
Abstract
Calcium is a versatile element that participates in cell signaling for a wide range of cell processes such as death, cell cycle, division, migration, invasion, metabolism, differentiation, autophagy, transcription, and others. Specificity of calcium in each of these processes is achieved through modulation of intracellular calcium concentrations by changing the characteristics (amplitude/frequency modulation) or location (spatial modulation) of the signal. Breast cancer utilizes calcium signaling as an advantage for survival and progression. This review integrates evidence showing that increases in expression of calcium channels, GPCRs, pumps, effectors, and enzymes, as well as resulting intracellular calcium signals, lead to high calcium and/or an elevated calcium- mobilizing capacity necessary for malignant functions such as migratory, invasive, proliferative, tumorigenic, or metastatic capacities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Ja Kalzium, das ist alles
When calcium ions (Ca2+) flow into a cell, they can bind to calcium-binding proteins to ultimately initiate cell functions. Paradoxically, however, this versatile single element universally participates in almost every single cell process: death (Boehning et al. 2003; Orrenius et al. 2003), cell cycle (Colomer et al. 1994; Kahl and Means 2004), division (Rasmussen and Means 1989), migration (Brundage et al. 1991; Giannone et al. 2004; Hahn et al. 1992; Yang and Huang 2005), invasion (Kato et al. 2007; Sun et al. 2014), metabolism (Cardenas et al. 2010), differentiation (Carey and Matsumoto 1999; Hennings et al. 1980; Holliday et al. 1991), autophagy (Cardenas et al. 2010; Medina et al. 2015), and transcription (Dolmetsch et al. 1998, 2001), as well as participating in many specialized cell functions: angiogenesis (Dragoni et al. 2011), fertilization (Miao et al. 2012; Saunders et al. 2002; Steinhardt et al. 1977), insulin secretion (Grodsky and Bennett 1966; Prentki and Wollheim 1984), synaptic transmission (Brose et al. 1992; Fernandez-Chacon et al. 2001), muscle contraction (Gergely et al. 1993; Sorenson et al. 1995), and immune response (Bhakta et al. 2005). This is perhaps why scientists quote the Nobel prize winning Otto Loewi’s proclamation, “Ja Kalzium, das ist alles” (Carafoli 2002), which literally translates to “Yes calcium, that’s all,” but instead is often interpreted as “calcium is everything” or “calcium is universal” (Brini and Carafoli 2000; Kaestner 2013).
The paradox of calcium in biology is that the divalent cation remains unchanged yet can still achieve specificity in initiating various cell processes. In other words, when calcium enters a cell, it is not bringing forth cell death, cell cycle, division, migration, invasion, metabolism, differentiation, autophagy, and transcription all at once, rather each process can be individually initiated by calcium while the other calcium-sensitive functions remain at rest. Before we discuss how calcium can discriminate between these various cell functions, let us begin with a simple binary (on/off) introduction to calcium signaling.
Introduction to calcium signaling
The plasma membrane is a lipid bilayer barrier that separates the outside and inside of the cell. However, contained within the cell are separate lipid barriers that serve to further compartmentalize cell contents. These intracellular membrane-bound areas, known as organelles, perform different cell functions and include the nucleus, Golgi apparatus, endoplasmic reticulum (ER), mitochondria, and various vesicles. The general non-membrane bound areas within the cell are collectively known as the cytoplasm. Calcium ions are differentially concentrated across these cell membranes between the outside of the cell, the cytoplasm, and within organelles. This separation of calcium is key to calcium signaling.
Calcium is a metallic element and fifth in abundance in the earth’s crust (2015). In the human body, most of the calcium exists in both a bound-form (bones, teeth or bound to extra- and intracellular proteins) and an ionized free form. When considering cells at rest, free calcium is present in relatively high concentrations outside the cell (~ 1.3 mM) (Kratz et al. 2004), and with respect to mammary glands, there is ~ 10 mM total calcium (Neville 2005) and ~ 2–4 mM free calcium in human milk (Neville et al. 1994). In contrast, free calcium is at very low concentrations in cytoplasm (~ 0.05–0.15 mM (McDonough and Button 1989; Ratto et al. 1988)). While resting free calcium concentrations within the cellular organelles vary (nucleus (~ 0.03–0.2 mM, (al-Mohanna et al. 1994; Brini et al. 1993; Ikeda et al. 2003; Przywara et al. 1991; Williams et al. 1985)), Golgi apparatus (~ 0.3 mM (Pinton et al. 1998)), ER (~ 0.5–0.7 mM (Launikonis et al. 2005)), mitochondria ~ 0.2 mM (Ivannikov and Macleod 2013)), lysosomes (~ 0.4 mM (Christensen et al. 2002)) (note that these are approximations)), there is still a concentration gradient between the cytoplasm and Golgi/ER/lysosomes, similar to that between the cytoplasm and extracellular space. The high vs. low concentrations are actively maintained by the cell using different transport mechanisms such as adenosine triphosphate (ATP)-driven calcium pumps that drive calcium within the ER (via sarco-/endoplasmic reticulum Ca2+-ATPase, SERCA) or Golgi apparatus (via Secretory Pathway Ca2+-ATPase, SPCA), or drive calcium outside of the cell (via plasma membrane Ca2+-ATPase, PMCA and sodium–calcium exchanger, NCX). In exchange for ATP consumption, the stored energy that is present in the ion’s electrochemical potential gradient can be utilized. If permitted across cell membranes, ions will passively move down an electrochemical potential gradient (from high to low) until equilibrium is achieved. Thus, calcium will flow from the outside of the cell or from within internally membrane-bound compartments (the ER is the major store) to the cytoplasm or even into the nucleus and mitochondria. Even though ATP pumps continually drive calcium against its electrochemical potential gradient (to areas of high calcium concentrations), equilibrium cannot practically be achieved; however, a steady state of cytoplasmic and organelle calcium concentrations can be.
Calcium signaling refers to the mobilization of calcium ions down these electrochemical potential gradients and this can be initiated in various ways (for in depth reviews on calcium signaling, see Berridge et al. 2000, 2003; Carafoli 2002; Carafoli et al. 2001; Clapham 2007, and see mitochondrial (Rizzuto et al. 2012) and nuclear (Bootman et al. 2009) calcium signaling focused reviews). There are two major classes of membrane bound proteins that permit calcium mobilization, ion channels and G-protein coupled receptors (GPCRs), which can be present on the cell’s plasma membrane and on organelle membranes (Fig. 1). Ion channels, once activated, open their central pore regions allowing calcium ions to mobilize through the channel. In contrast, activated GPCRs typically release ER calcium using lipid signal transduction to target ER ion channels. The resulting movement of calcium ions to the cytoplasm increases the calcium concentration and the positively charged ions are free to bind to oppositely charged counterparts (see the review on calcium-binding domains and motifs (Carafoli et al. 2001)), known as buffers and effectors (further reviews on buffers (Schwaller 2010) and effectors (Berridge et al. 2003) are available). Most of this cytosolic calcium is bound by buffers, which can limit or tune the cytoplasmic calcium signal and thus limit/tune the calcium bound by effectors, depending on the amount, localization, and type of buffer (i.e., fixed vs. mobile) present within the cell. In contrast, calcium-bound effectors play a more direct role in cell function by initiating downstream signaling (i.e., cell death, cell cycle, division, migration, invasion, metabolism, differentiation, autophagy, or transcription). Calcium signaling is eventually finalized as calcium ion channels deactivate and close, thus no longer permitting calcium passage into the cytoplasm. The calcium pumps simultaneously restore low cytoplasmic calcium concentrations and high ER/Golgi calcium concentrations by pumping calcium outside of the cell and into the organelle stores respectively. An additional mechanism by which calcium is removed from the cytoplasm is through the sodium–calcium exchanger (NCX) and is particularly notable in excitable cell types. Buffers and effectors then become unbound by calcium as the concentration of cytoplasmic calcium continues to fall to resting concentrations, and thus effectors inactivate and effector-mediated downstream signaling ceases.

Mechanisms for cellular calcium mobilization. The plasma membrane and intracellular membrane proteins responsible for calcium mobilization are shown. Calcium ions are differentially concentrated across cell membranes between the outside of the cell, the cytoplasm, and within organelles. This separation of calcium is key to calcium signaling. In a resting cell, high free calcium concentrations are maintained outside the cell (~ 1.3 mM), while free calcium is at very low concentrations in cytoplasm (~ 0.1 mM), establishing an ~ 10:000:1 gradient across the plasma membrane. Resting free calcium concentrations within the cellular organelles vary (nucleus (~ 0.3–0.2 mM), Golgi apparatus (~ 0.3 mM), ER (~ 0.5–0.7 mM), mitochondria ~ 0.2 mM), lysosomes (~ 0.4 mM) (note that these are approximations)). Calcium can enter the cell into the cytoplasm through plasma membrane ion channels (VGCCs, P2X, TRPs, ORAI) or ER membrane channels (IP3Rs and RyRs). Alternatively, cytosolic calcium can be depleted through mitochondrial calcium uptake (via MCU channels), ATP-driven pumps (PMCA, SERCA, SPCA), or the sodium–calcium exchanger (NCX)
This binary initiation/termination of calcium signaling explanation is the basis of understanding calcium signaling. However, the complex versatility of calcium ions in the wide variety of cell functions, as outlined above, is not possible in a binary world. Otherwise, increases in cytosolic calcium would simultaneously trigger all cell functions. Rather, the versatility of calcium ions is possible on a spectrum of calcium signals, which is the topic of the next section. Calcium signals can be big or small, be fast or long lasting, and be global or local. These amplitude, frequency, and spatial spectra ultimately regulate the discrimination between different cell functions and give the cell the tools to appropriately “choose” its fate.
