
Abstract
A liquid chromatography tandem mass spectrometry (LC–MS/MS) multi-mycotoxin method was developed for the analysis of the Alternaria toxins alternariol (AOH), alternariol monomethyl ether (AME), tentoxin (TEN), altertoxin I (ATX I), altertoxin II (ATX II), alterperylenol (ALTP), and altenuene (ALT), as well as the modified toxins AOH-3-glucoside (AOH-3-G), AOH-9-glucoside (AOH-9-G), AME-3-glucoside (AME-3-G), AOH-3-sulfate (AOH-3-S), and AME-3-sulfate (AME-3-S) in barley and malt. The toxin tenuazonic acid (TeA) was analyzed separately as it could not be included into the multi-mycotoxin method. Quantitation was conducted by using a combination of stable isotope dilution analysis (SIDA) for AOH, AME, and TeA, and matrix-matched calibration for all other toxins. Limits of detection were between 0.05 µg/kg (AME) and 2.45 µg/kg (ALT), whereas limits of quantitation ranged from 0.16 µg/kg (AME) to 8.75 µg/kg (ALT). Recoveries between 96 and 107% were obtained for the analytes when SIDA was applied, while recoveries between 84 and 112% were found for analytes quantified by matrix-matched calibration. The method was applied for the analysis of 50 barley samples and their respective malts from the harvest years 2016–2020 for their mycotoxin content, showing the overall potential of toxin formation during the malting process. The toxins ALTP and ATX I were mainly found in the malt samples, but not in barley.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Black molds of the fungal species Alternaria spp. are ubiquitously present in the environment and are suspected to pose a risk to human health due to their ability to form various mycotoxins. Until now, over 100 different species of Alternaria have been identified with A. alternata, A. citri, A. solani, and A. brassicae being the most dominant ones (Woudenberg et al. 2013).
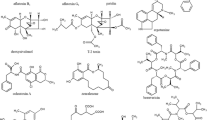
The most known Alternaria toxins are alternariol (AOH), alternariol monomethyl ether (AME), tenuazonic acid (TeA), tentoxin (TEN), altertoxin I (ATX I), altertoxin II (ATX II), alterperylenol (ALTP), altenuene (ALT), and stemphyltoxin III (STTX III) (Fig. 1). Also, the modified metabolites AOH-3-glucoside (AOH-3-G), AOH-9-glucoside (AOH-9-G), AME-3-glucoside (AME-3-G), AOH-3-sulfate (AOH-3-S), and AME-3-sulfate (AME-3-S) gained increased attention recently, as they might release their parent toxin during digestion and, therefore, contribute to the exposure level of AOH and AME (EFSA 2011).
Generally, the extent of mycotoxin production by fungi depends on external factors like temperature and water content (Lacey 1992), but also e.g. on the pH of the substrate, the infected kind of plant (Bottalico and Logrieco 1998; Lacey 1992), and the respective fungal species (Bottalico and Logrieco 1998; Grabarkiewicz-Szczesna and Chełkowski 1992). Concerning Alternaria, the species A. alternata was found to be the main mycotoxin producer whereas A. solani, A. brassicae, and A. dauci are mainly known to only produce AOH and AME (Bottalico and Logrieco 1998; Gotthardt et al. 2019a; Grabarkiewicz-Szczesna and Chełkowski 1992). Also, several studies have already demonstrated the ability of Alternaria spp. to grow in a temperature range between 1 and 35 °C and to produce mycotoxins between 10 and 35 °C, reaching maximal concentrations at 25–28 °C (Bottalico and Logrieco 1998; Lee et al. 2015; Magan and Lacey 1985). However, the exact temperature range can vary between different Alternaria species (Lacey 1992). In addition, higher humidity promotes both fungal growth and mycotoxin formation, with an aw value of 0.90 and 0.98 as ideal condition for the formation of TeA and AOH, respectively (Magan and Lacey 1985).
