
Abstract
Somatic maintenance and cell survival rely on proper protein homeostasis to ensure reliable functions across the cell and to prevent proteome collapse. Maintaining protein folding and solubility is central to proteostasis and is coordinated by protein synthesis, chaperoning, and degradation capacities. An emerging aspect that influences proteostasis is the dynamic protein partitioning across different subcellular structures and compartments. Here, we review recent literature related to nucleocytoplasmic partitioning of proteins, nuclear and cytoplasmic quality control mechanisms, and their impact on the development of age-related diseases. We also highlight new points of entry to modulate spatially-regulated proteostatic mechanisms to delay aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
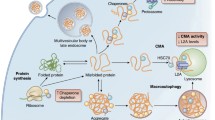
One of the key hallmarks of aging is the loss of protein homeostasis (Lopez-Otin et al. 2013; Moreno and Aldea 2020), which has global impact on cellular function, flexibility, resilience, and survival. Our current understanding of conserved molecular mechanisms of proteostasis and aging has greatly benefited from studies in model organisms including yeast (Saccharomyces cerevisiae), nematodes (Caenorhabditis elegans), and flies (Drosophila melanogaster) (He et al. 2018; Kenyon 2010; Yu and Hyun 2021). Several key lifespan-modulating pathways have been identified, including nutrient and germline signaling, mitochondrial respiration, and translation attenuation (Denzel et al. 2019). Different genetic and environmental longevity interventions have displayed proteostatic enhancements, but our understanding of the impact of these cellular improvements on proteome dynamics during aging remains incomplete. One of the major progressive changes associated with aging is the loss in solubility for numerous proteins, which jeopardizes the stability of the whole proteome (David et al. 2010; Reis-Rodrigues et al. 2012; Walther et al. 2015) and may form the basis of many age-related diseases. Protein chaperones can delay the collapse of the proteome by mitigating the impact of protein aggregation (Ben-Zvi et al. 2009) and by attempting to refold misfolded proteins. Cells have two major protein degradation processes, the 26S proteasome and the autophagy/lysosome pathway, that can preventively degrade misfolded, damaged, and aggregating proteins (Dikic 2017). Long-lived nematodes show enhanced proteasome function (Vilchez et al. 2012) and increased autophagic flux (Lapierre et al. 2015). Notably, 26S proteasomes may degrade up to 90% of intracellular proteins (Lee and Goldberg 1998). Organellar proteases can also contribute to proteostatic quality control (Quiros et al. 2015; Sun and Brodsky 2019). Altogether, the efficiency of these degradation pathways governs the ability of cells to prevent the accumulation of damaged and aggregating proteins, thereby maintaining protein solubility and function necessary for cell survival.
During the process of aging, protein solubility progressively wanes (Hipp et al. 2019; Vecchi et al. 2020) and several proteins aggregate (David et al. 2010; Reis-Rodrigues et al. 2012; Walther et al. 2015) as protein degradation efficiency fades and chaperoning systems are overwhelmed. Age-associated protein aggregation arises even in longevity models (Walther et al. 2015), but the types and properties of proteins aggregating as well as the quantity of aggregate-associated chaperones differ between wild-type and long-lived animals (Walther et al. 2015). This intriguing observation is in line with other studies showing that aggregation can serve a protective role (Cohen et al. 2009; Saad et al. 2017) and is part of the arsenal of tools cells employ to minimize cellular dysfunction associated with unstable proteomes. Subcellular protein repartitioning appears to underlie the ability of cells to withstand the proteome destabilization associated with heat stress (Domnauer et al. 2021). Subcellular localization of unstable proteins in the cell dictates their propensity to aggregate. Indeed, cytoplasmically accumulating proteins have a higher likelihood to aggregate than those accumulating in the nucleus (Samant et al. 2018), suggesting that supersaturation barriers and aggregation dynamics, as well as chaperoning capacity, are compartment-specific. Another example is the expression of unstable proteins in the ER reduces their propensity to aggregate (Vincenz-Donnelly et al. 2018). This location-specific nature of protein aggregation and toxicity provides a mechanism by which cells can regulate overall proteome stability by modulating subcellular protein partitioning.
Long-lived nematodes display a variety of nucleocytoplasmic proteostatic improvements that impact proteome stability and enable lifespan extension (Fig. 1). These include modulation of ribosomal function (Hansen et al. 2007; Schosserer et al. 2015; Tiku et al. 2017) as well as reduced protein export into the cytoplasm (Silvestrini et al. 2018). Transcriptional activation of proteasome (Li et al. 2011; Vilchez et al. 2012) and autophagy genes (Lapierre et al. 2013) as well as chaperones (Murphy et al. 2003) via longevity-associated transcription factors (including, but not limited to DAF-16/FOXO, HLH-30/TFEB, SKN-1/NRF2, HSF-1/HSF1) (Denzel et al. 2019) improve cytoplasmic proteostasis (Fig. 1). Altogether, these proteostatic changes prevent protein supersaturation and decrease the burden on chaperones and protein degradation machineries, which in turn delay the progressive solubility decline associated with neurodegenerative diseases and aging (Ben-Gedalya and Cohen 2012; Ciryam et al. 2013).

Key proteostatic mechanisms associated with longevity
From synthesis to degradation, proteins are constantly surveilled for proper folding and damage, but their dynamic subcellular partitioning, preponderance in different compartments, and association with various organelles leads to a variety of proteostatic outcomes that have important ramifications on disease onset and progression, and ultimately on aging itself. Therefore, spatio-temporal regulation of proteostasis is key in somatic maintenance and health (Sontag et al. 2017). This review highlights subcellular mechanisms of proteostasis and their impact on longevity and aging, with an emphasis on protein trafficking across the nuclear pore as well as specific nuclear and cytoplasmic proteostatic mechanisms.