Amplitude, frequency, and spatial modulation
One question when considering calcium ion flux into the cytoplasm is, how many ions enter? The amplitude of the calcium signal measured yields the answer (through the integral of the curve). The higher the amplitude, the more calcium occupies the cytoplasm, the lower the amplitude, less calcium. In a process known as amplitude modulation (AM) (Berridge 1997), these different amplitudes, or amounts of global cytosolic calcium, trigger different downstream signaling responses. For example, in B lymphocytes, changes in ~ 0.2–0.4 mM of calcium results in activation of the transcription factor, nuclear factor of activated T cells (NFAT) (i.e., nuclear translocation) without activating the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) or c-Jun N-terminal kinase 1 (JNK1) (i.e., IκBα degradation and JNK1 phosphorylation), whereas ~ 0.4–0.6 mM of calcium is required to activate NFκB/JNK1 (Dolmetsch et al. 1997). In this way, different downstream signaling pathways can be activated based on the amount of calcium permitted into the cell. This can be possible through an affinity-based mechanism whereby higher affinity calcium-binding proteins are activated with lower calcium concentrations and lower affinity calcium-binding proteins are activated with higher calcium concentrations, and/or possible via fractional occupancy-based mechanisms whereby proteins with one calcium-binding site are activated with lower calcium concentrations and proteins with up to four calcium-binding sites are activated with higher calcium concentrations (Parekh 2011). However, the limitation is that an increasing calcium signal will not activate proteins of differing affinities in a completely independent manner but will rather simply activate them sequentially. In other words, 0.6 mM of calcium will activate both NFAT and NFκB/JNK.
Calcium can also differentially activate downstream signaling pathways based on the temporal characteristics of the calcium signal. In a process known as frequency modulation (FM) (Berridge 1997), transient and repetitive elevations in cytosolic calcium, known as oscillating calcium signals, can determine different protein activation (Samanta and Parekh 2017). For example, the calcium-binding protein kinase C (PKC) can be partially activated with low-frequency calcium spikes, whereas maximal activation of PKC occurs during high-frequency calcium spikes (Oancea and Meyer 1998). Similarly, calcium- and calmodulin-dependent protein kinase II (CAMKII) (De Koninck and Schulman 1998) or calcium-sensitive mitochondrial dehydrogenases (CSMDHs) (Hajnoczky et al. 1995) can show greater activity with increasing frequency of calcium exposure. Oscillations can even discriminate between proteins within the same cell, low-frequency oscillations activate NFκB, while high frequencies activate NFAT (Dolmetsch et al. 1998). At least for NFAT, these oscillation-dependent mechanisms intriguingly seem to be entirely independent of total average calcium in the cell (i.e., amplitude) and rather specifically sensitive to a certain frequency of oscillations (Dolmetsch et al. 1998; Li et al. 1998).
Calcium-dependent activation of proteins can be further distinguished via spatial restriction of calcium signals (for more in depth reviews, see Parekh 2011; Samanta and Parekh 2017), which is in contrast to the global cytosolic calcium elevations discussed up until now, where the total cytosolic space is theoretically occupied. When considering ion flux through a single channel into the cytoplasm from the plasma membrane, free calcium concentrations indeed decrease with increasing distance from the channel (Neher 1998; Rizzuto and Pozzan 2006), which is largely due to the presence of buffers that limit the range of calcium diffusion. For example, during fast calcium influx, differences in calcium concentrations between the closest region to the inner leaflet of the plasma membrane, called the subplasma membrane, and the cytoplasm range from ~ 0.4 to 0.8 mM (Nagai et al. 2004; Pinton et al. 2002). Even at rest, there seems to be some graded calcium concentrations between the subplasma membrane and the cytoplasm (0.8 vs. 0.3 mM, respectively) (Nagai et al. 2004). Importantly, these microdomains of larger calcium concentrations relative to the cytoplasm have functional consequences. For example, cAMP response element-binding protein (CREB) phosphorylation via CAMK relies on calcium concentrations spanning 1–2 μm from the subplasma membrane (Deisseroth et al. 1996). Similarly, local signals can specifically trigger activation of transcription factors c-Fos (Di Capite et al. 2009), signal transducer and activator of transcription (STAT) 5 (Ng et al. 2009), and NFAT (Kar et al. 2011, 2016). These domain-dependent signaling pathways rely on the localization of effectors near calcium channels; therefore, re-localization of effectors away from the subplasma membrane would inhibit these pathways. Moreover, removal of cell buffering capacity may activate effectors not tethered near subplasma regions. Finally, spatial restriction of calcium signals independent of buffers are possible through elementary calcium events localized near calcium channels. The elementary calcium events are either derived from single ion channels, known as blips from inositol 1,4,5-trisphosphate receptors (IP3Rs) and quarks from ryanodine receptors (RYRs), or when groups of 10–20 channels collectively release calcium, called puffs (IP3R), and sparks (RYR) (Berridge 1997). Puffs and sparks release up to ~ 0.6 mM of calcium within a spatial spread of up to ~ 7 μm which can ultimately lead to larger calcium waves in the cell (Niggli 1999; Thomas et al. 2000).
These three means of calcium modulation are “clever” ways of getting around the calcium signaling paradox. The versatile calcium ion can be used for activating the diversity of cell functions without changing the ion itself, but rather changing the characteristics (amplitude/frequency modulation) or location (spatial modulation) of the signal. It is important to note, now that the characteristics of calcium signaling have been introduced, that these three modulation mechanisms are not necessarily mutually exclusive. For example, consider a low-affinity effector which requires a high concentration of ions for activation, but only a concentration of ions below the threshold for activation have entered a cell. Simply changing the effector’s location to near a channel where local calcium concentrations relative to the global cytosolic calcium is high enough would ensure activation, or vice versa. Thus, two or more means of modulation can be utilized in sync to add additional regulation of downstream signaling. Further considering all three, and that each sits on a spectrum of magnitude, gives the cell exponential combinations of tuning intracellular calcium signaling. These are the possibilities needed for an investigator to recognize when experimenting with calcium signaling and determining how calcium will affect the cell.
Calcium channels, buffers, and effectors
Now that the versatility of the calcium ion has been explained through spatial, amplitude, and frequency modulation, how calcium enters the cell and what it can do once inside the cell will now be discussed. There are many calcium channels, GPCRs, buffers, effectors, pumps, and calcium-sensitive enzymes that contribute the initiation of calcium signaling, the cell response, and the decay of the calcium signal. Berridge, Bootman, and Roderick have termed these elements as a cell’s “calcium signaling toolkit” (Berridge et al. 2003). As mentioned above, there are two major protein families that control the initiation of calcium signaling inside the cell: G-protein coupled receptors (GPCRs) and ion channels. GPCRs and ion channels are briefly introduced in this section, but there are extensive reviews available for the reader (Berridge et al. 2003; Tsien and Tsien 1990).
Ion channels are membrane bound proteins that form a central pore that can pass (i.e., conduct) ions when activated and open, or block ion flux when inactivated or deactivated and closed (inactivation describes a channel that has stopped conducting ions but has not fully closed, while deactivation describes a channel that has full closed). Ion conduction through an open channel is possible when there is an electrochemical potential gradient created by differential ion concentration across cell membranes, as discussed above. Some ion channels specifically permeate only one type of ion, such as potassium, sodium, calcium, or chloride; however, others can be more non-selective and can permit multiple ions. For example, transient receptor potential (TRP) channels can be permeable to multiple ions (reviewed in (Bouron et al. 2015)). The specificity in permeability not only is achieved through the charge of the residues lining the central pore region and its compatibility with an oppositely charged ion but is also governed by the geometry of the pore and its compatibility with ions of a specific size (Sun et al. 1997). These characteristics of protein structure, what is known as the selectivity filter of ion channels, are achieved on a molecular level via backbone carbonyls and specific amino acids. These specific structures of ion channels have evolved not only for specificity in ion selection (Doyle et al. 1998) but also for optimal rapid conductance of ions when the channels are open (Morais-Cabral et al. 2001). Ions are stabilized and dissolved in solution through hydration by water molecules; however, only ions and water molecules pass through channels in single file, and therefore, ions must shed their water molecules as they move through the pore (Doyle et al. 1998). Ions are then stabilized by the charged central pore as the ion passes through and water on the opposite side of the channel then re-hydrates the ion. While this may seem like a time-consuming process, ion permeation through channels is rather quite rapid because it minimizes energy constraints (Morais-Cabral et al. 2001) when compared with other forms of ion flow such as transport through pumps.
Ion channel activation can occur through a large variety of mechanisms (Fig. 1). Voltage-gated calcium channels (VGCCs) open their central pore regions (i.e., are gated) in response to changes in membrane voltage and specifically permeate calcium ions. VGCCs are typically present in excitable cells such as neurons and muscle and play roles in synaptic transmission and muscle contraction. Purinergic receptor ion channels, known as P2X channels, bind nucleotides (AMP, ADP, ATP) and permeate calcium (Valera et al. 1994). Inositol 1,4,5-trisphosphate receptors (IP3Rs) are present on the endoplasmic reticulum (ER) and permeate calcium upon the binding of inositol 1,4,5-trisphosphate (IP3). Ryanodine receptors (RYR) are present in muscle on the sarcoplasmic reticulum (SR) and in non-excitable cells on the endoplasmic reticulum (ER) and are activated by calcium in a process known as calcium-induced calcium release. RYRs can also be activated by depolarization-induced calcium release in skeletal muscle. In a process known as store-operated calcium entry (SOCE) (but has in the past been referred to as capacitative calcium entry), stromal interaction molecule (STIM)/ORAI complexes serve to replenish intracellular calcium stores after calcium release, from locations such as the ER, since some of the calcium gets pumped to the extracellular space instead of entirely back into internal stores. While both STIM/ORAI form what is known as the calcium release-activated calcium (CRAC) channel, ORAI is the pore-forming subunit that resides in the plasma membrane, while STIM is an ER calcium-sensing and ORAI activating protein in the ER membrane. Finally, there are the transient receptor potential (TRP) family of ion channels that range in selectivity but generally permit calcium and magnesium ions to pass through their central pore regions. TRP channels are composed of seven subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), TRPA (ankyrin), and TRPN (NOMPC-like) (reviewed in (Zheng 2013)). A very interesting family of channels, TRP channels can be activated in response to cold/menthol, stretch-activation, pH, calcium, and voltage (reviewed in (Clapham et al. 2001; Zheng 2013)).
