
Abstract
The KEAP1-NRF2 system is a sulfur-employing defense mechanism against oxidative and electrophilic stress. NRF2 is a potent transcription activator for genes mediating sulfur-involving redox reactions, and KEAP1 controls the NRF2 activity in response to the stimuli by utilizing reactivity of sulfur atoms. In many human cancer cells, the KEAP1-mediated regulation of NRF2 activity is abrogated, resulting in the persistent activation of NRF2. Persistently activated NRF2 drives malignant progression of cancers by increasing therapeutic resistance and promoting aggressive tumorigenesis, a state termed as NRF2 addiction. In NRF2-addicted cancer cell, NRF2 contributes to metabolic reprogramming in cooperation with other oncogenic pathways. In particular, NRF2 strongly activates cystine uptake coupled with glutamate excretion and glutathione synthesis, which increases consumption of intracellular glutamate. Decreased availability of glutamate limits anaplerosis of the TCA cycle, resulting in low mitochondrial respiration, and nitrogen source, resulting in the high dependency on exogenous non-essential amino acids. The highly enhanced glutathione synthesis is also likely to alter sulfur metabolism, which can contribute to the maintenance of the mitochondrial membrane potential in normal cells. The potent antioxidant and detoxification capacity supported by abundant production of glutathione is achieved at the expense of central carbon metabolism and requires skewed metabolic flow of sulfur. These metabolic features of NRF2 addiction status provide clues for novel therapeutic strategies to target NRF2-addicted cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
KEAP1-NRF2 system as a sulfur-utilizing defense mechanism
KEAP1-NRF2 system plays a central role in the defense mechanism from oxidative and electrophilic stresses (Yamamoto et al. 2018) (Fig. 1). NRF2 is a potent transcriptional activator regulating many cytoprotective genes that are involved in detoxification and anti-oxidant function. KEAP1 is a negative regulator of NRF2, serving as a substrate-recognizing subunit of CUL3-based ubiquitin E3 ligase for NRF2 (Yamamoto et al. 2018).

KEAP1-NRF2 system for oxidative stress response. NRF2 is a transcription activator and regulates many cytoprotective genes. In unstressed condition, NRF2 is bound by KEAP1 and ubiquitinated for degradation. In response to oxidative stress, NRF2 is stabilized, translocates into the nucleus, heterodimerizes with small Maf (sMAF), and activates transcription
One of the most important features of the KEAP1-NRF2 system is its transient and inducible nature (Yamamoto et al. 2018). Under unstressed condition, NRF2 is constantly ubiquitinated by KEAP1-CUL3 ubiquitin E3 ligase, in which KEAP1 serves as a substrate recognition subunit. When cells are exposed to oxidative and/or electrophilic stress, KEAP1 thiol residues are directly modified, which inactivates the ubiquitin E3 ligase activity of KEAP1-CUL3 complex, leading to stabilization of NRF2. Namely, KEAP1 is a redox biosensor utilizing reactivity of sulfur atoms, switching on the NRF2-mediated transcriptional activation of cytoprotective genes in response to the redox disturbance.
A major output of the NRF2 transcriptional program is involved in redox regulation in which sulfur atoms play a central role, such as glutathione synthesis and reduction, thioredoxin synthesis and reduction, and cystine uptake (Yamamoto et al. 2018). Thus, whereas KEAP1 is a sulfur-utilizing sensor, NRF2 is a sulfur-regulating effector, both together comprising a sulfur-employing defense mechanism.
NRF2 addiction in cancer cells
In many human cancers, the transient and inducible nature of the KEAP1-NRF2 system is lost. NRF2 is persistently stabilized, resulting in increased amplitude and duration of NRF2 pathway activation. NRF2 pathway activation promotes tumorigenesis by enabling aggressive proliferation and metastasis (Romero et al. 2017; Lignitto et al. 2019) and conferring therapeutic resistance to current standards of care, including chemotherapy and immunotherapy (Jeong et al. 2020; Arbour et al. 2018). These cancer cells are highly dependent on NRF2 activity for their survival and proliferation, a state termed NRF2 addiction (Kitamura et al. 2017; Kitamura and Motohashi 2018).