The main food commodities that provide good growing conditions for Alternaria spp. are fruits, vegetables, and grains (Bottalico and Logrieco 1998; Grabarkiewicz-Szczesna and Chełkowski 1992; Logrieco et al. 2009; Scott 2001; Strandberg 1992). As barley is mainly used in its malted form in the brewing and bakery industry, Alternaria toxins should also be targeted in malt due to the elevated water content and temperatures during the malting process, which can promote fungal growth and mycotoxin production (Noots et al. 1999). The possibility of mycotoxin formation during this processing step was already shown for some Fusarium toxins (Habler et al. 2016), and therefore should be analyzed for other fungi as well.
To our knowledge, there is only limited data about Alternaria mycotoxins in malt published, yet. Therefore, we developed and validated a multi-mycotoxin liquid chromatography tandem mass spectrometry (LC–MS/MS) method for the analysis of 13 Alternaria mycotoxins in barley and malt. We then analyzed 50 barley samples and the respective standardized produced malts prepared from them to get a first insight into mycotoxin occurrence in barley and their possible formation during the malting process. In this study, the modified toxins AOH-3-G, AOH-9-G, AME-3-G, AOH-3-S, and AME-3-S were analyzed as well, which is quite important as data about those modified forms are still scarce and are only available for beer, sunflower oil, tomato products, cereals, fruits, and vegetables so far (Puntscher et al. 2018; Scheibenzuber et al. 2021; Walravens et al. 2014, 2016).
Materials and methods
Chemicals and reference standards
Water was purchased from Th. Geyer (Renningen, Germany), methanol (MeOH) from Honeywell Riedel-de Haën (Seelze, Germany), and acetonitrile (ACN) and the ammonia solution (25%) were purchased from VWR (Ismaning, Germany), all at least in analytical grade. Reference standards for TEN, ALT, and TeA were bought from Merck (Darmstadt, Germany), while AOH, AME, ATX I, ATX II, STTX III, ALTP, AOH-3-G, and AOH-9-G, AME-3-G as well as AOH-3-S and AME-3-S were either isolated out of fungal extracts or synthesized at our chair as described previously (Liu and Rychlik 2015; Scheibenzuber et al. 2020, 2021). The stable isotope–labelled standards [2H4]-AOH, [2H4]-AME, and [13C6, 15 N]-TeA were synthesized in our laboratory as reported in literature (Asam et al. 2009, 2011a).

Chemical structures of the six most frequent Alternaria mycotoxins AOH, AME, TeA, TEN, ALTP, and ATX I
Preparation of stock solutions
All reference compounds were quantified by quantitative nuclear magnetic resonance (qNMR) measurements after their purchase, synthesis, or isolation as described by Frank et al. (2014). Afterwards, different stock solutions were prepared in either acetonitrile (TEN, ATX I, ATX II, ALT, ALTP, AOH-3-G, AOH-9-G, AME-3-G, STTX III) or methanol (AOH, AME, TeA, AOH-3-S, AME-3-S) in a concentration range from 1 to 100 µg/mL. To regularly verify the respective concentrations of the stock solutions, diluted standards were transferred into precision cells made out of quartz glass (Hellma GmbH & Co. KG, Müllheim, Germany) and measured with a UV spectrophotometer (Genesys, 10S, UV–Vis spectrophotometer, Thermo Fisher Scientific, Madison, WI, USA). Concentrations were then calculated by the respective molar extinction coefficients, which were either obtained from literature or determined in previous studies (Cole et al. 2003; Fleck 2014; Scheibenzuber et al. 2020; Scheibenzuber et al. 2021). Due to the instability of the analyte STTX III even when stored at − 80 °C, this toxin was only included qualitatively into the LC–MS/MS method.
Samples
Fifty industrial malting barley samples from the harvest years 2016 to 2020 were analyzed before and after malting. Barley and malt samples from 2016 were finely ground and stored at − 18 °C prior to analysis in 2018, and all other samples were analyzed within a few weeks after sample receipt.