In contrast to ion channels, GPCRs do not facilitate calcium movement through a central pore, but rather indirectly activate calcium signaling. GPCRs are membrane-bound proteins that are typically activated by ligands such as hormones, peptides, and neurotransmitters, but there are other activation mechanisms such as pH or mechanical stimuli. GPCRs are coupled to the G-proteins Gα, Gβ, and Gγ on the cytoplasmic side (Gα and Gγ are tethered to the plasma membrane by lipid anchors). This complex represents the inactivated state of GPCRs. Once activated, the exchange of GTP with GDP on the Gα subunit occurs and the GTP-bound Gα and Gβ/Gγ dimer decouple from the receptor and each other. The GTP-bound Gα and Gβ/Gγ dimer move on separately to activate downstream signaling. For calcium, it is the Gα subunit that activates phospholipase C (PLC) which in turn converts the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 in turn diffuses from the plasma membrane and binds to and opens IP3R channels located on the ER resulting in calcium efflux from the ER to the cytoplasm. Then Gα unbinds GTP and re-binds GDP, and Gα and Gβ/Gγ reassociate with each other as well as with the GPCR.
Once calcium has entered the cell, it quickly associates with calcium-binding proteins, which contain negatively charged and geometrically compatible protein domains or motifs (i.e., structures) that are capable of binding calcium (reviewed in (Carafoli et al. 2001)). For example, the EF-hand motifs bind two calcium ions per molecule. EF-hand motif containing calcium-binding proteins include calmodulin and the S100 protein family. There are also non-EF-hand calcium-binding proteins such as annexins, gelsolin, calreticulin, and those with C2-domains (protein kinase C is a very common enzyme with a C2-domain). Calcium-binding proteins can be generally categorized into two broad categories: buffers and effectors/sensors. While buffers/effectors are briefly introduced in the next paragraph, there are extensive reviews on buffers (Schwaller 2010; Yanez et al. 2012) and effectors (Berridge et al. 2003; Carafoli et al. 2001; Yanez et al. 2012) available for the reader.
Buffers quickly chelate calcium ions that enter the cell and ultimately can change the amplitude, frequency, and spatial characteristics of the free cytosolic calcium available to bind to effector proteins. The role of buffers in spatial modulation has already been outlined, and one can imagine how buffers can similarly affect the amplitude and frequency of calcium signaling. Therefore, buffers limit the amount of calcium freely available to a cell to activate functions (i.e., cell death, cell cycle, division, migration, invasion, metabolism, differentiation, autophagy, or transcription). Major buffers include parvalbumins, calbindin-D9k, calbindin-D28k, calretinin, calreticulin, calnexin, calsequestrin, and GRP78/94. In contrast, effectors/sensors serve to initiate downstream cell signaling pathways which ultimately lead to turning on cell functions. Several effectors have already been introduced with respect to activation under amplitude, frequency, and spatial modulation. Major effectors include the calmodulin and S100 protein families, but there are also troponin C, synaptotagmin, the annexin protein family, myosin light chain kinase, protein kinase C family, calcineurin, calmodulin-dependent protein kinases (CAMKs), calpain proteases, nitric oxide synthases, nuclear factor of activated T cell transcription factor family, cyclic AMP response element-binding protein (CREB) transcription factor, and downstream regulatory element modular (DREAM) transcription factor.
Methods for measuring intracellular calcium
The most common method for measuring calcium, and especially calcium inside cells, is through microscopy-based visualization of calcium using calcium-binding fluorescent indicators. These indicators can be either exogenous (artificially introduced within the cell) or endogenous (genetically introduced within the cell) (Rudolf et al. 2003; Tsien 1980, 1981). Exogenous indicators are commonly referred to as dyes and include green fluorescing Fluo-4, red fluorescing Rhod-2, and UV-excited Fura-2, and Indo-1. These dyes are introduced into the cell by diluting them into the extracellular media for an incubation time. By way of ester groups, they remain as uncharged molecules that can therefore freely diffuse across the cell plasma membrane. However, once inside the cell, intracellular esterases cleave these groups from the molecule, they become charged, are thus impermeable to the cell membrane, and become trapped inside (Fig. 2). Moreover, the charged molecules become sensitive to binding calcium and will become brightly fluorescent only in the calcium-bound state.

Mechanism of Fluo-4 cellular uptake. The most common method for measuring intracellular calcium is through fluorometric microscopy-based visualization of calcium using calcium-binding fluorescent indicators such as Fluo-4. Dye structures for Fluo-4 AM and Fluo-4 were derived from https://pubchem.ncbi.nlm.nih.gov/. The Fluo-4 AM molecule contains ester groups that renders the molecule uncharged and can thus freely diffuse across the cell plasma membrane into the cell (left). Once inside the cell, intracellular esterases cleave these groups from the molecule, and the molecule becomes charged and is thus impermeable to the cell membrane (right). The trapped intracellular charged Fluo-4 is also sensitive to binding calcium and will become brightly fluorescent only in the calcium-bound state
Exogenous dyes fall into two general categories, ratiometric and non-ratiometric dyes. Non-ratiometric dyes fluoresce in one color when bound by calcium. As calcium concentrations increase in the cytoplasm during calcium mobilization, calcium binds to the dye, the calcium-bound dye can then fluoresce, and the fluorescent light can be captured by a light-detecting device (photodiode, photomultiplier tube, or camera such as CCD) attached to a microscope during this process. Therefore, the changes in calcium signaling (i.e., increasing calcium, decreasing calcium, or calcium oscillations) can be visualized because changes in fluorescent light correlate with changes in calcium concentration. Calcium signaling can be qualitatively measured (i.e., relative amplitude, time course) using calculations for ΔF/F = (F − F0)/F0, where F = the fluorescence of the calcium dye when calcium signaling is occurring, and F0 = baseline fluorescence of the calcium dye before the initiation of calcium signaling. Non-ratiometric dyes are easy to use and do not require very special equipment. However, there are limitations to consider. When comparing sample to sample in an experiment, a user cannot exactly control the amount of calcium dye that gets into a single cell or on a cell to cell basis. This is a problem, because more dye will result in more fluorescence, which artificially reflects more calcium signaling. Therefore, one might be artificially recording differences in calcium signaling between samples. Yet, calculations for ΔF/F generally allow one to compare relative changes in calcium signaling between samples, since the ΔF/F = (F − F0)/F0 equation normalizes for sample to sample differences in dye concentration. The other major limitation is that users cannot measure exact calcium concentrations within a cell, or calcium concentrations between sample to sample, and they can only measure relative changes in calcium during calcium signaling (because of normalization steps). However, semi-quantitative approaches are available for non-ratiometric dyes (Maravall et al. 2000). Ratiometric dyes on the other hand can allow for one to directly compare calcium concentrations within a cell and between samples. This is because these dyes change color or excitation spectrum as calcium is bound to the dye. Therefore, users can compare the ratio of background fluorescence of the dye in one color/spectrum and changes in calcium concentration in another color/spectrum, within the same sample. In this way, differences in dye concentration can be normalized within a sample. Furthermore, the calcium concentration can then be extrapolated after the dyes are calibrated to known calcium concentrations.
Finally, there are endogenous calcium indicators that come from genetically encoded proteins. These have been engineered and are artificially introduced into cells, but once the DNA encoding the calcium indicator has been introduced, the protein-based indicators are produced by the cell from DNA and can remain intracellular. For example, GCaMP6 is a combination of green-fluorescent protein and calmodulin, which makes it a calcium-sensitive protein-based indicator that can be used to measure intracellular calcium signaling much like that of Fluo-4 (Chen et al. 2013).
Calcium signaling and cancer
So far, the detailed complexities of calcium signaling have been explained under normal contexts. However, as with many other cell signaling pathways, cancer cells can disfigure what is considered “normal” and change the prevailing choreography of calcium channel/receptor activation, intracellular calcium signaling, and downstream calcium-sensitive signaling. Moreover, overall calcium signaling modifications can be the result of adjusting any step of the calcium signaling pathway; it is possible for cancer cells to change upstream channel/receptor activation signals, cytoplasmic/organellar calcium concentrations available for calcium release, or the state or number of calcium channels, GPCRs, buffers, and effectors. Through these mechanisms, a cancer cell may overactivate, inhibit, or otherwise adjust its intracellular calcium signaling, which might serve as a survival advantage to the cell. The literature and current knowledge on how calcium signaling pathways are altered in breast cancer will be reviewed and the outcome of these changes with respect to cell function and survival will be discussed.