The aberrant activation of NRF2 is found in various cancers including the lung, gallbladder, esophagus, breast, head and neck, and renal cancers (Shibata et al. 2008; Wang et al. 2010; Shibata et al. 2011; Inoue et al. 2012; Onodera et al. 2014; Kanamori et al. 2015; Martinez et al. 2015; Cancer Genome Atlas Research Network 2016). One of the major causes of aberrant activation of NRF2 in cancer cells is somatic mutation in KEAP1 or NRF2 (NFE2L2) genes. In particular, KEAP1 and NRF2 mutations are encountered in 20–30% of non-small cell lung cancers (Cancer Genome Atlas Research Network 2012; Imielinski et al. 2012; Cancer Genome Atlas Research Network 2014; Campbell et al. 2016). In most cases, they are loss-of-function mutations of KEAP1 or gain-of-function mutations of NRF2, causing the persistent stabilization of NRF2.
Of note, in normal cells, persistent activation of NRF2 due to functional defects of KEAP1-mediated NRF2 degradation is sometimes deleterious, while transient activation of NRF2 in response to various stimuli can be beneficial for our health. Constitutive activation of NRF2 in hematopoietic stem cells in mice promotes proliferation and eventual exhaustion (Murakami et al. 2017) and shortens lifespan of drosophila (Tsakiri et al. 2019). It seems that persistent NRF2 activation increases anti-oxidant and detoxification capacities at the expense of juvenescence. Moreover, from a view point of cell competition, persistent activation of NRF2 primes cells for their elimination by wild-type neighbors, conferring the loser status (Kucinski et al. 2017). These results suggest that NRF2 addiction in cancer cells is established only after overcoming disadvantages and liabilities accompanying the persistent transcriptional activation mediated by NRF2.
“NRF2 addiction” of many cancer cells highlights that NRF2 itself or its target genes should be the most effective therapeutic target from a cell-autonomous point of view. NRF2 is very pleiotropic and there is redundancy in its transcriptional output, therefore targeting of specific NRF2 targets has been therapeutically ineffective. Furthermore, like most transcription factors, direct targeting of NRF2 itself has been extremely challenging. However, if an NRF2 inhibitor existed, administration of such an inhibitors to cancer-bearing patients may not be so advantageous considering the important role of NRF2 in normal cells for stress response and cytoprotection. For example, NRF2 inhibition in cancer-bearing hosts, especially in myeloid cell lineage, promotes metastatic colonization of cancer cells in the lung by augmenting the activity of myeloid derived suppressor cells (Satoh et al. 2010; Hiramoto et al. 2014). On the other hand, oxidative stress in regulatory T cells strengthen their suppressor activity, and administration of an NRF2-inducing reagent sulforaphane inhibits tumor growth in allograft experiment (Maj et al. 2017), suggesting that NRF2 activation in cancer-bearing hosts is beneficial. Therefore, alternative therapeutic targets other than NRF2 itself are required for controlling NRF2-addicted cancer cells.
Trans-omics approach for understanding molecular basis of NRF2-driven cancer malignancy
Several studies have demonstrated that NRF2 addiction of cancer cells is supported by unique metabolic activities (Fig. 2) (Mitsuishi et al. 2012; DeNicola et al. 2015; Romero et al. 2017). Mitsuishi et al. examined NRF2-dependent transcriptome and NRF2 cistrome (genome-wide NRF2 binding sites) using one of the KEAP1-mutant non-small cell lung cancer (NSCLC) cell lines, A549 cells, and found that several metabolic genes, those involved in the pentose phosphate pathway and NADPH production as well as glutathione synthesis, are directly regulated by NRF2. The NRF2-dependent transcriptome data and NRF2 antibody ChIP-seq data were combined with NRF2-dependent metabolome data to verify the functional contribution of the NRF2 target genes to the metabolic regulation. 13C-labeled glucose and glutamine were used to trace the destiny of the labeled carbons, which was an effective strategy to show that NRF2 activation skewed the metabolite flow in the cells. Enhancement of purine nucleotide synthesis via the pentose phosphate pathway was found to be advantageous for cell proliferation and tumorigenesis of NRF2-addicted cancer cells (Mitsuishi et al. 2012).

