096
Jacobson MR, Cash VL, Weiss MC, Laird NF, Newton WE, Dean DR (1989) Biochemical and genetic analysis of the nifUSVWZM cluster from Azotobacter vinelandii. Mol Gen Genet MGG 219(1):49–57. https://doi.org/10.1007/BF00261156
Jewett MC, Swartz JR (2004) Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol Bioeng 86(1):19–26. https://doi.org/10.1002/bit.20026
Ji S, Zhou Y, Chen J, Yang M, Li C, Liu M, Liu Y, Jiang L (2024) An ATP “Synthase” derived from a single structural domain of bacterial histidine kinase. Angew Chem Int Ed 63(13):e202318503. https://doi.org/10.1002/anie.202318503
Jia Q, Zheng YC, Li HP, Qian XL, Zhang ZJ, Xu JH (2022) Engineering isopropanol dehydrogenase for efficient regeneration of nicotinamide cofactors. Appl Environ Microbiol 88(9):e0034122. https://doi.org/10.1128/aem.00341-22
Jiang J, Bi H, Zhuang Y, Liu S, Liu T, Ma Y (2016) Engineered synthesis of rosmarinic acid in Escherichia coli resulting production of a new intermediate, caffeoyl-phenyllactate. Biotechnol Lett 38(1):81–88. https://doi.org/10.1007/s10529-015-1945-7
Jin JS, Ho SI, Hyun PD (2005) Electrochemical regeneration of FAD by catalytic electrode without electron mediator and biochemical reducing power. J Microbiol Biotechnol 15(2):281–286. https://koreascience.kr/article/JAKO200502636832890.page
Kameda A, Shiba T, Kawazoe Y, Satoh Y, Ihara Y, Munekata M, Ishige K, Noguchi T (2001) A novel ATP regeneration system using polyphosphate-AMP phosphotransferase and polyphosphate kinase. J Biosci Bioeng 91(6):557–563. https://doi.org/10.1263/jbb.91.557
Karim AS, Jewett MC (2016) A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab Eng 36:116–126. https://doi.org/10.1016/j.ymben.2016.03.002
Kelly WL, Pan L, Li C (2009) Thiostrepton biosynthesis: prototype for a new family of bacteriocins. J Am Chem Soc 131(12):4327–4334. https://doi.org/10.1021/ja807890a
Kim B, Gadd G (2008) Bacterial physiology and metabolism. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511790461. Accessed 15 May 2024
Kim HC, Kim DM (2009) Methods for energizing cell-free protein synthesis. J Biosci Bioeng 108(1):1–4. https://doi.org/10.1016/j.jbiosc.2009.02.007
Kim HJ, Winge DR (2013) Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim Biophys Acta 1827(5):627–636. https://doi.org/10.1016/j.bbabio.2013.01.012
Kim TW, Keum JW, Oh IS, Choi CY, Kim HC, Kim DM (2007) An economical and highly productive cell-free protein synthesis system utilizing fructose-1,6-bisphosphate as an energy source. J Biotechnol 130(4):389–393. https://doi.org/10.1016/j.jbiotec.2007.05.002
Kim DM, Swartz JR (1999) Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotechnol Bioeng 66(3):180–188. https://www.ncbi.nlm.nih.gov/pubmed/10577472
Kim DM, Swartz JR (2001) Regeneration of adenosine triphosphate from glycolytic intermediates for cell-free protein synthesis. Biotechnol Bioeng 74(4):309–316. https://www.ncbi.nlm.nih.gov/pubmed/11410855. Accessed 15 May 2024
Kohlmann C, Greiner L, Leitner W, Wandrey C, Lütz S (2009) Ionic liquids as performance additives for electroenzymatic syntheses. Chem–A Eur J 15(43):11692–11700. https://doi.org/10.1002/chem.200901046
Korman TP, Opgenorth PH, Bowie JU (2017) A synthetic biochemistry platform for cell free production of monoterpenes from glucose. Nat Commun 8:15526. https://doi.org/10.1038/ncomms15526
Krutsakorn B, Honda K, Ye X, Imagawa T, Bei X, Okano K, Ohtake H (2013) In vitro production of n-butanol from glucose. Metab Eng 20:84–91. https://doi.org/10.1016/j.ymben.2013.09.006
Lee C-Y, Chen AF (1982) Immobilized coenzymes and derivatives. In: Everse J, Anderson BM, You K-S (eds) The pyridine nucleotide coenzymes. Academic Press, New York, pp 189–224. https://doi.org/10.1016/B978-0-12-244750-1.50015-4