Nucleocytoplasmic protein trafficking
Proteome partitioning between the cytoplasm and the nucleus is mediated by passive and facilitated transport of proteins across the nuclear pore (Knockenhauer and Schwartz 2016; Timney et al. 2016). The nuclear pore is a massive complex (120 MDa in humans) in the nuclear membrane consisting of about 30 different nuclear pore proteins, or nucleoporins, in numerous copies (D’Angelo and Hetzer 2008). Altogether, the nuclear pore structure includes a ring-like pore, a nuclear basket, and cytoplasmic filaments (Solmaz et al. 2013), and integrates between 500 and 1000 nucleoporin proteins (Beck and Hurt 2017; Knockenhauer and Schwartz 2016; Schwartz 2016). Some of the nuclear pore proteins have particularly long lifespan in the nuclear pore and are exchanged at a low rate (Toyama et al. 2013), suggesting that damage in these proteins may result in lasting destabilization of the nuclear pore. Indeed, with age, nuclear pore complex instability and permeability progressively increases, leading to mislocalization of several proteins, a phenomenon that is prevented in long-lived nematodes (Doucet et al. 2010). Altogether, these studies suggest that maintenance of the nuclear pore integrity is essential for longevity (Toyama and Hetzer 2013).
The accepted passive threshold across the nuclear pore is 40 kDa (Knockenhauer and Schwartz 2016; Schmidt and Gorlich 2016) and transporters called karyopherins can recognize and facilitate the traffic of larger proteins across the nuclear pore, a process involving Ran GTPases (Cavazza and Vernos 2015). The partitioning of large proteins (> 40 kDa) between the nucleus and the cytoplasm involves a specific recognition of sequences within cargo proteins by karyopherins. The karyopherin family of proteins consists of trafficking receptors named importins (18 in humans) and exportins (6 in humans). Importins recognize nuclear localization sequences of cytoplasmic proteins and mediate their transit from cytoplasm to nucleus. Exportins recognize nuclear export sequences of nuclear-localized proteins and facilitate their transport from the nucleus to the cytoplasm. To maintain transport capacities across the nuclear pore, karyopherins are returned to their relevant site of action after trafficking. This dynamic cycle of import and export governs the temporal specification of nuclear and cytoplasmic proteomes, and ultimately impacts an array of key cellular processes including pathways associated with aging (Fig. 2).

Subcellular locations relevant for proteostasis
Several diseases have a fundamental basis in nucleocytoplasmic transport dysfunction, including cancer (Gandhi et al. 2018), neurodegeneration (Zhang et al. 2015), and age-related diseases (Kim and Taylor 2017). Many neurodegenerative diseases are characterized by impairments in nucleocytoplasmic protein partitioning (Kim and Taylor 2017) and nucleolar dynamics (White et al. 2019), in addition to autophagic defects due to lysosomal dysfunction (Wong and Cuervo 2010). Since intracellular mislocalization of proteins can lead to deleterious compartmental loss-of-function(s) or predispose mislocalized cargo proteins to aggregate and impair proteostatic mechanisms, nuclear transport dysfunction may be a factor underling the onset of neurodegeneration (Kim and Taylor 2017). Defective nucleocytoplasmic partitioning has been linked to the development of ALS as RNA processing protein, TDP43, aberrantly distributes in the cytoplasm (Solomon et al. 2018). In nuclei of cells from Hutchinson-Gilford progeria syndrome patients, mutated lamins that normally would provide structural support aberrantly accumulate, resulting in genomic instability, a feature exacerbated by dysfunctional nuclear protein transport (Kelley et al. 2011). Phosphorylated form of the protein Tau (Bejanin et al. 2017), a pathologically relevant agent in AD, was recently found to interact with nuclear pore components and disrupt nuclear protein transport leading to cytosolic protein mislocalization, which in turn facilitated cytosolic Tau aggregate formation (Eftekharzadeh et al. 2018). Studies of different proteinopathies have identified key importins and exportins as modifiers of the onset of these diseases (Fig. 3).

Coordination of nucleocytoplasmic protein partitioning and proteostatic mechanisms
Karyopherin β2, an importin involved in the import of several nuclear localization sequence-containing RNA-binding proteins (Chook and Suel 2011), was recently shown to display chaperoning and disaggregase functions for unstable proteins (FUS, dipeptide repeats, etc.) that agglomerate in the cytoplasm and are relevant in ALS and FTD (Guo et al. 2018; Hutten et al. 2020; Robinson et al. 2020). This pre-import disaggregation function demonstrates that repartitioning of certain RNA-binding proteins from the cytosol to the nucleus may improve the solubility and function of proteins that are bound to aggregate and mediate proteotoxicity in the cytoplasm. In turn, this chaperone-like function likely reduces the proteostatic burden in the cytoplasm, in addition to refolding and compartmentalization of unstable proteins in order to restore their native structure and function. Alternatively, unstable protein import in the nucleus may facilitate their degradation via nuclear 26S proteasomes (Albert et al. 2017).
Exportin 1 (XPO1/CRM1), an exportin involved in the recognition and transport of potentially hundreds of nuclear export sequence-containing proteins (Kirli et al. 2015), has been linked to the control of the longevity-associated pathway of autophagy in C. elegans (Kumar et al. 2018; Silvestrini et al. 2018). XPO1 is a highly conserved nuclear export receptor involved in trafficking several proteins including translation factors, vesicle coat proteins, centrosomal and autophagy proteins, ubiquitin pathway proteins, and ribosomal subunits out of the nucleus (Kirli et al. 2015). Notably, XPO1 levels are elevated in various cancers leading to nuclear depletion of tumor-suppressing proteins (Gandhi et al. 2018). XPO1 is also involved in snoRNA (Boulon et al. 2004) and snRNP trafficking (Sleeman 2007) and can localize to the nucleolus where it plays a role in rRNA processing (Bai et al. 2013). Recently developed XPO1 inhibitors showed success in slowing tumorigenesis in a variety of cancers and the selective inhibitor of nuclear export (SINE), Selinexor (KPT-330, Karyopharm Therapeutics), was approved for relapsed multiple myelomas in 2020 (Chen et al. 2018). Inhibition of XPO1 using SINE leads to nucleocytoplasmic repartitioning of several proteins and a corresponding reduction in translation rate (Wahba et al. 2018). This is associated with improvement in the process of autophagy and lysosomal biogenesis via the nuclear enrichment of the autophagy transcriptional regulator TFEB (Silvestrini et al. 2018), improving proteostasis and increasing lifespan in nematodes and ALS-afflicted flies (Silvestrini et al. 2018; Zhang et al. 2015). XPO1 inhibition was also recently shown to mitigate the nuclear defects of progeria (Garcia-Aguirre et al. 2019), suggesting that reducing protein export can foster healthy nuclear structure and proteome.