Sampling of barley
Barley sampling at the various sample locations was conducted as specified by the Mitteleuropäische Brautechnische Analysenkommission (R-110.00.001 [2016–03], Jacob and Mitteleuropäische Brautechnische Analysenkommission 2011): using grain samplers, individual barley samples were taken from at least 10% of the grain bags and were combined to create a composite barley sample (CBS) of at least 5 kg, which was then sent to TUM. After arrival, a sample divider (Pfeuffer GmbH, Kitzingen, Germany) was used for all CBS to create representative laboratory barley samples of 0.5–1 kg (LBS). From each LBS, 50 g was taken as a subsample for mycotoxin analysis (mycotoxin analysis barley sample, MABS), respectively.
Malting process
Malting was conducted every year shortly after sample receipt. One kilogram of each LBS was malted in a micro-malting procedure at the chair of brewing and beverage technology (TUM) according to MEBAK R-110.00.008 [2016–03], and standard malt parameters were analyzed based on the isothermal 65 C laboratory mashing regime R-207.00.002 [2016–03] (Jacob and Mitteleuropäische Brautechnische Analysenkommission 2011). In short, steeping was carried out at 14 °C for 48 h according to the following scheme: 5-h steeping, 19-h air rest, 4-h steeping, and 20-h air rest; the degree of steeping was 45%. After steeping, germination took place for 96 h at 14 °C, resulting in a green malt with a water content of 45%. Afterwards, kilning was conducted as follows: 16 h at 50 °C, 1 h at 60 °C, 1 h at 70 °C, and 5 h at 80 °C. The kilned malt was then cleaned to remove the rootlets from the grains. From each obtained malt, 50 g was taken as subsample for mycotoxin analysis (mycotoxin analysis malt sample, MAMS), respectively.
Sample grinding
The whole amount (50 g) of each MABS and MAMS was ground in a laboratory mill (Grindomix GM200, Retsch GmbH, Germany) to a fine powder and then stored at − 18 °C until further use.
Sample preparation for mycotoxin analysis
One gram of each sample was weighted in duplicate into a 50 mL centrifuge tube and spiked with 100 µL of a 0.1 µg/mL [2H4]-AOH solution and 100 µL of a 0.1 µg/mL [13C6, 15 N]-TeA solution, as well as with 100 µL of a 0.01 µg/mL [2H4]-AME solution. Then, 15 mL ACN/H2O (84/16, v/v) was added to the centrifuge tubes and samples were extracted on a horizontal shaker at 225 rpm for 1 h. After centrifugation for 5 min at 3220 g, the supernatant was transferred into a 50 mL pear-shaped flask. To the remaining residue, 15 mL of ACN/H2O (84/16, v/v) and 200 µL of formic acid were added, and samples were extracted on a horizontal shaker at 225 rpm for 30 min. Again, samples were centrifuged at 3220 g for 5 min and the supernatants were transferred into their corresponding flasks. After a third extraction, which was identical to the second one, solvents were evaporated using a rotary evaporator (40 °C). The remaining residue was taken up in 12 mL of water before further clean-up of the extracts. For that, Discovery® DSC-18 cartridges (500 mg/6 mL, Sigma-Aldrich, Bellefonte, PA, USA) were preconditioned with 6 mL MeOH and 6 mL of water, followed by loading the sample onto the column. After two washing steps, one with 6 mL H2O and one with 6 mL ACN/H2O (1.5/8.5, v/v), analytes were eluted with 6 mL MeOH and 9 mL MeOH/2% NH4OH, successively. All solvents were evaporated using a rotary evaporator (40 °C). Samples were taken up in 1 mL ACN/H2O (3/7, v/v), membrane-filtered (PVDF, 0.2 µm), and stored at − 18 °C until LC–MS/MS measurements.