Contribution of NRF2 to metabolism in proliferating cells. Whereas NRF2 mainly contributes to antioxidant function and detoxification in normal quiescent cells, NRF2 contributes to metabolic reprogramming in proliferating cells, facilitating pentose phosphate pathway, and serine synthesis.
DeNicola et al. examined serine/glycine synthesis activity of multiple NSCLC cell lines by flux analysis using 13C-labeled glucose, which were then compared with transcriptome data of these cell lines. The serine/glycine synthesis activities were found positively correlated with expression levels of NRF2 target genes. Although NRF2 does not directly regulate serine synthesizing enzyme genes, ATF4, a key regulator of serine synthesis, was shown to be activated by NRF2. This comprehensive study revealed that serine synthesis branching from the glycolytic intermediates 3-phosphoglycerate is enhanced in NSCLC cells with NRF2 activation (DeNicola et al. 2015).
While multiple advantageous aspects of the metabolic reprogramming for NRF2-addicted cancer cells have been described, metabolic liabilities resulting from NRF2-dependent reprogramming have also been investigated. Romero et al. conducted a CRISPR/Cas9 genetic screen with KEAP1-mutant lung cancer cells and found that NRF2-addicted cancer cells are highly dependent on glutamine uptake, and that, consequently, inhibition of glutamine-derived glutamate by glutaminase inhibition effectively suppresses NRF2-addicted lung cancers (Romero et al. 2017; Mukhopadhyay et al. 2020). Satisfying the increased demand for glutamine/glutamate is likely to be one of the critical requirements for the establishment of NRF2-addiction in cancer cells.
Potent antioxidant function at the expense of central carbon metabolism
The dependency of NRF2-addicted cancer cells on exogenous glutamine is attributable to the elevated consumption and excretion of glutamate as a result of two major transcriptional outputs of NRF2 (Fig. 3, lower panel). One is glutamate excretion coupled with cystine uptake through xCT, an antiporter of cystine and glutamate. The other is glutamate incorporation into glutathione catalyzed by gamma-glutamylcysteine ligase (γGCL), a rate limiting enzyme of glutathione synthesis (Sayin et al. 2017). Indeed, combination of exome, transcriptome, and metabolome analyses of multiple NSCLC cell lines also clearly demonstrated that remarkable enhancement of glutamate excretion, cystine uptake, and glutathione synthesis is a striking and reproducible feature of NRF2-addicted cancer cells (Saigusa et al. 2019).

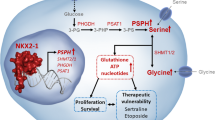
Unique metabolic signature of NRF2-addicted cancer cells. Cells with intact KEAP1-NRF2 system, regardless of normal cells and cancer cells, transiently activated NRF2 promotes glutathione synthesis. Glucose and glutamine are catabolized in mitochondria and fuel TCA cycle. Cysteine is converted to cysteine persulfide (Cys-SSH) by CARS2 and also contributes to the maintenance of the mitochondrial membrane potential. Cysteine is also converted to cysteine sulfinic acid by CDO1. Glutathione is effective to quench reactive oxygen species (ROS) generated from mitochondria. In contrast, NRF2-addicted cancer cells exhibit remarkable enhancement of cystine uptake via xCT and glutathione synthesis, which is coupled with enhanced glutamine uptake to supplement glutamate and CDO1 suppression to further increase the cysteine availability. Glucose-derived serine synthesis also supports glutathione synthesis. Mitochondrial respiration is limited due to redirection of metabolites, which instead flow into glutathione synthesis and other anabolic pathways
In cells with intact KEAP1-NRF2 system, transiently stabilized NRF2 induces transcriptional activation of genes encoding xCT, SLC7A11, and two subunits of gamma-glutamylcysteine ligase (γGCL), GCLC and GCLM, in response to oxidative and electrophilic stresses. Because the induction of these genes is only transient, the glutamate consumption is likely to be buffered by steady-state central carbon metabolism, under which glucose and glutamate fuel mitochondria to support cellular bioenergetic demands (Fig. 3, upper panel). In contrast, persistently stabilized NRF2 in response to KEAP1 or NRF2 mutations causes sustained activation of these genes, leading to the accumulation of xCT/SLC7A11, GCLC, and GCLM that cause a depletion of intracellular glutamate (Sayin et al. 2017). In addition, NRF2-addicted cells exhibit further elevated expression of SLC7A11, GCLC, and GCLM due to additional cooperative input to the NRF2-mediated transcriptional activation. Namely, sustained activation of PI3K-AKT pathway dramatically promotes the accumulation of NRF2 by inhibiting KEAP1-independent degradation mechanism of NRF2, resulting in the augmentation of NRF2-mediated transcriptional activation (Mitsuishi et al. 2012; Taguchi et al. 2014). This is because the sustained activation of PI3K-AKT pathway inhibits GSK3, which phosphorylates NRF2 and allows its recognition and ubiquitination by βTrCP-CUL1 ubiquitin E3 ligase for degradation. As for another cooperative factor, ATF4 activation has a synergistic effect with NRF2 activation on xCT expression (Ye et al. 2014; Mimura et al. 2019). Consequently, cystine uptake coupled with glutamate excretion and glutathione synthesis are enhanced (Fig. 3, lower panel).