Overall, the dynamics of nucleocytoplasmic protein partitioning is an emerging field with promising potential to markedly enhance our understanding of the process of aging and the onset of age-related diseases. Moreover, the development of new selective inhibitors of karyopherins is bound to improve our ability to pharmacologically modify the partitioning of proteins in cells in order to modulate proteostasis by leveraging different proteostatic mechanisms in the nucleus and the cytoplasm.
Nuclear proteostasis: nexus of ribosomal subunit and protein quality control
The nuclear proteome is diverse and requires proper protein surveillance in order to maintain nuclear structure and dynamic processes that characterize this essential organelle (Enam et al. 2018; Shibata and Morimoto 2014). As cellular proteome is specified by ribosomes, proper assembly of pre-ribosome subunits in the nucleus ultimately governs the rate of mRNA translation. Ribosome assembly originates inside the nucleus in the membraneless nucleolus (Boisvert et al., 2007; Iarovaia et al. 2019), where different ribosomal RNAs (rRNA) are transcribed by RNA polymerases (Paule and White 2000) and processed into the 40S (18S rRNA + 33 ribosomal proteins) and 60S (5S, 5.8S, and 28S rRNA + 46 ribosomal proteins) ribosomal subunits (Pena et al. 2017). Processing of pre-rRNA is required for proper ribosomal subunit assembly and is promoted by the highly conserved rRNA 2’O-methyltransferase fibrillarin (FIB-1/FBL) (Pereira-Santana et al. 2020). The nucleolus can expand or retract to address cellular needs for ribosomal biogenesis, and fibrillarin levels have been correlated with nucleolar expansion (Weber and Brangwynne 2015), which stimulates the rate of ribosome assembly (Tollervey et al. 1993). Interestingly, proteins that become unstable in the nucleus can accumulate inside nucleoli (Frottin et al. 2019). Ribosomal subunits that are translated in the cytoplasm require nuclear import to assemble with processed rRNAs. Subsequently, newly assembled rRNA-containing ribosomal subunits are exported out of the nucleus and combine to form large 80S ribosomes for mRNA translation. Notably, when exported in the cytoplasm, supernumerous ribosomal subunits (An and Harper 2020; Sung et al. 2016a, b), mislocalized (Yanagitani et al. 2017) and stalled ribosomes (Matsuo et al. 2017) can be sent for proteasomal and lysosomal degradation. Several long-lived nematodes display smaller nucleoli, in part via lower FIB-1, rRNA, and ribosomal protein levels, and specifically silencing fib-1 extends lifespan in C. elegans (Tiku et al. 2017). High levels of FBL expression are found in several cancers (Koh et al. 2011; Marcel et al. 2013; Su et al. 2014) and nucleolar hypertrophy is a hallmark of poor tumor prognosis (Derenzini et al. 2009). An E3 ubiquitin ligase, NCL-1/TRIM2, negatively regulates FIB-1 levels (Tiku and Antebi 2018; Tiku et al. 2017; Yi et al. 2015). TRIM2 mutations in humans are linked to axonal neurodegeneration (Ylikallio et al. 2013), and mutating ncl-1 in long-lived nematodes restores their nucleoli to wild-type size and significantly impairs their longevity (Tiku et al. 2017).
Different environmental stresses, including nucleotide depletion, heat shock, hypoxia, or UV, generate a nucleolar stress response (Rubbi and Milner 2003; Yang et al. 2018). This response elicits a signaling cascade mediated in part by p53 (Nicolas et al. 2016), which results in nucleolar fragmentation and disruption, and is associated with issues in ribosome biogenesis. Another environmental stress, starvation, results in chaperones (heat shock proteins) repartitioning into the nucleus (Chughtai et al. 2001; Nollen et al. 2001). Aggregated nucleoplasmic proteins can accumulate in the nucleolus (Latonen 2019), in particular when proteasome function is compromised (Latonen et al. 2011). In yeast, acute heat stress leads to the reversible formation of nucleolar protein aggregates (Gallardo et al. 2020). The nucleolus is also a temporary store for epigenetic regulators during heat shock, which are subsequently functionally restored after recovery from heat stress (Azkanaz et al. 2019). Aggregates in the nucleus have also been found in depots called intranuclear quality control compartment (INQ) (Miller et al. 2015). Notably, nuclear aggregate accumulation has been linked to polyglutamine-induced disease such as Huntington’s disease (Klement et al. 1998; Schilling et al. 2004). Mutated ⍺-synuclein was also shown to trigger nucleolar stress in a murine model of Parkinson’s disease (Evsyukov et al. 2017). Interestingly, there are mechanistic links between nucleolar stress and autophagy (Pfister 2019), and nucleolar proteins can be degraded via nucleophagy (Mostofa et al. 2018).
Heat shock proteins serve as chaperones and are found in both the nucleus and the cytoplasm (Echtenkamp and Freeman 2014; Vabulas et al. 2010). They modulate protein aggregation by converting unstable proteins into their native fold or into manageable proteasome targets (den Brave et al. 2020). Cryo-electron microscopy imaging of the nuclear pore in the green alga Chlamydomonas reinhardtii demonstrated tethering and enrichment of 26S proteasomes at the nuclear basket side (Albert et al. 2017), suggesting that a quality control checkpoint for proteins exists for nuclear proteins that are trafficked across the nuclear pore (Fig. 3). Studies in the yeast S. cerevisiae showed that quality control of cytoplasmic and nuclear proteins is mediated by spatially specific E3 ubiquitin ligases (Gardner et al. 2005) with different preferences for ubiquitin linkages (Samant et al. 2018). Recently, a study demonstrated that the accumulation of selective autophagy receptor SQSTM1 in nuclear condensates, brought about by reducing nuclear protein export, improves proteasomal function and degradation of c-myc, a key regulator of ribosome biogenesis and nucleolar dynamics (Fu et al. 2021). Thus, nuclear localization of autophagy-related factors can modulate different proteostatic mechanisms and impact proteostasis globally. Altogether, these studies highlight the ability of cells to sequester nuclear proteins into condensates or around the nuclear pore in order to determine their fate.