Altered calcium handling has indeed been implicated in cancer and there are many reviews available (Anguita and Villalobo 2018; Azimi and Monteith 2016; Azimi et al. 2014; Berchtold and Villalobo 2014; Bhargava and Saha 2019; Bong and Monteith 2018; Busselberg and Florea 2017; Chalmers and Monteith 2018; Cross et al. 2014; Cui et al. 2017; Deliot and Constantin 2015; Frisch et al. 2019; Grimm et al. 2018; Haworth and Brackenbury 2019; Hempel and Trebak 2017; Humeau et al. 2018; Iamshanova et al. 2017; Kadio et al. 2016; Kim and Wysolmerski 2016; Makena and Rao 2020; Maly and Hofmann 2018; Marchi and Pinton 2016; Martinez-Delgado and Felix 2017; Mignen et al. 2017; Missiroli et al. 2017; Mo and Yang 2018; Monteith et al. 2007; Monteith et al. 2017; Orrenius et al. 2003; Pierro et al. 2019; Prevarskaya et al. 2010, 2011; Ritaine et al. 2017; Roberts-Thomson et al. 2019; Sallan et al. 2018; So et al. 2019; Sterea and El Hiani 2020; Stewart et al. 2015; Tajbakhsh et al. 2018; Terrie et al. 2019; Tsai et al. 2015; Venkateswaran et al. 2018; Villalobos et al. 2017; Xu et al. 2018), including some excellent breast cancer-focused reviews (Azimi et al. 2014; Cross et al. 2014; Lee et al. 2006; So et al. 2019; Tajbakhsh et al. 2018). The data from the literature can be generally categorized into three groups. First, there are quantified cancer-mediated changes in the components of the calcium signaling toolkit: expression differences of calcium channels, pumps, GPCRs, and calcium-sensitive proteins from patient samples and cancer cell lines are measured against normal tissue or non-tumorigenic cell lines. Measured expression changes are often correlated with tumor grade, patient outcome, or tumorigenic capacity of cancer cells. Second, overexpression or knockout of components of the calcium signaling toolkit are used to enhance or inhibit the tumorigenic behavior of cells, which help determine how observed expression changes seen in patients and cancer cell lines affect cancer in a more mechanistic way. Third, there are direct measurements of intracellular calcium signals under different contexts, (i.e., in relation to normal vs. cancer cells or overexpression/knockout cells). It is useful to organize the data in this way as an attempt to more comprehensively understand the detailed mechanisms underlying cancer-mediated changes in calcium signaling (from receptor/channel to calcium signaling to cell function), the advantages these alterations confer to cancer, and the best approaches for designing new calcium-focused therapies.
High expression levels of calcium channels, calcium pumps, and GPCRs (Table 1; protein or mRNA measurements) have been reported in patient breast cancer tissues over normal tissue (STIM1/2 (Miao et al. 2019), ORAI3 (Faouzi et al. 2011), SPCA2 pumps (Feng et al. 2010), P2X7 channels (Tan et al. 2015), TRPA1/TRPC1/TRPC3/TRPC6/TRPC7/TRPV6/TRPM7/TRPM8 channels (Aydar et al. 2009; Bolanz et al. 2008; Chodon et al. 2010; Dhennin-Duthille et al. 2011; Guilbert et al. 2008, 2009; Liu et al. 2014; Meng et al. 2013; Takahashi et al. 2018; Tsavaler et al. 2001), IP3Rs2/3 (Singh et al. 2017b), S100 proteins (Cross et al. 2005)) and high expression is correlated with breast tumor grade (RYRs (Abdul et al. 2008), TRPV6 channels (Dhennin-Duthille et al. 2011; Peters et al. 2012), TRPM8 channels (Yapa et al. 2018), TRPV4 channels (Peters et al. 2017), SPCA1 pumps (Grice et al. 2010), ORAI1 (McAndrew et al. 2011), P2Y6 GPCRs (Azimi et al. 2016), PMCA2 pumps (Peters et al. 2016; VanHouten et al. 2010), mitochondrial calcium uniporter (MCU) (Curry et al. 2013), S100 proteins (McKiernan et al. 2011)). Cancer patient samples over normal samples have also shown increased expression of CREB1/2 (Chhabra et al. 2007; Fan et al. 2012; Sofi et al. 2003), PKCζ (Paul et al. 2015; Smalley et al. 2019), and CAMKII (Chi et al. 2016), increased nuclear localization of NFAT2 (Quang et al. 2015), and increased phosphorylation of PKCζ (Paul et al. 2015), CAMKII (Chi et al. 2016), and CREB2 (Fan et al. 2012). High expression was also associated with poor survival for S100 (McKiernan et al. 2011), CAMKII (Chi et al. 2016), CREB1 (Chhabra et al. 2007), PKCα (Lonne et al. 2010), PMCA2 pumps (VanHouten et al. 2010), STIM1 (McAndrew et al. 2011), P2Y6 (Azimi et al. 2016), and TRPV6/TRPM7 channels (Middelbeek et al. 2012; Peters et al. 2012). In contrast, other reports find low expression in breast cancer tissues over normal tissue for VGCCs (Phan et al. 2017) and SERCA3 (Papp and Brouland 2011) or low expression correlated with breast tumor grade for VGCCs (Pera et al. 2016). Other evidence shows that high expression in breast cancer over normal tissue (Wang et al. 2015) and poor survival for VGCC gene alterations (Jacquemet et al. 2016), highlighting conflicting data for VGCCs.
Similarly, human breast cancer cell lines were tested for expression differences. High expression (protein or mRNA measurements) has been reported in breast cancer cell lines compared with normal breast cell lines for SPCA2 pumps (Feng et al. 2010), PMCA1/2 pumps (Lee et al. 2002, 2005), ORAI1/3 channels (Faouzi et al. 2011; McAndrew et al. 2011), TRPC3/TRPC6/TRPM8/TRPV6 channels (Aydar et al. 2009; Liu et al. 2014; Peters et al. 2012), P2X4/5/7 channels (Jelassi et al. 2011, 2013), and P2Y2/P2Y6 GPCRs (Jin et al. 2014; Zhang et al. 2017), as well as increased phosphorylation of CAMKII (Chi et al. 2016) and CREB2 (Fan et al. 2012). Furthermore, NFATc2 (Kim et al. 2018), CREB (Son et al. 2010), and calreticulin (Lwin et al. 2010) showed higher expression in highly metastatic and tumorigenic cells over mildly tumorigenic cells. Expression differences are less clear for IP3Rs due to conflicting reports (Mound et al. 2017; Singh et al. 2017a), for RYRs and STIM due to lack of data, and for VGCCs due to variable expression patterns (Jacquemet et al. 2016).
In general, the patient and cell line data suggest that breast cancer elevates intracellular calcium concentration and signaling based on the overexpression of various plasma membrane calcium channels (P2X, TRP), intracellular release mechanisms (P2Y, RYR, IP3R), intracellular calcium store re-fill proteins (STIM/ORAI), and decreased expression of intracellular calcium store pumps (SPCA, SERCA). This notion of overactive calcium signaling is further supported by over expression and activation of calcium effectors (NFAT, CREB, CAMK, PKC). Of note, overexpression of PMCA may either conflict with these ideas (since it pumps cytoplasmic calcium outside of the cell) or it may serve to protect cancer cells from calcium overload. However, such broad conclusions would need to be supported by actual measurements of intracellular calcium concentrations or signaling. More detailed information such as spatial and frequency characteristics of the calcium signal may also be needed. In addition, whether these associations between increased expression and breast cancer are mere correlations or whether they have functional impact on breast cancer is necessary to test.
Indeed, some studies have investigated the functional impact of expression differences by targeting expression experimentally in breast cancer cells (Table 2). Increased expression of ORAI3 proteins found in cancer cells over normal cells was observed, and experimentally reducing the expression of ORAI3 inhibited cell proliferation and cell viability in cancer cells but not normal cells (Faouzi et al. 2011). Moreover, knockdown of ORAI1 or STIM1 in breast cancer cells led to reductions in in vitro migration and invasion and in vivo metastasis (Yang et al. 2009). Highly metastatic and tumorigenic cells show greater expression of IP3R3s over mildly tumorigenic cells, and knockdown of IP3R3s inhibited migration to a much greater extent in the aggressively tumorigenic and metastatic cells (Mound et al. 2017). Similarly, IP3Rs expression differences between cancer cells and normal cells was observed, but effects of downregulation of IP3R2/3s was only tested in cancer cells which reduced cancer cell viability (Singh et al. 2017a). Observations that cancer cells show increased phospho-CAMKII over normal cells guided investigators to test further overexpression of WT or phosphomimic CAMKII in cancer cells which enhanced colony formation, migration, and invasion (Chi et al. 2016). The increased expression of SPCA2 measured in cancer cells over normal cells was targeted for knockdown resulting in reduced cancer cell proliferation and colony formation (Feng et al. 2010). Likewise, high expression of TRPM8 channels in cancer cells over normal cells was targeted for knockdown to show reductions in cancer cell migration (Liu et al. 2014).
Experimental reductions in many other different calcium signaling related proteins can inhibit the tumorigenic and invasive capacity for breast cancer cells (Table 2). The knockdown of P2Y2 GPCRs was able to reduce in vivo primary tumor growth and metastatic lesions (Jin et al. 2014; Zhang et al. 2017) and in vitro invasion and migration (Jin et al. 2014), STIM1 could inhibit tumor growth and metastasis (Miao et al. 2019), PKCζ reduced in vitro migration and invasion and in vivo metastasis (Paul et al. 2015; Smalley et al. 2019), TRPM7 channels led to decreased in vitro migration and in vivo metastasis (Middelbeek et al. 2012), NFAT was able to reduce in vitro invasion (Kim et al. 2018) and in vivo tumor growth (Quang et al. 2015), TRPV6 (Bolanz et al. 2008) or TRPM7 (Guilbert et al. 2009) reduced cell viability, VGCCs reduced cell invasion (Jacquemet et al. 2016), TRPV6 reduced migration/invasion (Dhennin-Duthille et al. 2011), ORAI3 inhibited in vivo tumor growth (Motiani et al. 2013), MCU decreased in vivo tumor growth and metastasis (Tosatto et al. 2016), and proliferation of breast cancer cells was reduced when VGCCs (Taylor et al. 2008) or PMCA2 (Peters et al. 2016) were targeted for knockdown. These data show that many calcium channels, pumps, GPCRs, and effectors are necessary for the migratory, invasive, proliferative, tumorigenic, or metastatic capacity of cancer cells. These data also bolster the expression data previously outlined for breast cancer patients and cells, which suggests that high expression of calcium channels, pumps, GPCRs, and effectors is utilized by breast cancer for survival advantages.