LC–MS/MS analysis
Liquid chromatography was conducted on a Shimadzu Nexera X2 UHPLC system (Shimadzu, Kyoto, Japan) as previously published (Scheibenzuber et al. 2021). Chromatographic separation was performed on a Hyperclone BDS C18 column (150 × 3.2 mm, 3 µm, 130 Å, Phenomenex, Aschaffenburg, Germany) for all toxins except TeA, which was analyzed separately on a Gemini-NX C18 column (150 × 4.6 mm, 3 µm, 110 Å, Phenomenex, Aschaffenburg, Germany). The binary gradient for the multi-mycotoxin method was set as follows: 0–2 min 10% B, 2–2.5 min 10–18% B, 2.5–10.5 min 18% B, 10.5–14 min 18–40% B, 14–20 min 40% B, 20–23 min 40–100% B, 23–25 min 100% B, 25–27 min 100–10% B, and 27–32 min 10% B. The flow rate was set to 0.3 mL/min, solvents were water (A) and acetonitrile (B), the injection volume was 10 µL, and the column oven was tempered to 40 °C. For the analysis of TeA, solvent A was a 5 mM ammonium formate solution (adjusted to pH 9 with ammonia solution), and solvent B was methanol as published previously (Asam et al. 2013). The flow rate was 0.5 mL/min, the injection volume 10 µL, and the oven temperature was set to 40 °C. Here, the binary gradient was as follows: 0–3 min 5% B, 5–8 min 5–100% B, 8–10 min 100% B, 10–13 min 100–5% B, and 13–24 min 5% B. Due to fourfold solvent selection for each pump and a column oven for up to six columns, both methods could be run in sequence by using the automated column switching function.
The LC system was interfaced with a Shimadzu 8050 triple quadrupole mass spectrometer (Shimadzu Corporation, Kyoto, Japan). The ion source parameters are listed in the following: heat block temperature 400 °C, interface temperature 300 °C, desolvation temperature 250 °C, interface voltage 4 kV, drying gas flow 10 L/min, heating gas flow 10 L/min, nebulizing gas flow 3 L/min, and collision-induced dissociation gas pressure 270 kPa. All measurements were operated in the negative electrospray ionization (ESI) mode and the multiple reaction monitoring (MRM) mode was used. To optimize MS parameters for each analyte, standard solutions of every toxin (0.01 µg/mL to 1 µg/mL) were directly injected into the source. From the six optimized mass transition, the two most dominant ones were chosen for quantification and for identification. Mass transitions for each toxin as well as their retention times, final collision energies, and optimized voltages are listed in Table 1. For data acquisition and data analysis, the LabSolutions Software (Shimadzu, Kyoto, Japan) was used.
Calibration and quantitation
For toxins that were quantified by SIDA, response curves were measured by mixing constant amounts of the stable isotope–labelled standards (S) [2H4]-AOH, [2H4]-AME and [13C6,15 N]-TeA with different amounts of their non-labelled forms (A) to obtain molar ratios in the range from 0.001 to 100 (1:100, 1:50, 1:20, 1:10, 1:5, 1:2, 1:1, 2:1, 5:1, 10:1, 50:1, and 100:1). After measuring those standard mixtures with the LC–MS/MS method, peak area ratios [A(A)/A(S)] were calculated and plotted against the corresponding molar ratios [n(A)/n(S)] to obtain a response function after linear regression of the data.
For all other toxins, matrix-matched calibration curves were measured, using a mycotoxin-free barley sample from this study. Here, 8–10 matrix calibration points were prepared for each toxin, ranging from 2 to 10 µg/kg (ATX I), 2.2 to 22 µg/kg (ALTP), 0.2 to 13 µg/kg (TEN), 3.6 to 17.5 µg/kg (ATX II), 2.2 to 20 µg/kg (AOH-3-S), 1.5 to 15 µg/kg (AME-3-S), 3 to 20 µg/kg (AOH-3-G), 4 to 20 µg/kg (AOH-9-G), and 4.9 to 15 µg/kg (AME-3-G). After measuring all points with LC–MS/MS, peak areas [A(A)] were plotted against the concentration of the analytes [c(A)], and calibration curves were calculated by linear regression. Mandel’s fitting test was applied to all calibration curves to check for linearity (Mandel and Mansfield 1964).
For quantification of the barley and malt samples, toxin concentrations were either calculated by the respective response curves of AOH, AME, or TeA, or by matrix-matched calibration. To compensate for day to day variations and intensity variabilities of the LC–MS/MS system, two matrix calibration points were prepared with each sample batch, which were then used to check for validity of the priorly determined calibration curves. In addition, all samples were prepared in pairs, meaning that both barley and its respective malt were analyzed on the same day to ensure that both samples were measured with the same instrumental conditions.