Cytoplasmic proteostasis: organelle-specific and bulk protein quality control
The cytoplasm encompasses several membrane-bound organelles that interact with each other and mediate and integrate key cellular functions (Cohen et al. 2018). As organisms age, organelles accumulate damage and need to be degraded. Bulk degradation of these organelles is mediated by the recycling process of autophagy and lysosomal degradation (Galluzzi et al. 2017; Lapierre et al. 2015). Selective sequestration of organelles is mediated by selective autophagy receptors that recognize damaged organelles and facilitate their degradation (Zaffagnini and Martens 2016). For instance, efficient degradation of mitochondria via mitophagy is required in the lifespan extension of long-lived nematodes (Palikaras et al. 2015). Concomitantly, cytoplasmic 26S proteasomes degrade a vast array of damaged and ubiquitinated proteins. When proteostatic and protein degradation machineries are overwhelmed, aggregating proteins can accumulate in specific sites in the cytoplasm called the insoluble protein depot (IPOD) and the juxtanuclear quality control (JUNQ) compartments (Samant et al. 2018), akin to the originally described aggresomes (Johnston et al. 1998). In specific proteostatic challenges, cells can activate organelle-specific unfolded protein responses (UPRER or UPRMT) (Shpilka and Haynes 2018; Walter and Ron 2011), which results in enhancement in protein folding in order to ensure solubility and function. Aging leads to dysfunction in UPR at the endoplasmic reticulum (UPRER) (Frakes and Dillin, 2017) and mitochondrial UPR (UPRMT), which can affect stem cells and tissue aging (Mohrin et al. 2015). The ER and mitochondria also possess lumenal proteases that directly degrade proteins (synthesized or imported) (Quiros et al. 2015; Sun and Brodsky 2019). In yeast, mitochondria can degrade resident proteins (Hughes et al. 2016) and aggregating proteins imported from the cytoplasm (Zhou et al. 2014). The ER can also send proteins to the proteasome via ER-associated degradation (ERAD) where polypeptides are recognized and threaded back into the cytosol via a retro-translocon (Brodsky 2012; Qi et al. 2017). Lipid droplets can serve as an intermediary organelle for ERAD where cargo bound for degradation transit on the lipid droplet surface before being degraded by the proteasome (Olzmann and Carvalho 2019).
The endosomal sorting complexes required for transport (ESCRT) is a multisubunit complex tasked with sorting ubiquitinated proteins and multi-vesicular bodies toward lysosomal degradation (Schmidt and Teis 2012). Compromised ESCRT leads to the autophagic dysfunction and accumulation of aggregating proteins relevant to neurodegeneration (Oshima et al. 2016). Notably, proteins associated with the lysosomal membrane can be degraded by lysosomes via the ESCRT machinery (Zhu et al. 2017) or intraluminal fragments (McNally and Brett 2018). Overall, the cytoplasm possesses several options to stabilize or degrade proteins, but aging systematically decreases the ability of this compartment to properly manage proteostasis, resulting in molecular crowding and aggregated protein deposition.
Conclusion
Cells employ an arsenal of mechanisms to maintain protein homeostasis in order to ensure cell survival and to adapt to changing environments. In addition to compartment-specific proteostatic processes, the integration of different mechanisms (such as nucleolar dynamics and autophagy (Pfister 2019)) generates a global response against proteotoxic stress associated with aging. Signaling between different organelles, such as mitochondria and nucleus (Fang et al. 2016), and also between tissues may serve to generate organismal response to stress and aging (Zhang et al. 2018). Signaling pathways that can coordinate a proteostatic response, such as nutrient signaling mediated by mTOR complexes (Laplante and Sabatini 2012) and the integrated stress response via the eIF2 complex (Costa-Mattioli and Walter 2020), are important mechanisms to balance protein synthesis and degradation. These processes modify ribosome biogenesis and function, protein specification, and localization, and ultimately affect the stability of the proteome. An important mechanism of proteostasis that potentially fails during aging is the proper partitioning of proteins across the nuclear pore (Fig. 4). Mislocalization of proteins fosters aggregation, but concomitant aberrant DNA release into the cytoplasm can also lead to inflammation and neurodegeneration (Paul et al. 2021). Therefore, pharmacologically modulating the nucleocytoplasmic partitioning of proteins is emerging as an attractive strategy to impact the stability of the whole proteome and delay aging.