Finally, some experiments set out to determine differences in breast cancer calcium signaling by directly measuring intracellular calcium (Table 2). Basal intracellular calcium concentrations are higher in breast cancer cells over normal cells (Jonathan Pottle et al. 2013). Cancer cells over normal cells also exhibit a reduced capacity to release calcium from intracellular stores but a more robust calcium entry during SOCE (Baldi et al. 2003; Motiani et al. 2010). Experimental reductions in expression of ORAI1/3 not only reduced cell proliferation and viability but also inhibited SOCE in cancer cells (Faouzi et al. 2011; McAndrew et al. 2011). Similarly, SOCE could be reduced with knockdown of ORAI1 or STIM1 in breast cancer cells, but which also led to reductions in in vitro migration and invasion and in vivo metastasis (Yang et al. 2009). Breast cancer cell viability could be reduced by targeting TRPM7, which was linked to reductions in resting intracellular calcium concentrations (Guilbert et al. 2009). SPCA2 knockdown resulted in reduced resting calcium concentrations as well as reduced cancer cell proliferation and colony formation (Feng et al. 2010). Knockdown of MCU channels led to decreases in cancer cell migration and SOCE (Tang et al. 2015). Reductions in cell viability and calcium influx via TRPV6 occurred after TRPV6 knockdown (Peters et al. 2012). IP3R3s knockdown led to decreased cancer cell migration that was also associated with a switch from an ATP-stimulated global intracellular calcium signal to an oscillating one (Mound et al. 2017). P2Y2 GPCR knockdown was able to reduce ATP-stimulated cytosolic calcium signaling, as well as in vivo primary tumor growth and metastatic lesions, and in vitro invasion and migration (Jin et al. 2014). Reductions in cancer cell MCU expression was able to reduce ATP-stimulated mitochondrial calcium uptake and decreased in vivo tumor growth and metastasis (Tosatto et al. 2016). Overexpression of PMCA2 was able to protect cancer cells from ionomycin-stimulated calcium overload and apoptosis (VanHouten et al. 2010). In contrast to the many examples cited above, reductions in cancer cell proliferation via PMCA2 knockdown could not be attributed to any changes in cytosolic calcium signaling (Peters et al. 2016). In general, these data suggest that breast cancer cells not only rely on these calcium-related proteins for migratory, invasive, proliferative, tumorigenic, or metastatic behaviors but also rely on the associated intracellular calcium signals. However, future studies may reveal even more complexity beyond mere changes in protein expression, as genomically unstable cancer cells inherently have a high probability for mutagenesis that could alter protein activity and calcium signaling. Indeed, some studies report that cancer cell lines which harbor many different mutations, as well as specific cancer mutations such as KRas, can alter breast epithelial calcium signaling responses (Pratt et al. 2018, 2020).
Conclusion
The collective data reviewed here highlights the potential significance for cancer-dependent calcium signaling alterations in the progression of cancer. In conjunction, the patient and cell line data support an idea that breast cancer overexpresses calcium channels, pumps, GPCRs, and effectors, which are altering intracellular calcium signaling and ultimately aiding in migratory, invasive, proliferative, tumorigenic, or metastatic behaviors. The data suggest that breast cancer tumors and cells have high concentrations of intracellular calcium and/or an elevated capacity to mobilize calcium (Fig. 3). Still, more questions remain. While numerous components of the calcium signaling pathway are disrupted in cancer cells, the specific targets that will improve cancer treatment have not yet been fully clarified. It is also worth remembering that preclinical studies in mice do not always translate to success in the treatment of human disease. Clinical trials using therapies targeting calcium signaling or calcium-related proteins will be the clearest tests. Furthermore, there may be an opportunity to repurpose existing drugs, as combination therapy of simvastatin and doxorubicin show promising results in preclinical work, which additionally establishes a calcium-based therapeutic mechanism (Abdoul-Azize et al. 2018). Still, it is unclear which therapeutic approach in targeting calcium signaling for treating human breast cancer patients will be effective, since there are many potential targets as outlined above. Finally, the measured changes in calcium signaling with genetic ablation of calcium-related proteins are still somewhat correlative. More direct approaches are needed for modulating intracellular calcium and establish whether it can affect cancer cell behavior or tumor growth, in order to fully comprehend how breast cancer is altering intracellular calcium signaling as a selective advantage for survival and progression.

Calcium signaling is altered in breast cancer. The general hypothesis of abnormally elevated calcium signaling in the pathogenesis of breast cancer is illustrated. In general, human patient and cell line data suggest that breast cancer tumors and cells have high concentrations of intracellular calcium and/or an elevated capacity to mobilize calcium. This is based on the overexpression of various plasma membrane calcium channels (P2X, TRP), intracellular release mechanisms (P2Y, RYR, IP3R), intracellular calcium store re-fill proteins (STIM/ORAI), and calcium store pumps (PMCA, SPCA). This is further supported by overexpression and activation of calcium effectors (NFAT, CREB, CAMK, PKC). Moreover, experimental data using overexpression or knockdown of many calcium channels, pumps, GPCRs, and effectors in cells show that they are necessary for the migratory, invasive, proliferative, tumorigenic, or metastatic capacity of breast cancer cells. Finally, some data measuring intracellular calcium signaling directly suggest that breast cancer cells rely not only on the calcium-related proteins but also the associated intracellular calcium signals
References
2015 CRC handbook of chemistry and physics, 96th ed. CRC Press, 2015
Abdoul-Azize S, Buquet C, Li H, Picquenot JM, Vannier JP (2018) Integration of Ca(2+) signaling regulates the breast tumor cell response to simvastatin and doxorubicin. Oncogene 37:4979–4993
Abdul M, Ramlal S, Hoosein N (2008) Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol Oncol Res 14:157–160
al-Mohanna FA, Caddy KW, Bolsover SR (1994) The nucleus is insulated from large cytosolic calcium ion changes. Nature 367:745–750
Anguita E, Villalobo A (2018) Ca(2+) signaling and Src-kinases-controlled cellular functions. Arch Biochem Biophys 650:59–74
Aydar E, Yeo S, Djamgoz M, Palmer C (2009) Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: a potential target for breast cancer diagnosis and therapy. Cancer Cell Int 9:23
Azimi I, Monteith GR (2016) Plasma membrane ion channels and epithelial to mesenchymal transition in cancer cells. Endocr Relat Cancer 23:R517–R525
Azimi I, Roberts-Thomson SJ, Monteith GR (2014) Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol 171:945–960
Azimi I, Beilby H, Davis FM, Marcial DL, Kenny PA, Thompson EW, Roberts-Thomson SJ, Monteith GR (2016) Altered purinergic receptor-Ca(2)(+) signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol Oncol 10:166–178
Baldi C, Vazquez G, Boland R (2003) Capacitative calcium influx in human epithelial breast cancer and non-tumorigenic cells occurs through Ca2+ entry pathways with different permeabilities to divalent cations. J Cell Biochem 88:1265–1272
Berchtold MW, Villalobo A (2014) The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta 1843:398–435
Berridge MJ (1997) The AM and FM of calcium signalling. Nature 386:759–760
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529
Bhakta NR, Oh DY, Lewis RS (2005) Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol 6:143–151
Bhargava A, Saha S (2019) T-type voltage gated calcium channels: a target in breast cancer? Breast Cancer Res Treat 173:11–21
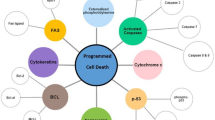
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH (2003) Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5:1051–1061
Bolanz KA, Hediger MA, Landowski CP (2008) The role of TRPV6 in breast carcinogenesis. Mol Cancer Ther 7:271–279
Bong AHL, Monteith GR (2018) Calcium signaling and the therapeutic targeting of cancer cells. Biochim Biophys Acta Mol Cell Res 1865:1786–1794
Bootman MD, Fearnley C, Smyrnias I, MacDonald F, Roderick HL (2009) An update on nuclear calcium signalling. J Cell Sci 122:2337–2350
Bouron A, Kiselyov K, Oberwinkler J (2015) Permeation, regulation and control of expression of TRP channels by trace metal ions. Pflugers Arch 467:1143–1164
Brini M, Carafoli E (2000) Calcium signalling: a historical account, recent developments and future perspectives. Cell Mol Life Sci 57:354–370
Brini M, Murgia M, Pasti L, Picard D, Pozzan T, Rizzuto R (1993) Nuclear Ca2+ concentration measured with specifically targeted recombinant aequorin. EMBO J 12:4813–4819