In order to maintain intracellular glutamine-derived glutamate levels, NRF2-addicted cells exploit glutamine transporter, SLC1A5, to uptake more extracellular glutamine that is then converted to glutamate by glutaminase (Romero et al. 2017). NRF2 redirects glutamate to glutathione synthesis and cystine uptake and away from TCA cycle anaplerosis, therefore limiting glutamate as a carbon source for TCA cycle and mitochondrial activity (Sayin et al. 2017). Additionally, the enhancement of serine synthesis pathway in NRF2-addicted cancer cells (DeNicola et al. 2015) can promote glycine availability for glutathione synthesis (Yang and Vousden 2016; Rodriguez et al. 2019). Thus, NRF2-addicted cancers adopt highly specialized metabolism favoring glutathione synthesis that is essential for the potent anti-oxidant and detoxification capacities, which cannot be achieved by a simple activation of NRF2.
Cysteine catabolism for mitochondrial energy production
A recent study revealed that mitochondrial sulfur metabolism makes an important contribution to the cellular energy production (Akaike et al. 2017). A mitochondrial enzyme CARS2 generates cysteine persulfide from cysteine, which is regarded as one of the major processes for production of persulfides, i.e., molecular species containing more than one sulfur atoms in thiol moiety. Persulfides possess dual reactivities as electrophiles and nucleophiles (Fletcher and Robson 1963; Parker and Kharasch 1959; Abdolrasulnia and Wood 1980). This unique chemical property makes persulfides favorable substrates for the energy production that basically relies on multiple steps of redox reactions. Indeed, CARS2 inhibition in KEAP1-NRF2-intact cells decreased the mitochondrial membrane potential and oxygen consumption rate (Akaike et al. 2017).
CARS2 is originally known as a mitochondrial isoform of cysteinyl t-RNA synthetase, and the persulfide synthesizing activity is a moon-lighting function of CARS2. The cysteinyl t-RNA synthetases of various species possess four highly conserved motifs. Two of them are critical for the cysteinyl t-RNA synthesis and subsequent protein translation, and the other two are critical for the persulfide synthesis. CARS2 mutant lacking the persulfide synthesis activity did not rescue the decreased mitochondrial membrane potential of CASR2-deficient cells, whereas CARS2 mutant lacking cysteinyl t-RNA synthesis activity did. Thus, mitochondrial persulfide production is regarded necessary for the mitochondrial energy metabolism (Akaike et al. 2017).