Nucleocytoplasmic proteostatic inter-relationship during aging
References
Albert S, Schaffer M, Beck F, Mosalaganti S, Asano S, Thomas HF, Plitzko JM, Beck M, Baumeister W, Engel BD (2017) Proteasomes tether to two distinct sites at the nuclear pore complex. Proc Natl Acad Sci U S A 114:13726–13731. https://doi.org/10.1073/pnas.1716305114
An H, Harper JW (2020) Ribosome abundance control via the ubiquitin-proteasome system and autophagy. J Mol Biol 432:170–184. https://doi.org/10.1016/j.jmb.2019.06.001
Azkanaz M, Rodriguez Lopez A, de Boer B, Huiting W, Angrand PO, Vellenga E, Kampinga HH, Bergink S, Martens JH, Schuringa JJ, et al (2019) Protein quality control in the nucleolus safeguards recovery of epigenetic regulators after heat shock. eLife 8. https://doi.org/10.7554/eLife.45205
Bai B, Moore HM, Laiho M (2013) CRM1 and its ribosome export adaptor NMD3 localize to the nucleolus and affect rRNA synthesis. Nucleus 4:315–325. https://doi.org/10.4161/nucl.25342
Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18:73–89. https://doi.org/10.1038/nrm.2016.147
Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, Ayakta N, Cantwell A, Janabi M, Lauriola M et al (2017) Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140:3286–3300. https://doi.org/10.1093/brain/awx243
Ben-Gedalya T, Cohen E (2012) Quality control compartments coming of age. Traffic 13:635–642. https://doi.org/10.1111/j.1600-0854.2012.01330.x
Ben-Zvi A, Miller EA, Morimoto RI (2009) Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A 106:14914–14919. https://doi.org/10.1073/pnas.0902882106
Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI (2007) The multifunctional nucleolus. Nat Rev Mol Cell Biol 8:574–585. https://doi.org/10.1038/nrm2184
Boulon S, Verheggen C, Jady BE, Girard C, Pescia C, Paul C, Ospina JK, Kiss T, Matera AG, Bordonne R et al (2004) PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol Cell 16:777–787. https://doi.org/10.1016/j.molcel.2004.11.013
Brodsky JL (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151:1163–1167. https://doi.org/10.1016/j.cell.2012.11.012
Cavazza T, Vernos I (2015) The RanGTP pathway: from nucleo-cytoplasmic transport to spindle assembly and beyond. Front Cell Dev Biol 3:82. https://doi.org/10.3389/fcell.2015.00082
Chen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N, Baz R, Mau-Sorensen M, Berdeja JG, Savona M et al (2018) Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood 131:855–863. https://doi.org/10.1182/blood-2017-08-797886
Chook YM, Suel KE (2011) Nuclear import by karyopherin-betas: recognition and inhibition. Biochim Biophys Acta 1813:1593–1606. https://doi.org/10.1016/j.bbamcr.2010.10.014
Chughtai ZS, Rassadi R, Matusiewicz N, Stochaj U (2001) Starvation promotes nuclear accumulation of the hsp70 Ssa4p in yeast cells. J Biol Chem 276:20261–20266. https://doi.org/10.1074/jbc.M100364200
Ciryam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M (2013) Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep 5:781–790. https://doi.org/10.1016/j.celrep.2013.09.043
Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW et al (2009) Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139:1157–1169. https://doi.org/10.1016/j.cell.2009.11.014
Cohen S, Valm AM, Lippincott-Schwartz J (2018) Interacting organelles. Curr Opin Cell Biol 53:84–91. https://doi.org/10.1016/j.ceb.2018.06.003
Costa-Mattioli M, Walter P (2020) The integrated stress response: from mechanism to disease. Science 368. https://doi.org/10.1126/science.aat5314
D’Angelo MA, Hetzer MW (2008) Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol 18:456–466. https://doi.org/10.1016/j.tcb.2008.07.009
David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C (2010) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8:e1000450. https://doi.org/10.1371/journal.pbio.1000450
den Brave F, Cairo LV, Jagadeesan C, Ruger-Herreros C, Mogk A, Bukau B, Jentsch S (2020) Chaperone-mediated protein disaggregation triggers proteolytic clearance of intra-nuclear protein inclusions. Cell Rep 31:107680. https://doi.org/10.1016/j.celrep.2020.107680
Denzel MS, Lapierre LR, Mack HID (2019) Emerging topics in C. elegans aging research: transcriptional regulation, stress response and epigenetics. Mech Ageing Dev 177:4–21. https://doi.org/10.1016/j.mad.2018.08.001
Derenzini M, Montanaro L, Trere D (2009) What the nucleolus says to a tumour pathologist. Histopathology 54:753–762. https://doi.org/10.1111/j.1365-2559.2008.03168.x
Dikic I (2017) Proteasomal and autophagic degradation systems. Annu Rev Biochem 86:193–224. https://doi.org/10.1146/annurev-biochem-061516-044908
Domnauer M, Zheng F, Li L, Zhang Y, Chang CE, Unruh JR, Conkright-Fincham J, McCroskey S, Florens L, Zhang Y et al (2021) Proteome plasticity in response to persistent environmental change. Mol Cell 81:3294.e3212-3309.e3212. https://doi.org/10.1016/j.molcel.2021.06.028
Doucet CM, Talamas JA, Hetzer MW (2010) Cell cycle-dependent differences in nuclear pore complex assembly in metazoa. Cell 141:1030–1041. https://doi.org/10.1016/j.cell.2010.04.036
Echtenkamp FJ, Freeman BC (2014) Molecular chaperone-mediated nuclear protein dynamics. Curr Protein Pept Sci 15:216–224. https://doi.org/10.2174/1389203715666140331112230
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS et al (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99:925.e927-940.e927. https://doi.org/10.1016/j.neuron.2018.07.039
Enam C, Geffen Y, Ravid T, Gardner RG (2018) Protein quality control degradation in the nucleus. Annu Rev Biochem 87:725–749. https://doi.org/10.1146/annurev-biochem-062917-012730
Evsyukov V, Domanskyi A, Bierhoff H, Gispert S, Mustafa R, Schlaudraff F, Liss B, Parlato R (2017) Genetic mutations linked to Parkinson’s disease differentially control nucleolar activity in pre-symptomatic mouse models. Dis Model Mech 10:633–643. https://doi.org/10.1242/dmm.028092
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA (2016) Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 17:308–321. https://doi.org/10.1038/nrm.2016.14
Frakes AE, Dillin A (2017) The UPR(ER): sensor and coordinator of organismal homeostasis. Mol Cell 66:761–771. https://doi.org/10.1016/j.molcel.2017.05.031