Brose N, Petrenko AG, Sudhof TC, Jahn R (1992) Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256:1021–1025
Brundage RA, Fogarty KE, Tuft RA, Fay FS (1991) Calcium gradients underlying polarization and chemotaxis of eosinophils. Science 254:703–706
Busselberg D, Florea (2017) AM: Targeting Intracellular Calcium Signaling ([Ca(2+)]i) to Overcome Acquired Multidrug Resistance of Cancer Cells: A Mini-Overview. Cancers (Basel) 9(5)
Carafoli E (2002) Calcium signaling: a tale for all seasons. Proc Natl Acad Sci U S A 99:1115–1122
Carafoli E, Santella L, Branca D, Brini M (2001) Generation, control, and processing of cellular calcium signals. Crit Rev Biochem Mol Biol 36:107–260
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142:270–283
Carey MB, Matsumoto SG (1999) Spontaneous calcium transients are required for neuronal differentiation of murine neural crest. Dev Biol 215:298–313
Chalmers SB, Monteith GR (2018) ORAI channels and cancer. Cell Calcium 74:160–167
Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300
Chhabra A, Fernando H, Watkins G, Mansel RE, Jiang WG (2007) Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep 18:953–958
Chi M, Evans H, Gilchrist J, Mayhew J, Hoffman A, Pearsall EA, Jankowski H, Brzozowski JS, Skelding KA (2016) Phosphorylation of calcium/calmodulin-stimulated protein kinase II at T286 enhances invasion and migration of human breast cancer cells. Sci Rep 6:33132
Chodon D, Guilbert A, Dhennin-Duthille I, Gautier M, Telliez MS, Sevestre H, Ouadid-Ahidouch H (2010) Estrogen regulation of TRPM8 expression in breast cancer cells. BMC Cancer 10:212
Christensen KA, Myers JT, Swanson JA (2002) pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 115:599–607
Clapham DE (2007) Calcium signaling. Cell 131:1047–1058
Clapham DE, Runnels LW, Strubing C (2001) The TRP ion channel family. Nat Rev Neurosci 2:387–396
Colomer J, Lopez-Girona A, Agell N, Bachs O (1994) Calmodulin regulates the expression of cdks, cyclins and replicative enzymes during proliferative activation of human T lymphocytes. Biochem Biophys Res Commun 200:306–312
Cross SS, Hamdy FC, Deloulme JC, Rehman I (2005) Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology 46:256–269
Cross BM, Breitwieser GE, Reinhardt TA, Rao R (2014) Cellular calcium dynamics in lactation and breast cancer: from physiology to pathology. Am J Physiol Cell Physiol 306:C515–C526
Cui C, Merritt R, Fu L, Pan Z (2017) Targeting calcium signaling in cancer therapy. Acta Pharm Sin B 7:3–17
Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR (2013) Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 434:695–700
De Koninck P, Schulman H (1998) Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279:227–230
Deisseroth K, Bito H, Tsien RW (1996) Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16:89–101
Deliot N, Constantin B (2015) Plasma membrane calcium channels in cancer: alterations and consequences for cell proliferation and migration. Biochim Biophys Acta 1848:2512–2522
Dhennin-Duthille I, Gautier M, Faouzi M, Guilbert A, Brevet M, Vaudry D, Ahidouch A, Sevestre H, Ouadid-Ahidouch H (2011) High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: correlation with pathological parameters. Cell Physiol Biochem 28:813–822
Di Capite J, Ng SW, Parekh AB (2009) Decoding of cytoplasmic Ca(2+) oscillations through the spatial signature drives gene expression. Curr Biol 19:853–858
Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386:855–858
Dolmetsch RE, Xu K, Lewis RS (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392:933–936
Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294:333–339
Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77
Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Berra-Romani R, Carlo Bongio G, Cinelli MP, Guerra G, Pedrazzoli P, Rosti V, Tanzi F, Moccia F (2011) Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 29:1898–1907
Fan CF, Mao XY, Wang EH (2012) Elevated p-CREB-2 (ser 245) expression is potentially associated with carcinogenesis and development of breast carcinoma. Mol Med Rep 5:357–362
Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H (2011) Down-regulation of ORAI3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol 226:542–551
Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJ, Monteith GR, Rao R (2010) Store-independent activation of ORAI1 by SPCA2 in mammary tumors. Cell 143:84–98
Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410:41–49
Frisch J, Angenendt A, Hoth M, Prates Roma L, Lis A (2019) STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment. Cancers (Basel) 11(4)
Gergely J, Grabarek Z, Tao T (1993) The molecular switch in troponin C. Adv Exp Med Biol 332:117–123
Giannone G, Ronde P, Gaire M, Beaudouin J, Haiech J, Ellenberg J, Takeda K (2004) Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J Biol Chem 279:28715–28723
Grice DM, Vetter I, Faddy HM, Kenny PA, Roberts-Thomson SJ, Monteith GR (2010) Golgi calcium pump secretory pathway calcium ATPase 1 (SPCA1) is a key regulator of insulin-like growth factor receptor (IGF1R) processing in the basal-like breast cancer cell line MDA-MB-231. J Biol Chem 285:37458–37466
Grimm C, Bartel K, Vollmar AM, Biel M (2018) Endolysosomal Cation Channels and Cancer-A Link with Great Potential. Pharmaceuticals (Basel) 11(1)
Grodsky GM, Bennett LL (1966) Cation requirements for insulin secretion in the isolated perfused pancreas. Diabetes 15:910–913
Guilbert A, Dhennin-Duthille I, Hiani YE, Haren N, Khorsi H, Sevestre H, Ahidouch A, Ouadid-Ahidouch H (2008) Expression of TRPC6 channels in human epithelial breast cancer cells. BMC Cancer 8:125
Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H (2009) Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol 297:C493–C502
Hahn K, DeBiasio R, Taylor DL (1992) Patterns of elevated free calcium and calmodulin activation in living cells. Nature 359:736–738
Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82:415–424
Haworth AS, Brackenbury WJ (2019) Emerging roles for multifunctional ion channel auxiliary subunits in cancer. Cell Calcium 80:125–140
Hempel N, Trebak M (2017) Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 63:70–96
Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH (1980) Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell 19:245–254
Holliday J, Adams RJ, Sejnowski TJ, Spitzer NC (1991) Calcium-induced release of calcium regulates differentiation of cultured spinal neurons. Neuron 7:787–796
Humeau J, Bravo-San Pedro JM, Vitale I, Nunez L, Villalobos C, Kroemer G, Senovilla L (2018) Calcium signaling and cell cycle: progression or death. Cell Calcium 70:3–15
Iamshanova O, Fiorio Pla A, Prevarskaya N (2017) Molecular mechanisms of tumour invasion: regulation by calcium signals. J Physiol 595:3063–3075
Ikeda M, Sugiyama T, Wallace CS, Gompf HS, Yoshioka T, Miyawaki A, Allen CN (2003) Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron 38:253–263
Ivannikov MV, Macleod GT (2013) Mitochondrial free Ca(2)(+) levels and their effects on energy metabolism in Drosophila motor nerve terminals. Biophys J 104:2353–2361
Jacquemet G, Baghirov H, Georgiadou M, Sihto H, Peuhu E, Cettour-Janet P, He T, Perala M, Kronqvist P, Joensuu H, Ivaska J (2016) L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat Commun 7:13297
Jelassi B, Chantome A, Alcaraz-Perez F, Baroja-Mazo A, Cayuela ML, Pelegrin P, Surprenant A, Roger S (2011) P2X(7) receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 30:2108–2122
Jelassi B, Anchelin M, Chamouton J, Cayuela ML, Clarysse L, Li J, Gore J, Jiang LH, Roger S (2013) Anthraquinone emodin inhibits human cancer cell invasiveness by antagonizing P2X7 receptors. Carcinogenesis 34:1487–1496
Jin H, Eun SY, Lee JS, Park SW, Lee JH, Chang KC, Kim HJ (2014) P2Y2 receptor activation by nucleotides released from highly metastatic breast cancer cells increases tumor growth and invasion via crosstalk with endothelial cells. Breast Cancer Res 16:R77
Jonathan Pottle CS, Gray L, Li M (2013) Exploiting MCF-7 cells’ calcium dependence with interlaced therapy. J Cancer Ther 4(7):32–40
Kadio B, Yaya S, Basak A, Dje K, Gomes J, Mesenge C (2016) Calcium role in human carcinogenesis: a comprehensive analysis and critical review of literature. Cancer Metastasis Rev 35:391–411
Kaestner L (2013) Perspective, calcium signalling: approaches and findings in the heart and blood. Springer Berlin Heidelberg, Berlin, pp 29–34
Kahl CR, Means AR (2004) Calcineurin regulates cyclin D1 accumulation in growth-stimulated fibroblasts. Mol Biol Cell 15:1833–1842
Kar P, Nelson C, Parekh AB (2011) Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem 286:14795–14803
Kar P, Mirams GR, Christian HC, Parekh AB (2016) Control of NFAT isoform activation and NFAT-dependent gene expression through two coincident and spatially segregated intracellular Ca(2+) signals. Mol Cell 64:746–759
Kato Y, Ozawa S, Tsukuda M, Kubota E, Miyazaki K, St-Pierre Y, Hata R (2007) Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J 274:3171–3183
Kim W, Wysolmerski JJ (2016) Calcium-sensing receptor in breast physiology and cancer. Front Physiol 7:440