The sulfur atoms in persulfides participate in the energy metabolism in mitochondria serving as electron donors as well as electron acceptors and are mostly likely to be excreted as thiosulfate eventually. While the cysteine decomposition is coupled with mitochondrial bioenergetics in the cells with intact KEAP1-NRF2 system, in NRF2-addicted cancer cells, most of the available cysteine appears to flow into the anabolic pathway, namely, glutathione synthesis and leaves the catabolic pathway in mitochondria, which is consistent with the observation that mitochondrial respiration is limited in NRF2-addicted cancer cells (Sayin et al. 2017). Moreover, an alternative pathway of cysteine catabolism, which is mediated by CDO1, has been shown to be inactive in NRF2-addicted cancer cells due to DNA methylation at the CDO1 locus (Kang et al. 2019). CDO1 converts cysteine to cysteine sulfinic acid, resulting in the limited availability of cysteine for the anabolic pathway. Thus, by silencing CDO1, NRF2-addicted cancer cells maintain a large intracellular pool of cysteine for the glutathione synthesis, resulting from the increased cysteine uptake by xCT and decreased cysteine decomposition.
Future therapeutic strategy for controlling NRF2-addicted cancers based on their metabolic features
The dependency on glutamine and glutamate is regarded as an Achilles’ heel of NRF2-addicted cancer cells. Therefore, targeting glutaminolysis using glutaminase inhibitors presents a promising therapeutic approach for aggressive subtypes of lung cancer with mutations in KEAP1/NRF2 (Romero et al. 2017). Another interesting approach emerges by focusing on an important role of glutamine and glutamate in the synthesis of non-essential amino acids (NEAAs) (LeBoeuf et al. 2019). Although NEAAs can be synthesized by cells, many cancer cells tend to be dependent on the exogenous supply of NEAAs due to their highly proliferative nature and increased demand of amino acids (Tsun and Possemato 2015). Given that glutamate is a critical nitrogen donor for transamination reactions that generate most NEAAs, limited availability of glutamate in NRF2-addicted cancer cells results in increased dependency on exogenous NEAAs (LeBoeuf et al. 2019). Therefore, restriction of NEAAs, such as serine, glycine, and asparagine, in the tumor microenvironment can effectively inhibit tumorigenesis of NRF2-addicted cancer cells.
To overcome the difficulties in targeting NRF2 for controlling NRF2-addicted cancers, targeting metabolic liabilities unique to NRF2-addicted cancer cells, such as dependency on exogenous NEAAs, including glutamine, is a very sensible way to achieve selective toxicity toward cancers that have fallen in NRF2 addiction. Importantly, persistent activation of NRF2 that is induced pharmacologically in KEAP1-NRF2-intact cancer cells also decreases NEAA synthesis and makes them rely on exogenous NEAAs (LeBoeuf et al. 2019). Administration of NRF2 activator, which would be favorable for reinvigorating anti-tumor immunity and conferring the metabolic liabilities on cancer cells, in combination with inhibitors of glutamate and/or NEAA synthesis may be an effective anti-tumor therapy from the viewpoint of interaction between cancer cells and their microenvironment.
NRF2-addicted cancer cells are very well adapted to the greatly enhanced generation of antioxidant and detoxification capacity by establishing unique metabolism. With abundant synthesis of glutathione and NADPH, a redox balance in NRF2-addicted cancer cells is likely to be shifted toward the reducing condition. An excessive shift in the balance would generate reductive stress leading to the protein aggregates and proteotoxicity as was reported in myocardium (Rajasekaran et al. 2011; Kannan et al. 2013). Further enhancement of the reducing condition might be an alternative approach for the selective toxicity to NRF2-addicted cancer cells by provoking the reductive stress.