Method validation
LODs and LOQs
Limits of detection (LODs) and limits of quantifications (LOQs) were determined as described by Vogelgesang and Hädrich (1998). Therefore, an Alternaria toxin–free barley sample was used as blank matrix, which was spiked with the unlabelled analytes in four different concentration levels, each in triplicate (see Table 1 in the supplementary material for detailed information). Isotope-labelled standards were added and samples were further prepared for LC–MS/MS measurements following the developed sample workup procedure.
Precision
The blank matrix was spiked in triplicate with all analytes (see Table 1 in the supplementary material for detailed information), and was then subjected to sample preparation for intra-day (n = 3) and inter-day (n = 9, triplicate measurement every week within 3 weeks) precision measurements.
Recovery
Three different concentration levels of each toxin were spiked in triplicate into the blank matrix (see Table 1 in the supplementary material for detailed information). After sample preparation and LC–MS/MS measurement, recovery was calculated as ratio of the found value divided through the spiked value times 100.
Sample homogeneity
To check for homogeneity of the sampling procedure, the following experiment was performed: three CBS were chosen and from two respective LBS thereof the MABS were prepared, extracted in duplicate and analyzed with double injections. The variation of the complete sample set was calculated for each toxin. The same scheme was followed for MAMS from the same CBS.
Results and discussion
Sample preparation
Sample preparation was based on a method described by Gotthardt et al. (2019b). However, some adaptions had to be made for the integration of the modified Alternaria toxins into the method. First, extraction solvents containing methanol were avoided, as more matrix components were extracted with methanol-based than with acetonitrile-based extraction solvents. Also, the addition of 0.02% formic acid reduced the capability to extract modified toxins, while at the same time showing the highest recoveries for AOH, AME, ATX I, and ALTP. Therefore, the first extraction step was conducted with acetonitrile/water (84/16, v/v) without acid to mainly extract the modified mycotoxins, while a second and third extraction was done with acetonitrile/water (84/16, v/v) and 0.02% formic acid for better recoveries of AOH, AME, ATX I, and ALTP. The usage of the azeotropic mixture acetonitrile/water (84/16, v/v) facilitated the evaporation in the following step. To prevent signal suppression in the ESI source and contamination of the LC–MS/MS by matrix components, a purification step using solid-phase extraction (SPE) was necessary. Using C18 material was effective for matrix removal in accordance to literature (Asam et al. 2011b, 2012; Gotthardt et al. 2019b; Scheibenzuber et al. 2021); however, some alterations had to be made for barley and malt matrices. Here, especially the second washing step with ACN/H20 (1.5/8.5, v/v) reduced the matrix burden due to the increased polarity, while at the same time not being polar enough to elute the modified toxins from the column.
Calibration and quantitation
Linearity of the calculated response functions of AOH, AME, and TeA towards their corresponding isotope-labelled standards was checked with Mandel’s fitting test (Mandel and Mansfield 1964) and was confirmed for molar ratios between 0.01 and 100 for all three toxins.
Matrix-matched calibration curves were obtained for all other analytes by spiking a blank matrix with at least eight different concentrations. Here, the lowest spiking level was identical to the LOQ, while the highest concentration level was at least ten times higher than the LOQ. Again, linearity was checked with Mandel’s fitting test, followed by reducing the chosen range when necessary, resulting in the following linear ranges: 0.4–13 µg/kg for TEN, 2.1–10 µg/kg for ATX I, 3.6–17.5 µg/kg for ATX II, 2.1–22 µg/kg for ALTP, 8.8–100 µg/kg for ALT, 2.2–20 µg/kg for AOH-3-S, 2.2–15 µg/kg for AME-3-S, 2.9–20 µg/kg for AOH-3-G, 3.9–20 µg/kg for AOH-9-G, and 4.9–15 µg/kg AME-3-G.