Frottin F, Schueder F, Tiwary S, Gupta R, Korner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS (2019) The nucleolus functions as a phase-separated protein quality control compartment. Science 365:342–347. https://doi.org/10.1126/science.aaw9157
Fu A, Cohen-Kaplan V, Avni N, Livneh I, Ciechanover A (2021) p62-containing, proteolytically active nuclear condensates, increase the efficiency of the ubiquitin-proteasome system. Proc Natl Acad Sci U S A 118. https://doi.org/10.1073/pnas.2107321118
Gallardo P, Real-Calderon P, Flor-Parra I, Salas-Pino S, Daga RR (2020) Acute heat stress leads to reversible aggregation of nuclear proteins into nucleolar rings in fission yeast. Cell Rep 33:108377. https://doi.org/10.1016/j.celrep.2020.108377
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI et al (2017) Molecular definitions of autophagy and related processes. EMBO J 36:1811–1836. https://doi.org/10.15252/embj.201796697
Gandhi UH, Senapedis W, Baloglu E, Unger TJ, Chari A, Vogl D, Cornell RF (2018) Clinical implications of targeting XPO1-mediated nuclear export in multiple myeloma. Clin Lymphoma Myeloma Leuk 18:335–345. https://doi.org/10.1016/j.clml.2018.03.003
Garcia-Aguirre I, Alamillo-Iniesta A, Rodriguez-Perez R, Velez-Qguilera G, Amaro-Encarnacion E, Jimenez-Gutierrez E, Vasquez-Limeta A, Samuel Laredo-Cisneros M, Morales-Lazaro SL, Tiburcio-Felix R, et al (2019) Enhanced nuclear protein export in premature aging rescue of the progeria phenotype by modulation of CRM1 activity. Aging Cell 18:e130002. https://doi.org/10.1111/acel.13002
Gardner RG, Nelson ZW, Gottschling DE (2005) Degradation-mediated protein quality control in the nucleus. Cell 120:803–815. https://doi.org/10.1016/j.cell.2005.01.016
Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N et al (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173:677.e620-692.e620. https://doi.org/10.1016/j.cell.2018.03.002
Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6:95–110. https://doi.org/10.1111/j.1474-9726.2006.00267.x
He C, Zhou C, Kennedy BK (2018) The yeast replicative aging model. Biochim Biophys Acta Mol Basis Dis 1864:2690–2696. https://doi.org/10.1016/j.bbadis.2018.02.023
Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20:421–435. https://doi.org/10.1038/s41580-019-0101-y
Hughes AL, Hughes CE, Henderson KA, Yazvenko N, Gottschling DE (2016) Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife 5. https://doi.org/10.7554/eLife.13943
Hutten S, Usluer S, Bourgeois B, Simonetti F, Odeh HM, Fare CM, Czuppa M, Hruska-Plochan M, Hofweber M, Polymenidou M et al (2020) Nuclear import receptors directly bind to arginine-rich dipeptide repeat proteins and suppress their pathological interactions. Cell Rep 33:108538. https://doi.org/10.1016/j.celrep.2020.108538
Iarovaia OV, Minina EP, Sheval EV, Onichtchouk D, Dokudovskaya S, Razin SV, Vassetzky YS (2019) Nucleolus: a central hub for nuclear functions. Trends Cell Biol 29:647–659. https://doi.org/10.1016/j.tcb.2019.04.003
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898. https://doi.org/10.1083/jcb.143.7.1883
Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang CS, Cubenas-Potts C, Matunis MJ, Paschal BM (2011) The defective nuclear lamina in Hutchinson-Gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol Cell Biol 31:3378–3395. https://doi.org/10.1128/MCB.05087-11
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512. https://doi.org/10.1038/nature08980
Kim HJ, Taylor JP (2017) Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96:285–297. https://doi.org/10.1016/j.neuron.2017.07.029
Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Gorlich D (2015) A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. eLife 4. https://doi.org/10.7554/eLife.11466
Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT (1998) Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95:41–53. https://doi.org/10.1016/s0092-8674(00)81781-x
Knockenhauer KE, Schwartz TU (2016) The nuclear pore complex as a flexible and dynamic gate. Cell 164:1162–1171. https://doi.org/10.1016/j.cell.2016.01.034
Koh CM, Gurel B, Sutcliffe S, Aryee MJ, Schultz D, Iwata T, Uemura M, Zeller KI, Anele U, Zheng Q et al (2011) Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am J Pathol 178:1824–1834. https://doi.org/10.1016/j.ajpath.2010.12.040
Kumar AV, Thakurta TG, Silvestrini MJ, Johnson JR, Reenan RA, Lapierre LR (2018) Give me a SINE: how selective inhibitors of nuclear export modulate autophagy and aging. Molecular & cellular oncology 5:e1502511. https://doi.org/10.1080/23723556.2018.1502511
Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE et al (2013) The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4:2267. https://doi.org/10.1038/ncomms3267
Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M (2015) Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11:867–880. https://doi.org/10.1080/15548627.2015.1034410
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. https://doi.org/10.1016/j.cell.2012.03.017
Latonen L (2019) Phase-to-phase with nucleoli - stress responses, protein aggregation and novel roles of RNA. Front Cell Neurosci 13:151. https://doi.org/10.3389/fncel.2019.00151
Latonen L, Moore HM, Bai B, Jaamaa S, Laiho M (2011) Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 30:790–805. https://doi.org/10.1038/onc.2010.469
Lee DH, Goldberg AL (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 8:397–403. https://doi.org/10.1016/s0962-8924(98)01346-4
Li X, Matilainen O, Jin C, Glover-Cutter KM, Holmberg CI, Blackwell TK (2011) Specific SKN-1/Nrf stress responses to perturbations in translation elongation and proteasome activity. PLoS Genet 7:e1002119. https://doi.org/10.1371/journal.pgen.1002119
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano-Gonzalez E, Vendrell JA, Hacot S, Mertani HC, Albaret MA et al (2013) p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24:318–330. https://doi.org/10.1016/j.ccr.2013.08.013
Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K et al (2017) Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun 8:159. https://doi.org/10.1038/s41467-017-00188-1