Kim GC, Kwon HK, Lee CG, Verma R, Rudra D, Kim T, Kang K, Nam JH, Kim Y, Im SH (2018) Upregulation of Ets1 expression by NFATc2 and NFKB1/RELA promotes breast cancer cell invasiveness. Oncogenesis 7:91
Kratz A, Ferraro M, Sluss PM, Lewandrowski KB (2004) Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Laboratory reference values. N Engl J Med 351:1548–1563
Launikonis BS, Zhou J, Royer L, Shannon TR, Brum G, Rios E (2005) Confocal imaging of [Ca2+] in cellular organelles by SEER, shifted excitation and emission ratioing of fluorescence. J Physiol 567:523–543
Lee WJ, Roberts-Thomson SJ, Holman NA, May FJ, Lehrbach GM, Monteith GR (2002) Expression of plasma membrane calcium pump isoform mRNAs in breast cancer cell lines. Cell Signal 14:1015–1022
Lee WJ, Roberts-Thomson SJ, Monteith GR (2005) Plasma membrane calcium-ATPase 2 and 4 in human breast cancer cell lines. Biochem Biophys Res Commun 337:779–783
Lee WJ, Monteith GR, Roberts-Thomson SJ (2006) Calcium transport and signaling in the mammary gland: targets for breast cancer. Biochim Biophys Acta 1765:235–255
Li W, Llopis J, Whitney M, Zlokarnik G, Tsien RY (1998) Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 392:936–941
Liu J, Chen Y, Shuai S, Ding D, Li R, Luo R (2014) TRPM8 promotes aggressiveness of breast cancer cells by regulating EMT via activating AKT/GSK-3beta pathway. Tumour Biol 35:8969–8977
Lonne GK, Cornmark L, Zahirovic IO, Landberg G, Jirstrom K, Larsson C (2010) PKCalpha expression is a marker for breast cancer aggressiveness. Mol Cancer 9:76
Lwin ZM, Guo C, Salim A, Yip GW, Chew FT, Nan J, Thike AA, Tan PH, Bay BH (2010) Clinicopathological significance of calreticulin in breast invasive ductal carcinoma. Mod Pathol 23:1559–1566
Makena MR, Rao R (2020) Subtype specific targeting of calcium signaling in breast cancer. Cell Calcium 85:102109
Maly IV, Hofmann WA (2018) Calcium and Nuclear Signaling in Prostate Cancer. Int J Mol Sci 19(4)
Maravall M, Mainen ZF, Sabatini BL, Svoboda K (2000) Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J 78:2655–2667
Marchi S, Pinton P (2016) Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol 29:1–6
Martinez-Delgado G, Felix R (2017) Emerging role of CaV1.2 channels in proliferation and migration in distinct cancer cell lines. Oncology 93:1–10
McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, Monteith GR (2011) ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther 10:448–460
McDonough PM, Button DC (1989) Measurement of cytoplasmic calcium concentration in cell suspensions: correction for extracellular Fura-2 through use of Mn2+ and probenecid. Cell Calcium 10:171–180
McKiernan E, McDermott EW, Evoy D, Crown J, Duffy MJ (2011) The role of S100 genes in breast cancer progression. Tumour Biol 32:441–450
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17:288–299
Meng X, Cai C, Wu J, Cai S, Ye C, Chen H, Yang Z, Zeng H, Shen Q, Zou F (2013) TRPM7 mediates breast cancer cell migration and invasion through the MAPK pathway. Cancer Lett 333:96–102
Miao YL, Stein P, Jefferson WN, Padilla-Banks E, Williams CJ (2012) Calcium influx-mediated signaling is required for complete mouse egg activation. Proc Natl Acad Sci U S A 109:4169–4174
Miao Y, Shen Q, Zhang S, Huang H, Meng X, Zheng X, Yao Z, He Z, Lu S, Cai C, Zou F (2019) Calcium-sensing stromal interaction molecule 2 upregulates nuclear factor of activated T cells 1 and transforming growth factor-beta signaling to promote breast cancer metastasis. Breast Cancer Res 21:99
Middelbeek J, Kuipers AJ, Henneman L, Visser D, Eidhof I, van Horssen R, Wieringa B, Canisius SV, Zwart W, Wessels LF, Sweep FC, Bult P, Span PN, van Leeuwen FN, Jalink K (2012) TRPM7 is required for breast tumor cell metastasis. Cancer Res 72:4250–4261
Mignen O, Constantin B, Potier-Cartereau M, Penna A, Gautier M, Gueguinou M, Renaudineau Y, Shoji KF, Felix R, Bayet E, Buscaglia P, Debant M, Chantome A, Vandier C (2017) Constitutive calcium entry and cancer: updated views and insights. Eur Biophys J 46:395–413
Missiroli S, Danese A, Iannitti T, Patergnani S, Perrone M, Previati M, Giorgi C, Pinton P (2017) Endoplasmic reticulum-mitochondria Ca(2+) crosstalk in the control of the tumor cell fate. Biochim Biophys Acta Mol Cell Res 1864:858–864
Mo P, Yang S (2018) The store-operated calcium channels in cancer metastasis: from cell migration, invasion to metastatic colonization. Front Biosci (Landmark Ed) 23:1241–1256
Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ (2007) Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 7:519–530
Monteith GR, Prevarskaya N, Roberts-Thomson SJ (2017) The calcium-cancer signalling nexus. Nat Rev Cancer 17:367–380
Morais-Cabral JH, Zhou Y, MacKinnon R (2001) Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature 414:37–42
Motiani RK, Abdullaev IF, Trebak M (2010) A novel native store-operated calcium channel encoded by ORAI3: selective requirement of ORAI3 versus ORAI1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem 285:19173–19183
Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M (2013) ORAI3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis. FASEB J 27:63–75
Mound A, Vautrin-Glabik A, Foulon A, Botia B, Hague F, Parys JB, Ouadid-Ahidouch H, Rodat-Despoix L (2017) Downregulation of type 3 inositol (1,4,5)-trisphosphate receptor decreases breast cancer cell migration through an oscillatory Ca(2+) signal. Oncotarget 8:72324–72341
Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A (2004) Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A 101:10554–10559
Neher E (1998) Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20:389–399
Neville MC (2005) Calcium secretion into milk. J Mammary Gland Biol Neoplasia 10:119–128
Neville MC, Keller RP, Casey C, Allen JC (1994) Calcium partitioning in human and bovine milk. J Dairy Sci 77:1964–1975
Ng SW, Nelson C, Parekh AB (2009) Coupling of Ca(2+) microdomains to spatially and temporally distinct cellular responses by the tyrosine kinase Syk. J Biol Chem 284:24767–24772
Niggli E (1999) Localized intracellular calcium signaling in muscle: calcium sparks and calcium quarks. Annu Rev Physiol 61:311–335
Oancea E, Meyer T (1998) Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 95:307–318
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552–565
Papp B, Brouland JP (2011) Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer (Auckl) 5:163–174
Parekh AB (2011) Decoding cytosolic Ca2+ oscillations. Trends Biochem Sci 36:78–87
Paul A, Danley M, Saha B, Tawfik O, Paul S (2015) PKCzeta promotes breast cancer invasion by regulating expression of E-cadherin and zonula occludens-1 (ZO-1) via NFkappaB-p65. Sci Rep 5:12520
Pera E, Kaemmerer E, Milevskiy MJG, Yapa K, O'Donnell JS, Brown MA, Simpson F, Peters AA, Roberts-Thomson SJ, Monteith GR (2016) The voltage gated Ca(2+)-channel Cav3.2 and therapeutic responses in breast cancer. Cancer Cell Int 16:24
Peters AA, Simpson PT, Bassett JJ, Lee JM, Da Silva L, Reid LE, Song S, Parat MO, Lakhani SR, Kenny PA, Roberts-Thomson SJ, Monteith GR (2012) Calcium channel TRPV6 as a potential therapeutic target in estrogen receptor-negative breast cancer. Mol Cancer Ther 11:2158–2168
Peters AA, Milevskiy MJ, Lee WC, Curry MC, Smart CE, Saunus JM, Reid L, da Silva L, Marcial DL, Dray E, Brown MA, Lakhani SR, Roberts-Thomson SJ, Monteith GR (2016) The calcium pump plasma membrane Ca(2+)-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci Rep 6:25505
Peters AA, Jamaludin SYN, Yapa K, Chalmers S, Wiegmans AP, Lim HF, Milevskiy MJG, Azimi I, Davis FM, Northwood KS, Pera E, Marcial DL, Dray E, Waterhouse NJ, Cabot PJ, Gonda TJ, Kenny PA, Brown MA, Khanna KK, Roberts-Thomson SJ, Monteith GR (2017) Oncosis and apoptosis induction by activation of an overexpressed ion channel in breast cancer cells. Oncogene 36:6490–6500
Phan NN, Wang CY, Chen CF, Sun Z, Lai MD, Lin YC (2017) Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol Lett 14:2059–2074
Pierro C, Sneyers F, Bultynck G, Roderick HL (2019) ER Ca(2+) release and store-operated Ca(2+) entry - partners in crime or independent actors in oncogenic transformation? Cell Calcium 82:102061
Pinton P, Pozzan T, Rizzuto R (1998) The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J 17:5298–5308
Pinton P, Tsuboi T, Ainscow EK, Pozzan T, Rizzuto R, Rutter GA (2002) Dynamics of glucose-induced membrane recruitment of protein kinase C beta II in living pancreatic islet beta-cells. J Biol Chem 277:37702–37710
Pratt SJP, Hernandez-Ochoa EO, Lee RM, Ory EC, Lyons JS, Joca HC, Johnson A, Thompson K, Bailey P, Lee CJ, Mathias T, Vitolo MI, Trudeau M, Stains JP, Ward CW, Schneider MF, Martin SS (2018) Real-time scratch assay reveals mechanisms of early calcium signaling in breast cancer cells in response to wounding. Oncotarget 9:25008–25024
Pratt SJP, Lee RM, Chang KT, Hernández-Ochoa EO, Annis DA, Ory EC, Thompson KN, Bailey PC, Mathias TJ, Ju JA, Vitolo MI, Schneider MF, Stains JP, Ward CW, Martin SS (2020) Mechanoactivation of NOX2-generated ROS elicits persistent TRPM8 Ca2+ signals that are inhibited by oncogenic KRas. Proc Natl Acad Sci U S A 117(42):26008–26019. https://doi.org/10.1073/pnas.2009495117