References
Abdolrasulnia R, Wood JL (1980) Persulfide properties of thiocystine and related trisulfides. Bioorg Chem 9:253–260
Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, Ihara H, Sawa T, Matsunaga T, Kasamatsu S, Nishimura A, Morita M, Tomizawa K, Nishimura A, Watanabe S, Inaba K, Shima H, Tanuma N, Jung M, Fujii S, Watanabe Y, Ohmuraya M, Nagy P, Feelisch M, Fukuto JM, Motohashi H (2017) Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun 8:1177
Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, Lito P, Berger M, Solit DB, Hellmann M, Kris MG, Rudin CM, Ni A, Arcila M, Ladanyi M, Riely GJ (2018) Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res 24:334–340
Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, Imielinski M, Hu X, Ling S, Akbani R, Rosenberg M, Cibulskis C, Ramachandran A, Collisson EA, Kwiatkowski DJ, Lawrence MS, Weinstein JN, Verhaak RG, Wu CJ, Hammerman PS, Cherniack AD, Getz G, Cancer Genome Atlas Research Network, Artyomov MN, Schreiber R, Govindan R, Meyerson M (2016) Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607–616
Cancer Genome Atlas Research Network (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489:519–525
Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543–550
Cancer Genome Atlas Research Network (2016) Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 374:135–145
DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, Wistuba II, Minna JD, DeBerardinis RJ, Cantley LC (2015) NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47:1475–1481
Fletcher JC, Robson A (1963) The occurrence of bis-(2-amino-2-carboxyethyl) trisulphide in hydrolysates of wool and other proteins. Biochem J 87:553–559
Hiramoto K, Satoh H, Suzuki T, Moriguchi T, Pi J, Shimosegawa T, Yamamoto M (2014) Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev Res (Phila) 7:835–844
Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansén S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Jänne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M (2012) Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150:1107–1120
Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, Sasano H, Nakagawa T, Satoh K, Tanaka N, Kubo H, Motohashi H, Yamamoto M (2012) Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci 103:760–766
Jeong Y, Hellyer JA, Stehr H, Hoang NT, Niu X, Das M, Padda SK, Ramchandran K, Neal JW, Wakelee H, Diehn M (2020) Role of KEAP1/NFE2L2 mutations in the chemotherapeutic response of patients with non-small cell lung cancer. Clin Cancer Res 26:274–281
Kanamori M, Higa T, Sonoda Y, Murakami S, Dodo M, Kitamura H, Taguchi K, Shibata T, Watanabe M, Suzuki H, Shibahara I, Saito R, Yamashita Y, Kumabe T, Yamamoto M, Motohashi H, Tominaga T (2015) Activation of the NRF2 pathway and its impact on the prognosis of anaplastic glioma patients. Neuro-Oncology 17:555–565
Kang YP, Torrente L, Falzone A, Elkins CM, Liu M, Asara JM, Dibble CC, DeNicola GM (2019) Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 8. https://doi.org/10.7554/eLife.45572
Kannan S, Muthusamy VR, Whitehead KJ, Wang L, Gomes AV, Litwin SE, Kensler TW, Abel ED, Hoidal JR, Rajasekaran NS (2013) Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc Res 100:63–73
Kitamura H, Motohashi H (2018) NRF2 addiction in cancer cells. Cancer Sci 109:900–911
Kitamura H, Onodera Y, Murakami S, Suzuki T, Motohashi H (2017) IL-11 contribution to tumorigenesis in an NRF2 addiction cancer model. Oncogene 36:6315–6324
Kucinski I, Dinan M, Kolahgar G, Piddini E (2017) Chronic activation of JNK JAK/STAT and oxidative stress signalling causes the loser cell status. Nat Commun 8:136
LeBoeuf SE, Wu WL, Karakousi TR, Karadal B, Jackson SR, Davidson SM, Wong KK, Koralov SB, Sayin VI, Papagiannakopoulos T (2019) Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab 31(2):339–350
Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B, Sayin VI, Papagiannakopoulos T, Pagano M (2019) Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 178:316–329.e18
Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, Lyssiotis C, Liu JR, Kryczek I, Zou W (2017) Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol 18:1332–1341
Martinez VD, Vucic EA, Thu KL, Pikor LA, Lam S, Lam WL (2015) Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: association with poor prognosis in head and neck cancer. Head Neck 37:727–734
Mimura J, Inose-Maruyama A, Taniuchi S, Kosaka K, Yoshida H, Yamazaki H, Kasai S, Harada N, Kaufman RJ, Oyadomari S, Itoh K (2019) Concomitant Nrf2- and ATF4-activation by carnosic acid cooperatively induces expression of cytoprotective genes. Int J Mol Sci 20. https://doi.org/10.3390/ijms20071706
Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22:66–79
Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, Kozlov SV, Nissley DV, McCormick F (2020) Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-19-1363
Murakami S, Suzuki T, Harigae H, Romeo PH, Yamamoto M, Motohashi H (2017) NRF2 activation impairs quiescence and bone marrow reconstitution capacity of hematopoietic stem cells. Mol Cell Biol 37. https://doi.org/10.1128/MCB.00086-17
Onodera Y, Motohashi H, Takagi K, Miki Y, Shibahara Y, Watanabe M, Ishida T, Hirakawa H, Sasano H, Yamamoto M, Suzuki T (2014) NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr Relat Cancer 21:241–252
Parker AJ, Kharasch N (1959) The scission of the sulfur-sulfur bond. Chem Rev 59:583–628
Rajasekaran NS, Varadharaj S, Khanderao GD, Davidson CJ, Kannan S, Firpo MA, Zweier JL, Benjamin IJ (2011) Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid Redox Signal 14:957–971
Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, Friedman A, Manfredi MG, Weinberg SE, Rabinowitz JD, Chandel NS (2019) Serine metabolism supports macrophage IL-1β production. Cell Metab 29:1003–1011.e4
Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez-Rivera FJ, Subbaraj L, Martinez B, Bronson RT, Prigge JR, Schmidt EE, Thomas CJ, Goparaju C, Davies A, Dolgalev I, Heguy A, Allaj V, Poirier JT, Moreira AL, Rudin CM, Pass HI, Vander Heiden MG, Jacks T, Papagiannakopoulos T (2017) Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23:1362–1368
Saigusa D, Motoike IN, Saito S, Zorzi M, Aoki Y, Kitamura H, Suzuki M, Katsuoka F, Ishii H, Kinoshita K, Motohashi H, Yamamoto M (2019) Impacts of NRF2 activation in non-small-cell lung cancer cell lines on extracellular metabolites. Cancer Sci 111(2):667–678
Satoh H, Moriguchi T, Taguchi K, Takai J, Maher JM, Suzuki T, Winnard PT Jr, Raman V, Ebina M, Nukiwa T, Yamamoto M (2010) Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 31:1833–1843
Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, Ubriaco J, Muir A, Karagiannis D, Morris PJ, Thomas CJ, Possemato R, Vander Heiden MG, Papagiannakopoulos T (2017) Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6. https://doi.org/10.7554/eLife.28083.001
Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S (2008) Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A 105:13568–13573
Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T, Yamamoto M (2011) NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 13:864–873
Taguchi K, Hirano I, Itoh T, Tanaka M, Miyajima A, Suzuki A, Motohashi H, Yamamoto M (2014) Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol Cell Biol 34:900–913
Tsakiri EN, Gumeni S, Iliaki KK, Benaki D, Vougas K, Sykiotis GP, Gorgoulis VG, Mikros E, Scorrano L, Trougakos IP (2019) Hyperactivation of Nrf2 increases stress tolerance at the cost of aging acceleration due to metabolic deregulation. Aging Cell 18:e12845
Tsun ZY, Possemato R (2015) Amino acid management in cancer. Semin Cell Dev Biol 43:22–32
Wang J, Zhang M, Zhang L, Cai H, Zhou S, Zhang J, Wang Y (2010) Correlation of Nrf2, HO-1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J Surg Res 164:e99–e105
Yamamoto M, Kensler TW, Motohashi H (2018) The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 98:1169–1203
Yang M, Vousden KH (2016) Serine and one-carbon metabolism in cancer. Nat Rev Cancer 16:650–662
Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, Ohyama C, Itoh K (2014) Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol 34:3421–3434
Acknowledgments
We thank current and past colleagues in our laboratories and collaborators for active contribution to the research on NRF2 and cancers. T.P. was supported by the National Institute of Health (1 K22 CA201088-01; 1R37CA222504-01A1; 1R01CA227649-01) and American Cancer Society Research Scholar Grant (RSG-17-200-01 – TBE). H.M. was supported by Japan Society for the Promotion of Science (18H02621; 18H04794), the Naito Foundation, a research grant from the Princess Takamatsu Cancer Research Fund 15-24728, the Uehara Memorial Foundation, and Japan Agency for Medical Research and Development (JP19gm5010002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to be disclosed in this review article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okazaki, K., Papagiannakopoulos, T. & Motohashi, H. Metabolic features of cancer cells in NRF2 addiction status. Biophys Rev 12, 435–441 (2020). https://doi.org/10.1007/s12551-020-00659-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-020-00659-8