McNally EK, Brett CL (2018) The intralumenal fragment pathway mediates ESCRT-independent surface transporter down-regulation. Nat Commun 9:5358. https://doi.org/10.1038/s41467-018-07734-5
Miller SB, Ho CT, Winkler J, Khokhrina M, Neuner A, Mohamed MY, Guilbride DL, Richter K, Lisby M, Schiebel E et al (2015) Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J 34:778–797. https://doi.org/10.15252/embj.201489524
Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, Chen D (2015) Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347:1374–1377. https://doi.org/10.1126/science.aaa2361
Moreno DF, Aldea M (2020) Proteostatic stress as a nodal hallmark of replicative aging. Exp Cell Res 394:112163. https://doi.org/10.1016/j.yexcr.2020.112163
Mostofa MG, Rahman MA, Koike N, Yeasmin AM, Islam N, Waliullah TM, Hosoyamada S, Shimobayashi M, Kobayashi T, Hall MN et al (2018) CLIP and cohibin separate rDNA from nucleolar proteins destined for degradation by nucleophagy. J Cell Biol 217:2675–2690. https://doi.org/10.1083/jcb.201706164
Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424:277–283. https://doi.org/10.1038/nature01789
Nicolas E, Parisot P, Pinto-Monteiro C, de Walque R, De Vleeschouwer C, Lafontaine DL (2016) Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat Commun 7:11390. https://doi.org/10.1038/ncomms11390
Nollen EA, Salomons FA, Brunsting JF, van der Want JJ, Sibon OC, Kampinga HH (2001) Dynamic changes in the localization of thermally unfolded nuclear proteins associated with chaperone-dependent protection. Proc Natl Acad Sci U S A 98:12038–12043. https://doi.org/10.1073/pnas.201112398
Olzmann JA, Carvalho P (2019) Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20:137–155. https://doi.org/10.1038/s41580-018-0085-z
Oshima R, Hasegawa T, Tamai K, Sugeno N, Yoshida S, Kobayashi J, Kikuchi A, Baba T, Futatsugi A, Sato I et al (2016) ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci Rep 6:24997. https://doi.org/10.1038/srep24997
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525–528. https://doi.org/10.1038/nature14300
Paul BD, Snyder SH, Bohr VA (2021) Signaling by cGAS-STING in neurodegeneration, neuroinflammation, and aging. Trends Neurosci 44:83–96. https://doi.org/10.1016/j.tins.2020.10.008
Paule MR, White RJ (2000) Transcription by RNA polymerases I and III. Nucleic Acids Res 28:1283–1298. https://doi.org/10.1093/nar/28.6.1283
Pena C, Hurt E, Panse VG (2017) Eukaryotic ribosome assembly, transport and quality control. Nat Struct Mol Biol 24:689–699. https://doi.org/10.1038/nsmb.3454
Pereira-Santana A, Gamboa-Tuz SD, Zhao T, Schranz ME, Vinuesa P, Bayona A, Rodriguez-Zapata LC, Castano E (2020) Fibrillarin evolution through the Tree of Life: comparative genomics and microsynteny network analyses provide new insights into the evolutionary history of Fibrillarin. PLoS Comput Biol 16:e1008318. https://doi.org/10.1371/journal.pcbi.1008318
Pfister AS (2019) Emerging role of the nucleolar stress response in autophagy. Front Cell Neurosci 13:156. https://doi.org/10.3389/fncel.2019.00156
Qi L, Tsai B, Arvan P (2017) New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol 27:430–440. https://doi.org/10.1016/j.tcb.2016.12.002
Quiros PM, Langer T, Lopez-Otin C (2015) New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 16:345–359. https://doi.org/10.1038/nrm3984
Reis-Rodrigues P, Czerwieniec G, Peters TW, Evani US, Alavez S, Gaman EA, Vantipalli M, Mooney SD, Gibson BW, Lithgow GJ et al (2012) Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell 11:120–127. https://doi.org/10.1111/j.1474-9726.2011.00765.x
Robinson E, Shorter J, Guo L (2020) Karyopherin-beta2 inhibits and reverses aggregation and liquid-liquid phase separation of the ALS/FTD-associated protein FUS. Bio Protoc 10:e3725. https://doi.org/10.21769/BioProtoc.3725
Rubbi CP, Milner J (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22:6068–6077. https://doi.org/10.1093/emboj/cdg579
Saad S, Cereghetti G, Feng Y, Picotti P, Peter M, Dechant R (2017) Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat Cell Biol 19:1202–1213. https://doi.org/10.1038/ncb3600
Samant RS, Livingston CM, Sontag EM, Frydman J (2018) Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 563:407–411. https://doi.org/10.1038/s41586-018-0678-x
Schilling G, Savonenko AV, Klevytska A, Morton JL, Tucker SM, Poirier M, Gale A, Chan N, Gonzales V, Slunt HH et al (2004) Nuclear-targeting of mutant huntingtin fragments produces Huntington’s disease-like phenotypes in transgenic mice. Hum Mol Genet 13:1599–1610. https://doi.org/10.1093/hmg/ddh175
Schmidt HB, Gorlich D (2016) Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem Sci 41:46–61. https://doi.org/10.1016/j.tibs.2015.11.001
Schmidt O, Teis D (2012) The ESCRT machinery. Curr Biol 22:R116-120. https://doi.org/10.1016/j.cub.2012.01.028
Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, Calle-Perez A, Pircher A, Gerstl MP, Pfeifenberger S et al (2015) Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun 6:6158. https://doi.org/10.1038/ncomms7158
Schwartz TU (2016) The structure inventory of the nuclear pore complex. J Mol Biol 428:1986-200010.1016/j.jmb.2016.03.015
Shibata Y, Morimoto RI (2014) How the nucleus copes with proteotoxic stress. Curr Biol 24:R463-474. https://doi.org/10.1016/j.cub.2014.03.033
Shpilka T, Haynes CM (2018) The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 19:109–120. https://doi.org/10.1038/nrm.2017.110
Silvestrini MJ, Johnson JR, Kumar AV, Thakurta TG, Blais K, Neill ZA, Marion SW, St Amand V, Reenan RA, Lapierre LR (2018) Nuclear export inhibition enhances HLH-30/TFEB activity, autophagy, and lifespan. Cell Rep 23:1915–1921. https://doi.org/10.1016/j.celrep.2018.04.063
Sleeman J (2007) A regulatory role for CRM1 in the multi-directional trafficking of splicing snRNPs in the mammalian nucleus. J Cell Sci 120:1540–1550. https://doi.org/10.1242/jcs.001529
Solmaz SR, Blobel G, Melcak I (2013) Ring cycle for dilating and constricting the nuclear pore. Proc Natl Acad Sci U S A 110:5858–5863. https://doi.org/10.1073/pnas.1302655110