Lee LG, Whitesides GM (1985) Enzyme-catalyzed organic synthesis: a comparison of strategies for in situ regeneration of NAD from NADH. J Am Chem Soc 107(24):6999–7008. https://doi.org/10.1021/ja00310a043
Lee S-G, Lee J-O, Yi J-K, Kim B-G (2002) Production of cytidine 5′-monophosphate N-acetylneuraminic acid using recombinant Escherichia coli as a biocatalyst. Biotechnol Bioeng 80(5):516–524. https://doi.org/10.1002/bit.10398
Lee J-H, Chung S-W, Lee H-J, Jang K-S, Lee S-G, Kim B-G (2010) Optimization of the enzymatic one pot reaction for the synthesis of uridine 5′-diphosphogalactose. Bioprocess Biosyst Eng 33(1):71–78. https://doi.org/10.1007/s00449-009-0365-2
Leonardi R, Zhang YM, Rock CO, Jackowski S (2005) Coenzyme A: back in action. Prog Lipid Res 44(2–3):125–153. https://doi.org/10.1016/j.plipres.2005.04.001
Li H, Ding W, Zhang Q (2024) Discovery and engineering of ribosomally synthesized and post-translationally modified peptide (RiPP) natural products. RSC Chem Biol 5(2):90–108. https://doi.org/10.1039/d3cb00172e
Lill R, Freibert SA (2020) Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem 89:471–499. https://doi.org/10.1146/annurev-biochem-013118-111540
Liu W-Q, Zhang L, Chen M, Li J (2019) Cell-free protein synthesis: recent advances in bacterial extract sources and expanded applications. Biochem Eng J 141:182–189. https://doi.org/10.1016/j.bej.2018.10.023
Metsa-Ketela M, Oja T, Taguchi T, Okamoto S, Ichinose K (2013) Biosynthesis of pyranonaphthoquinone polyketides reveals diverse strategies for enzymatic carbon-carbon bond formation. Curr Opin Chem Biol 17(4):562–570. https://doi.org/10.1016/j.cbpa.2013.06.032
Miyanaga A, Kudo F, Eguchi T (2022) Recent advances in the structural analysis of adenylation domains in natural product biosynthesis. Curr Opin Chem Biol 71:102212. https://doi.org/10.1016/j.cbpa.2022.102212
Moore SJ, Lai HE, Chee SM, Toh M, Coode S, Chengan K, Capel P, Corre C, de Los Santos EL, Freemont PS (2021) A streptomyces Venezuelae cell-free toolkit for synthetic biology. ACS Synth Biol 10(2):402–411. https://doi.org/10.1021/acssynbio.0c00581
Ng CY, Farasat I, Maranas CD, Salis HM (2015) Rational design of a synthetic Entner-Doudoroff pathway for improved and controllable NADPH regeneration. Metab Eng 29:86–96. https://doi.org/10.1016/j.ymben.2015.03.001
Nirenberg MW, Matthaei JH (1961) The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc Natl Acad Sci U S A 47(10):1588-1602. https://doi.org/10.1073/pnas.47.10.1588
Partipilo M, Ewins EJ, Frallicciardi J, Robinson T, Poolman B, Slotboom DJ (2021) Minimal pathway for the regeneration of redox cofactors. JACS Au 1(12):2280–2293. https://doi.org/10.1021/jacsau.1c00406
Petersen M, Abdullah Y, Benner J, Eberle D, Gehlen K, Hucherig S, Janiak V, Kim KH, Sander M, Weitzel C, Wolters S (2009) Evolution of rosmarinic acid biosynthesis. Phytochemistry 70(15–16):1663–1679. https://doi.org/10.1016/j.phytochem.2009.05.010
Piasecki SK, Taylor CA, Detelich JF, Liu J, Zheng J, Komsoukaniants A, Siegel DR, Keatinge-Clay AT (2011) Employing modular polyketide synthase ketoreductases as biocatalysts in the preparative chemoenzymatic syntheses of diketide chiral building blocks. Chem Biol 18(10):1331–1340. https://doi.org/10.1016/j.chembiol.2011.07.021
Poizat M, Arends IW, Hollmann F (2010) On the nature of mutual inactivation between [Cp* Rh (bpy)(H2O)] 2+ and enzymes–analysis and potential remedies. J Mol Catal B Enzym 63(3–4):149–156. https://doi.org/10.1016/j.molcatb.2010.01.006
Pollak A, Baughn RL, Whitesides GM (1977) Large-scale enzymic synthesis with cofactor regeneration: glucose 6-phosphate. J Am Chem Soc 99(7):2366–2367. https://doi.org/10.1021/ja00449a071