Method validation
Method validation was performed as suggested by Vogelgesang and Hädrich (1998). All results are summarized in Table 2. An analyte-free barley sample was spiked in triplicate with four different concentration levels to determine the limits of detections (LODs) and limits of quantification (LOQs). In this matrix, LOD values were between 0.05 µg/kg (AME) and 2.45 µg/kg (ALT), while LOQ values ranged from 0.16 µg/kg (AME) to 8.75 µg/kg (ALT). Measuring ALT in the ESI positive mode would have improved the sensitivity and most methods described in literature (Nguyen et al. 2018; Wang et al. 2016) do so. However, as all other toxins were most sensitive in the negative ESI mode and we did not want to measure ALT separately, we had to compromise and accept the higher LODs and LOQs for this analyte. Anyway, LODs and LOQs of other methods measuring ALT in ESI negative mode are similar to the values in our study (Puntscher et al. 2018).
To determine the recovery of every analyte, a blank matrix was spiked in triplicate with three different concentrations that ideally resembled the expected concentrations in the samples. As in real samples low or no mycotoxin contaminations were expected, the first level was the concentration of the LOQ, while the other two levels were chosen close to the first level. Only for TeA spiking concentrations up to 500 µg/kg were used as higher levels were expected for this toxin. Recoveries for all analytes laid within the acceptable range of 70 to 120% (Table 2), mean value of the lowest recovery was 84% for ALT and AOH-9-G, and the mean value of the highest recovery was 112% for TEN.
Inter-injection, intra-day, and inter-day precisions were determined as the relative standard deviation (RSD) of every analyte after a certain number of repeated measurements. All obtained precisions are listed in Table 2. For inter-injection precision measurements, one spiked sample was measured five times in a row with LC–MS/MS, resulting in precisions ranging from 2 to 5%, which demonstrates the stability of the used system. To determine the intra-day precision, one sample was prepared in triplicate, while for inter-day precisions one sample was prepared in triplicate every week within 3 weeks. RSD values for both were below 10%, which shows the good precision of the used method.
Sample homogeneity
The analysis of two respective individual LBS and MBS from the same CBS showed reasonable variations in the toxin contents, demonstrating the suitability of the used sampling method for this study. For AOH, ATX I and ALTP variations in these real samples were lower than 10% and are therefore comparable to the obtained values for inter-day precisions from the method validation with spiked samples. AME and TEN, both of which were often present in concentrations below 2 µg/kg, showed maximum variations of less than 30%, and TeA concentrations varied up to 20% for concentrations close to the LOQ, and less than 12% for concentrations over 10 µg/kg (see Table 2 in the supplementary information for more details). We concluded that sample heterogeneity could be handled satisfactorily within the general uncertainty of analytical methods in trace analysis, therefore.
Screening of 50 barley and malt samples
Fifty malting barley samples were analyzed before and after malting to get a first insight into Alternaria mycotoxin occurrence and formation during malting. An overview of the obtained results is shown in Table 3 and the complete dataset can be found in Table 5 in the supplementary information.
Out of all analyzed samples, only five barley samples and one malt sample were completely free of Alternaria toxins; the other samples were contaminated with at least one toxin.
For barley, 42 samples were contaminated with TeA with concentrations up to 165 ± 9 µg/kg. TEN was found in 29 and AOH in 13 barley samples. The toxins AME, ALTP, and ATX I were detected in less than ten samples and the modified toxin AOH-3-S could only be found in one sample. Generally, concentrations were rather low or even below the LOQ, and the samples with the highest concentration of each toxin do not seem critical in terms of exposure. Interestingly, samples from the 2017 harvest were noticeably more often contaminated than the samples from the other three harvest years, with every sample containing at least three Alternaria mycotoxins. A more detailed list with data for every harvest year can be found in the supplementary information (Tables 3 and 4).
In general, toxins that were detected in the barley samples were also found in their respective malts with only four exceptions in sample pairs no. 20, 22, 26, and 43 regarding the toxins ATX I, TEN, TeA, and TEN, respectively. In the latter case, concentrations of the respective toxins in all sample pairs except sample no. 20 were below the LOQ values in barley and the absence in malt could therefore be attributed to measurement uncertainty at low concentrations. However, in sample no. 20, 3.12 µg/kg ATX I could be found in barley, but was not detected in the respective malt. Chemical or enzymatic modification of this toxin during the malting process may be an explanation for this finding.