Prentki M, Wollheim CB (1984) Cytosolic free Ca2+ in insulin secreting cells and its regulation by isolated organelles. Experientia 40:1052–1060
Prevarskaya N, Skryma R, Shuba Y (2010) Ion channels and the hallmarks of cancer. Trends Mol Med 16:107–121
Prevarskaya N, Skryma R, Shuba Y (2011) Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer 11:609–618
Przywara DA, Bhave SV, Bhave A, Wakade TD, Wakade AR (1991) Stimulated rise in neuronal calcium is faster and greater in the nucleus than the cytosol. FASEB J 5:217–222
Quang CT, Leboucher S, Passaro D, Fuhrmann L, Nourieh M, Vincent-Salomon A, Ghysdael J (2015) The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis 6:e1658
Rasmussen CD, Means AR (1989) Calmodulin is required for cell-cycle progression during G1 and mitosis. EMBO J 8:73–82
Ratto GM, Payne R, Owen WG, Tsien RY (1988) The concentration of cytosolic free calcium in vertebrate rod outer segments measured with fura-2. J Neurosci 8:3240–3246
Ritaine A, Shapovalov G, Prevarskaya N (2017) Metabolic disorders and cancer: store-operated Ca(2+) entry in cancer: focus on IP3R-mediated Ca(2+) release from intracellular stores and its role in migration and invasion. Adv Exp Med Biol 993:623–637
Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86:369–408
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13:566–578
Roberts-Thomson SJ, Chalmers SB, Monteith GR (2019) The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb Perspect Biol 11(8)
Rudolf R, Mongillo M, Rizzuto R, Pozzan T (2003) Looking forward to seeing calcium. Nat Rev Mol Cell Biol 4:579–586
Sallan MC, Visa A, Shaikh S, Nager M, Herreros J, Canti C (2018) T-type Ca(2+) channels: T for targetable. Cancer Res 78:603–609
Samanta K, Parekh AB (2017) Spatial Ca(2+) profiling: decrypting the universal cytosolic Ca(2+) oscillation. J Physiol 595:3053–3062
Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM, Swann K, Lai FA (2002) PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 129:3533–3544
Schwaller B (2010) Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol 2:a004051
Singh A, Chagtoo M, Tiwari S, George N, Chakravarti B, Khan S, Lakshmi S, Godbole MM (2017a) Inhibition of inositol 1, 4, 5-trisphosphate receptor induce breast cancer cell death through deregulated autophagy and cellular bioenergetics. J Cell Biochem 118:2333–2346
Singh A, Sharma RK, Chagtoo M, Agarwal G, George N, Sinha N, Godbole MM (2017b) 1H NMR metabolomics reveals association of high expression of inositol 1, 4, 5 trisphosphate receptor and metabolites in breast cancer patients. PLoS One 12:e0169330
Smalley T, Islam SMA, Apostolatos C, Apostolatos A, Acevedo-Duncan M (2019) Analysis of PKC-zeta protein levels in normal and malignant breast tissue subtypes. Oncol Lett 17:1537–1546
So CL, Saunus JM, Roberts-Thomson SJ, Monteith GR (2019) Calcium signalling and breast cancer. Semin Cell Dev Biol 94:74–83
Sofi M, Young MJ, Papamakarios T, Simpson ER, Clyne CD (2003) Role of CRE-binding protein (CREB) in aromatase expression in breast adipose. Breast Cancer Res Treat 79:399–407
Son J, Lee JH, Kim HN, Ha H, Lee ZH (2010) cAMP-response-element-binding protein positively regulates breast cancer metastasis and subsequent bone destruction. Biochem Biophys Res Commun 398:309–314
Sorenson MM, da Silva AC, Gouveia CS, Sousa VP, Oshima W, Ferro JA, Reinach FC (1995) Concerted action of the high affinity calcium binding sites in skeletal muscle troponin C. J Biol Chem 270:9770–9777
Steinhardt R, Zucker R, Schatten G (1977) Intracellular calcium release at fertilization in the sea urchin egg. Dev Biol 58:185–196
Sterea AM, El Hiani Y (2020) The role of mitochondrial calcium signaling in the pathophysiology of cancer cells. Adv Exp Med Biol 1131:747–770
Stewart TA, Yapa KT, Monteith GR (2015) Altered calcium signaling in cancer cells. Biochim Biophys Acta 1848:2502–2511
Sun YM, Favre I, Schild L, Moczydlowski E (1997) On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel. Effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. J Gen Physiol 110:693–715
Sun J, Lu F, He H, Shen J, Messina J, Mathew R, Wang D, Sarnaik AA, Chang WC, Kim M, Cheng H, Yang S (2014) STIM1- and ORAI1-mediated Ca(2+) oscillation orchestrates invadopodium formation and melanoma invasion. J Cell Biol 207:535–548
Tajbakhsh A, Pasdar A, Rezaee M, Fazeli M, Soleimanpour S, Hassanian SM, FarshchiyanYazdi Z, Younesi Rad T, Ferns GA, Avan A (2018) The current status and perspectives regarding the clinical implication of intracellular calcium in breast cancer. J Cell Physiol 233:5623–5641
Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, Deraedt T, Cichowski K, Welm AL, Mori Y, Mills GB, Brugge JS (2018) Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell 33:985–1003.e1007
Tan C, Han LI, Zou L, Luo C, Liu A, Sheng X, Xi D (2015) Expression of P2X7R in breast cancer tissue and the induction of apoptosis by the gene-specific shRNA in MCF-7 cells. Exp Ther Med 10:1472–1478
Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F (2015) Mitochondrial Ca(2)(+) uniporter is critical for store-operated Ca(2)(+) entry-dependent breast cancer cell migration. Biochem Biophys Res Commun 458:186–193
Taylor JT, Huang L, Pottle JE, Liu K, Yang Y, Zeng X, Keyser BM, Agrawal KC, Hansen JB, Li M (2008) Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett 267:116–124
Terrie E, Coronas V, Constantin B (2019) Role of the calcium toolkit in cancer stem cells. Cell Calcium 80:141–151
Thomas D, Lipp P, Tovey SC, Berridge MJ, Li W, Tsien RY, Bootman MD (2000) Microscopic properties of elementary Ca2+ release sites in non-excitable cells. Curr Biol 10:8–15
Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C (2016) The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol Med 8:569–585
Tsai FC, Kuo GH, Chang SW, Tsai PJ (2015) Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res Int 2015:409245
Tsavaler L, Shapero MH, Morkowski S, Laus R (2001) Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res 61:3760–3769
Tsien RY (1980) New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry 19:2396–2404
Tsien RY (1981) A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290:527–528
Tsien RW, Tsien RY (1990) Calcium channels, stores, and oscillations. Annu Rev Cell Biol 6:715–760
Valera S, Hussy N, Evans RJ, Adami N, North RA, Surprenant A, Buell G (1994) A new class of ligand-gated ion channel defined by P2x receptor for extracellular ATP. Nature 371:516–519
VanHouten J, Sullivan C, Bazinet C, Ryoo T, Camp R, Rimm DL, Chung G, Wysolmerski J (2010) PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proc Natl Acad Sci U S A 107:11405–11410
Venkateswaran K, Verma A, Bhatt AN, Shrivastava A, Manda K, Raj HG, Prasad A, Len C, Parmar VS, Dwarakanath BS (2018) Emerging roles of calreticulin in cancer: implications for therapy. Curr Protein Pept Sci 19:344–357
Villalobos C, Sobradillo D, Hernandez-Morales M, Nunez L (2017) Calcium remodeling in colorectal cancer. Biochim Biophys Acta Mol Cell Res 1864:843–849
Wang CY, Lai MD, Phan NN, Sun Z, Lin YC (2015) Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PLoS One 10:e0125766
Williams DA, Fogarty KE, Tsien RY, Fay FS (1985) Calcium gradients in single smooth muscle cells revealed by the digital imaging microscope using Fura-2. Nature 318:558–561
Xu M, Seas A, Kiyani M, Ji KSY, Bell HN (2018) A temporal examination of calcium signaling in cancer - from tumorigenesis, to immune evasion, and metastasis. Cell Biosci 8:25
Yanez M, Gil-Longo J, Campos-Toimil M (2012) Calcium binding proteins. Adv Exp Med Biol 740:461–482
Yang S, Huang XY (2005) Ca2+ influx through L-type Ca2+ channels controls the trailing tail contraction in growth factor-induced fibroblast cell migration. J Biol Chem 280:27130–27137
Yang S, Zhang JJ, Huang XY (2009) ORAI1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 15:124–134
Yapa K, Deuis J, Peters AA, Kenny PA, Roberts-Thomson SJ, Vetter I, Monteith GR (2018) Assessment of the TRPM8 inhibitor AMTB in breast cancer cells and its identification as an inhibitor of voltage gated sodium channels. Life Sci 198:128–135
Zhang JL, Liu Y, Yang H, Zhang HQ, Tian XX, Fang WG (2017) ATP-P2Y2-beta-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci 108:1318–1327
Zheng J (2013) Molecular mechanism of TRP channels. Compr Physiol 3:221–242
Funding
This work was supported by the METAvivor Foundation (SSM) and Veterans Administration (SSM, BX002746), as well as grants from the National Institutes of Health to SSM (R01-CA124704, R01-CA154624), SJPP (5T32GM008181-30, 1F31CA232393-01).
Author information
Authors and Affiliations
Contributions
Drafted the manuscript: SJPP. Prepared the figures: SJPP and EHO. Revised the manuscript: SJPP, EHO, and SSM. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pratt, S.J., Hernández-Ochoa, E. & Martin, S.S. Calcium signaling: breast cancer’s approach to manipulation of cellular circuitry. Biophys Rev 12, 1343–1359 (2020). https://doi.org/10.1007/s12551-020-00771-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-020-00771-9