Pollegioni L, Molla G, Sacchi S, Rosini E, Verga R, Pilone MS (2008) Properties and applications of microbial D-amino acid oxidases: current state and perspectives. Appl Microbiol Biotechnol 78(1):1–16. https://doi.org/10.1007/s00253-007-1282-4
Resnick SM, Zehnder AJ (2000) In vitro ATP regeneration from polyphosphate and AMP by polyphosphate:AMP phosphotransferase and adenylate kinase from Acinetobacter johnsonii 210A. Appl Environ Microbiol 66(5):2045–2051. https://doi.org/10.1128/AEM.66.5.2045-2051.2000
Restiawaty E, Iwasa Y, Maya S, Honda K, Omasa T, Hirota R, Kuroda A, Ohtake H (2011) Feasibility of thermophilic adenosine triphosphate-regeneration system using Thermus thermophilus polyphosphate kinase. Process Biochem 46(9):1747–1752. https://doi.org/10.1016/j.procbio.2011.05.021
Shi Q, Yang D, Jiang Z, Li J (2006) Visible-light photocatalytic regeneration of NADH using P-doped TiO2 nanoparticles. J Mol Catal B Enzym 43(1):44–48. https://doi.org/10.1016/j.molcatb.2006.06.005
Shi T, Han P, You C, Zhang YPJ (2018) An in vitro synthetic biology platform for emerging industrial biomanufacturing: Bottom-up pathway design. Synth Syst Biotechnol 3(3):186–195. https://doi.org/10.1016/j.synbio.2018.05.002
Shin J, Jardine P, Noireaux V (2012) Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth Biol 1(9):408–413. https://doi.org/10.1021/sb300049p
Silverman AD, Karim AS, Jewett MC (2020) Cell-free gene expression: an expanded repertoire of applications. Nat Rev Genet 21(3):151–170. https://doi.org/10.1038/s41576-019-0186-3
Sitaraman K, Esposito D, Klarmann G, Le Grice SF, Hartley JL, Chatterjee DK (2004) A novel cell-free protein synthesis system. J Biotechnol 110(3):257–263. https://doi.org/10.1016/j.jbiotec.2004.02.014
Suryatin Alim G, Suzuki T, Honda K (2023) Cell-Free Production and Regeneration of Cofactors. Adv Biochem Eng Biotechnol 186:29–49. https://doi.org/10.1007/10_2023_222
Swartz J (2006) Developing cell-free biology for industrial applications. J Ind Microbiol Biotechnol 33(7):476–485. https://doi.org/10.1007/s10295-006-0127-y
Sword TT, Abbas GSK, Bailey CB (2024) Cell-free protein synthesis for nonribosomal peptide synthetic biology [Review]. Front Nat Prod 3. https://doi.org/10.3389/fntpr.2024.1353362
Taniguchi H, Okano K, Honda K (2017) Modules for in vitro metabolic engineering: pathway assembly for bio-based production of value-added chemicals. Synth Syst Biotechnol 2(2):65–74. https://doi.org/10.1016/j.synbio.2017.06.002
Travin DY, Metelev M, Serebryakova M, Komarova ES, Osterman IA, Ghilarov D, Severinov K (2018) Biosynthesis of translation inhibitor klebsazolicin proceeds through heterocyclization and N-Terminal amidine formation catalyzed by a single YcaO enzyme. J Am Chem Soc 140(16):5625–5633. https://doi.org/10.1021/jacs.8b02277
Tsujimura S (2019) From fundamentals to applications of bioelectrocatalysis: bioelectrocatalytic reactions of FAD-dependent glucose dehydrogenase and bilirubin oxidase. Biosci Biotechnol Biochem 83(1):39–48. https://doi.org/10.1080/09168451.2018.1527209
Ullah MW, Ul-Islam M, Khan S, Kim Y, Park JK (2015) Innovative production of bio-cellulose using a cell-free system derived from a single cell line. Carbohydr Polym 132:286–294. https://doi.org/10.1016/j.carbpol.2015.06.037
Ullah MW, Manan S, Ul-Islam M, Khattak WA, Khan KA, Liu J, Yang G, Sun J (2023) Cell-free systems for biosynthesis: towards a sustainable and economical approach. Green Chem 25(13):4912–4940. https://doi.org/10.1039/D3GC00985H
Unversucht S, Hollmann F, Schmid A, van Pée KH (2005) FADH2-dependence of tryptophan 7-halogenase. Adv Synth Catal 347(7–8):1163–1167. https://doi.org/10.1002/adsc.200505029
Voloshin AM, Swartz JR (2005) Efficient and scalable method for scaling up cell free protein synthesis in batch mode. Biotechnol Bioeng 91(4):516–521. https://doi.org/10.1002/bit.20528