In total, more malt than barley samples showed contaminations with Alternaria toxins, which indicates a mycotoxin formation during the malting process. Especially the number of samples containing ALTP and ATX I was noticeably higher with 30 and 24 positive (> LOD) malt samples, respectively, compared to 5 and 7 positive (> LOD) barley samples, respectively. However, half of the malt samples containing ALTP and almost all malt samples containing ATX I showed concentrations below the LOQ. Thus, for these two toxins, a clear evidence of formation during malting is hampered by measurement uncertainty, unfortunately. However, the increase of samples exceeding the LOD for ALTP and ATX I after malting seems to be more than a coincidence. As well as in barley, tenuazonic acid was the most dominant toxin in the malt samples and was detected in 48 out of 50 samples. However, an increase in TeA concentration was observed in almost every sample this time, reaching up to 247 µg/kg TeA as the maximal amount. For AOH, the number of contaminated samples increased from 13 to 23 after malting, and AME was found in 13 malt samples instead of 9 barley samples, but overall concentrations of AOH and AME stayed relatively stable or were only slightly higher in malt, which indicates only a low formation of both toxins during malting. For TEN, the number of contaminated barley and malt samples was rather stable, and concentrations did not show any major differences between the raw material and the malt.
Figure 2 shows the relative increase or decline of the toxins TeA, AOH, and AME of every malt sample. We chose a relative presentation of the data because of the different toxin contents of the barley samples and a logarithmic scale because of the large concentration differences. For TeA, an increase was observed in 44 samples, whereas six samples showed a reduced TeA concentration in malt. A hypothesis for the decrease of these six samples could be that more TeA was removed with the steeping water due to its high polarity than was formed by the Alternaria fungus. However, the steeping water was not analyzed in this study and more detailed studies of the malting process are required to confirm this theory, therefore. For AOH and AME, an increase in concentration was observed for 18 and 7 samples, respectively, and a decrease for 6 and 8 samples, respectively. Interestingly, the AOH concentration in sample no. 46 decreased from 11.89 µg/kg in barley to 6.94 µg/kg in malt while at the same time the modified toxin AOH-3-S increased from 6.23 µg/kg to 15.66 µg/kg. This indicates a transformation process during malting and a potential reason for the toxin decrease. However, two barley samples did neither contain AOH nor AOH-3-S, but both toxins were found in the respective malt samples, which shows that a simultaneous formation of both toxins during malting is also possible. In the malt samples that showed a decreased AME content, no AME-3-S or AME-3-G could be detected, but as the concentrations of AME were rather low, it could be possible that formed modified toxins were below the LOD and therefore not detected. Also, the formation of modified Alternaria toxins other than glucosides and sulfates cannot be excluded at this point.

Graphic visualization of the mycotoxin behavior of TeA A) and the sum of AOH and AME B) in barley and malt, calculated as the relative increase/decline of mycotoxins in malt, using mean values of each sample: \({\Delta }_{\mathrm{rel}}=\frac{{\overline{c} }_{\mathrm{malt}}-{\overline{c} }_{\mathrm{barley}}}{{\overline{c} }_{\mathrm{barley}}}\times 100\). A logarithmic scale was used for better display of data
Our results confirmed the hypothesis that Alternaria mycotoxin formation may take place during malting as the number of contaminated samples increased for almost every toxin. We did not perform significance tests at sample level due to limited data (duplicate analyses). However, to validate our observations, a one-tailed paired t-test was performed for all samples and all mycotoxins to see if mycotoxin concentrations are significantly higher in malt than in barley in general. For calculation, the middle-bound scenario for left-censored data was applied; i.e., concentrations of ½ LOD were used for all non-detected analytes (< LOD), and concentrations of ½ LOQ were used for all values between LOD and LOQ. The test revealed significantly higher concentrations (α = 0.05) for the mycotoxins TeA, AOH, AME, ATX I, and ALTP, which means that those toxins are most likely to be formed during malting. No significant difference (α = 0.05) could be found for the toxins TEN and AOH-3-S, which also confirmed our previous observations. Those results are also mostly in accordance with a study from Prusova et al. (2021), where AOH, AME, TEN, and TeA were included in a multi-mycotoxin mycotoxin method and analyzed during malting and brewing.