Solomon DA, Stepto A, Au WH, Adachi Y, Diaper DC, Hall R, Rekhi A, Boudi A, Tziortzouda P, LeeYB, et al (2018) A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-alpha mediates C9orf72-related neurodegeneration. Brain 141:2908–2924. https://doi.org/10.1093/brain/awy241
Sontag EM, Samant RS, Frydman J (2017) Mechanisms and functions of spatial protein quality control. Annu Rev Biochem 86:97–122. https://doi.org/10.1146/annurevbiochem-060815-014616
Su H, Xu T, Ganapathy S, Shadfan M, Long M, Huang TH, Thompson I, Yuan ZM (2014) Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 33:1348–1358. https://doi.org/10.1038/onc.2013.89
Sun Z, Brodsky JL (2019) Protein quality control in the secretory pathway. J Cell Biol 218:3171–3187. https://doi.org/10.1083/jcb.201906047
Sung MK, Porras-Yakushi TR, Reitsma JM, Huber FM, Sweredoski MJ, Hoelz A, Hesse S, Deshaies RJ (2016a) A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. eLife 5:e19105. https://doi.org/10.7554/eLife.19105
Sung MK, Reitsma JM, Sweredoski MJ, Hess S, Deshaies RJ (2016) Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol Biol Cell 27:2642–2652. https://doi.org/10.1091/mbc.E16-05-0290
Tiku V, Antebi A (2018) Nucleolar function in lifespan regulation. Trends Cell Biol 28:662–672. https://doi.org/10.1016/j.tcb.2018.03.007
Tiku V, Jain C, Raz Y, Nakamura S, Heestand B, Liu W, Spath M, Suchiman HED, Muller RU, Slagboom PE et al (2017) Small nucleoli are a cellular hallmark of longevity. Nat Commun 8:16083. https://doi.org/10.1038/ncomms16083
Timney BL, Raveh B, Mironska R, Trivedi JM, Kim SJ, Russel D, Wente SR, Sali A, Rout MP (2016) Simple rules for passive diffusion through the nuclear pore complex. J Cell Biol 215:57–76. https://doi.org/10.1083/jcb.201601004
Tollervey D, Lehtonen H, Jansen R, Kern H, Hurt EC (1993) Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell 72:443–457. https://doi.org/10.1016/0092-8674(93)90120-f
Toyama BH, Hetzer MW (2013) Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 14:55–61. https://doi.org/10.1038/nrm3496
Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, Hetzer MW (2013) Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154:971–982. https://doi.org/10.1016/j.cell.2013.07.037
Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU (2010) Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2:a004390. https://doi.org/10.1101/cshperspect.a004390
Vecchi G, Sormanni P, Mannini B, Vandelli A, Tartaglia GG, Dobson CM, Hartl FU, Vendruscolo M (2020) Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc Natl Acad Sci U S A 117:1015–1020. https://doi.org/10.1073/pnas.1910444117
Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A (2012) RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489:263–268. https://doi.org/10.1038/nature11315
Vincenz-Donnelly L, Holthusen H, Korner R, Hansen EC, Presto J, Johansson J, Sawarkar R, Hartl FU, Hipp MS (2018) High capacity of the endoplasmic reticulum to prevent secretion and aggregation of amyloidogenic proteins. EMBO J 37:337–350. https://doi.org/10.15252/embj.201695841
Wahba A, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ (2018) The XPO1 inhibitor selinexor inhibits translation and enhances the radiosensitivity of glioblastoma cells grown in vitro and in vivo. Mol Cancer Ther 17:1717–1726. https://doi.org/10.1158/1535-7163.MCT-17-1303
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. https://doi.org/10.1126/science.1209038
Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M et al (2015) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161:919–932. https://doi.org/10.1016/j.cell.2015.03.032
Weber SC, Brangwynne CP (2015) Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr Biol 25:641–646. https://doi.org/10.1016/j.cub.2015.01.012
White MR, Mitrea DM, Zhang P, Stanley CB, Cassidy DE, Nourse A, Phillips AH, Tolbert M, Taylor JP, Kriwacki RW (2019) C9orf72 Poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol Cell 74:713.e716-728.e716. https://doi.org/10.1016/j.molcel.2019.03.019
Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–811. https://doi.org/10.1038/nn.2575
Yanagitani K, Juszkiewicz S, Hegde RS (2017) UBE2O is a quality control factor for orphans of multiprotein complexes. Science 357:472–475. https://doi.org/10.1126/science.aan0178
Yang K, Yang J, Yi J (2018) Nucleolar stress: hallmarks, sensing mechanism and diseases. Cell Stress 2:125–140. https://doi.org/10.15698/cst2018.06.139
Yi YH, Ma TH, Lee LW, Chiou PT, Chen PH, Lee CM, Chu YD, Yu H, Hsiung KC, Tsai YT et al (2015) A genetic cascade of let-7-ncl-1-fib-1 modulates nucleolar size and rRNA pool in Caenorhabditis elegans. PLoS Genet 11:e1005580. https://doi.org/10.1371/journal.pgen.1005580
Ylikallio E, Poyhonen R, Zimon M, De Vriendt E, Hilander T, Paetau A, Jordanova A, Lonnqvist T, Tyynismaa H (2013) Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum Mol Genet 22:2975–2983. https://doi.org/10.1093/hmg/ddt149
Yu G, Hyun S (2021) Proteostasis-associated aging: lessons from a Drosophila model. Genes Genomics 43:1–9. https://doi.org/10.1007/s13258-020-01012-9
Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428:1714–1724. https://doi.org/10.1016/j.jmb.2016.02.004
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. https://doi.org/10.1038/nature14973
Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, Dillin A (2018) The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174:870.e817-883.e817. https://doi.org/10.1016/j.cell.2018.06.029
Zhou C, Slaughter BD, Unruh JR, Guo F, Yu Z, Mickey K, Narkar A, Ross RT, McClain M, Li R (2014) Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 159:530–542. https://doi.org/10.1016/j.cell.2014.09.026
Zhu L, Jorgensen JR, Li M, Chuang YS, Emr SD (2017) ESCRTs function directly on the lysosome membrane to downregulate ubiquitinated lysosomal membrane proteins. eLife 6. https://doi.org/10.7554/eLife.26403
Funding
L.R.L. is funded by grants from the National Institute of Health (R01 AG051810 and R21 AG068922).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, A.V., Lapierre, L.R. Location, location, location: subcellular protein partitioning in proteostasis and aging. Biophys Rev 13, 931–941 (2021). https://doi.org/10.1007/s12551-021-00890-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-021-00890-x