Wang Y, Huang W, Sathitsuksanoh N, Zhu Z, Zhang YH (2011) Biohydrogenation from biomass sugar mediated by in vitro synthetic enzymatic pathways. Chem Biol 18(3):372–380. https://doi.org/10.1016/j.chembiol.2010.12.019
Wang M, Chen B, Fang Y, Tan T (2017) Cofactor engineering for more efficient production of chemicals and biofuels. Biotechnol Adv 35(8):1032–1039. https://doi.org/10.1016/j.biotechadv.2017.09.008
Wang PH, Nishikawa S, McGlynn SE, Fujishima K (2023) One-pot de novo synthesis of [4Fe-4S] proteins using a recombinant SUF system under aerobic conditions. ACS Synth Biol 12(10):2887–2896. https://doi.org/10.1021/acssynbio.3c00155
Weckbecker A, Groger H, Hummel W (2010) Regeneration of nicotinamide coenzymes: principles and applications for the synthesis of chiral compounds. Adv Biochem Eng Biotechnol 120:195–242. https://doi.org/10.1007/10_2009_55
Wu H, Tian C, Song X, Liu C, Yang D, Jiang Z (2013) Methods for the regeneration of nicotinamide coenzymes. Green Chem 15(7):1773–1789. https://doi.org/10.1039/C3GC37129H
Xu Z, Jing K, Liu Y, Cen P (2007) High-level expression of recombinant glucose dehydrogenase and its application in NADPH regeneration. J Ind Microbiol Biotechnol 34(1):83–90. https://doi.org/10.1007/s10295-006-0168-2
Yan B, Ding Q, Ou L, Zou Z (2014) Production of glucose-6-phosphate by glucokinase coupled with an ATP regeneration system. World J Microbiol Biotechnol 30(3):1123–1128. https://doi.org/10.1007/s11274-013-1534-7
Yan Y, Jia P, Bai Y, Fan TP, Zheng X, Cai Y (2019) Production of rosmarinic acid with ATP and CoA double regenerating system. Enzyme Microb Technol 131:109392. https://doi.org/10.1016/j.enzmictec.2019.109392
Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, Gold DS, Heinsohn HG, Murray CJ (2011) Microscale to manufacturing scale-up of cell-free cytokine production–a new approach for shortening protein production development timelines. Biotechnol Bioeng 108(7):1570–1578. https://doi.org/10.1002/bit.23103
Zhang YH (2015) Production of biofuels and biochemicals by in vitro synthetic biosystems: Opportunities and challenges. Biotechnol Adv 33(7):1467–1483. https://doi.org/10.1016/j.biotechadv.2014.10.009
Zhao H, van der Donk WA (2003) Regeneration of cofactors for use in biocatalysis. Curr Opin Biotechnol 14(6):583–589. https://doi.org/10.1016/j.copbio.2003.09.007
Zhao S, Feng R, Gu Y, Han L, Cong X, Liu Y, Liu S, Shen Q, Huo L, Yan F (2024) Heterologous expression facilitates the discovery and characterization of marine microbial natural products. Eng Microbiol 4(2):100137. https://doi.org/10.1016/j.engmic.2023.100137
Zheng GW, Xu JH (2011) New opportunities for biocatalysis: driving the synthesis of chiral chemicals. Curr Opin Biotechnol 22(6):784–792. https://doi.org/10.1016/j.copbio.2011.07.002
Zheng L, Cash VL, Flint DH, Dean DR (1998) Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem 273(21):13264–13272. https://doi.org/10.1074/jbc.273.21.13264
Acknowledgements
This work was supported by the University of Sydney School of Chemistry, Faculty of Science. C.B.B. is a member of the University of Sydney Centre for Drug Discovery Innovation, the University of Sydney Infectious Diseases Institute, and the University of Sydney Nanoscience Institute.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
Y.Z. and C.B. wrote and conceptualized the manuscript. Y.Z. generated the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zou, Y., Bailey, C.B. Cofactor recycling strategies for secondary metabolite production in cell-free protein expression systems. Biophys Rev (2024). https://doi.org/10.1007/s12551-024-01234-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12551-024-01234-1