Our study revealed that concentrations of AOH, AME, TeA, ALTP, and ATX I were higher in malt than in barley, which indicates mycotoxin formation during malting. Especially the formation of ALTP and ATX I, which were only rarely found in barley, seems to be promoted by the conditions during malting. However, concentrations of all detected toxins were generally low and often below the LOQ in both barley and malt. Anyway, more data is needed for further risk assessments as fungal growth and mycotoxin concentrations in barley could be elevated in harvest years with unfortunate climate that leads to increased Alternaria infection in the field.
Data availability
The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Asam S, Konitzer K, Schieberle P, Rychlik M (2009) Stable isotope dilution assays of alternariol and alternariol monomethyl ether in beverages. J Agric Food Chem 57:5152–5160. https://doi.org/10.1021/jf900450w
Asam S, Liu Y, Konitzer K, Rychlik M (2011a) Development of a stable isotope dilution assay for tenuazonic acid. J Agric Food Chem 59:2980–2987. https://doi.org/10.1021/jf104270e
Asam S, Konitzer K, Rychlik M (2011b) Precise determination of the Alternaria mycotoxins alternariol and alternariol monomethyl ether in cereal, fruit and vegetable products using stable isotope dilution assays. Mycotoxin Res 27:23–28. https://doi.org/10.1007/s12550-010-0071-6
Asam S, Lichtenegger M, Liu Y, Rychlik M (2012) Content of the Alternaria mycotoxin tenuazonic acid in food commodities determined by a stable isotope dilution assay. Mycotoxin Res 28:9–15. https://doi.org/10.1007/s12550-011-0111-x
Asam S, Lichtenegger M, Muzik K, Liu Y, Frank O, Hofmann T, Rychlik M (2013) Development of analytical methods for the determination of tenuazonic acid analogues in food commodities. J Chromatogr A 1289:27–36. https://doi.org/10.1016/j.chroma.2013.03.015
Bottalico A, Logrieco A (1998) Toxigenic Alternaria species of economic importance. In: Marcel Dekker Inc. (ed) Mycotoxins in agriculture and food safety, New York, pp 65–96
Cole RJ, Schweikert MA, Jarvis BB (2003) Handbook of secondary fungal metabolites, vol I. Academic Press, Elsevier Science, USA, pp 697–714
EFSA – European Food Safety Authority (2011) Scientific opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J 9:2407. https://doi.org/10.2903/j.efsa.2011.2407
Fleck SC (2014) In vitro-Studien zum genotoxischen Potential und zum Metabolismus von Mykotoxinen mit Resorcylsäurelakton- und Perylenchinon-Struktur. Zugl.: Karlsruher Institut für Technologie, KIT, Dissertation, 2013. KIT Scientific Publishing, Karlsruhe, Baden
Frank O, Kreissl JK, Daschner A, Hofmann T (2014) Accurate determination of reference materials and natural isolates by means of quantitative (1)h NMR spectroscopy. J Agric Food Chem 62:2506–2515. https://doi.org/10.1021/jf405529b
Gotthardt M, Kanawati B, Schmidt F, Asam S, Hammerl R, Frank O, Hofmann T, Schmitt-Kopplin P, Rychlik M (2019a) Comprehensive analysis of the Alternaria mycobolome using mass spectrometry based metabolomics. Mol Nutr Food Res:e1900558. https://doi.org/10.1002/mnfr.2019a00558
Gotthardt M, Asam S, Gunkel K, Moghaddam AF, Baumann E, Kietz R, Rychlik M (2019b) Quantitation of six Alternaria toxins in infant foods applying stable isotope labeled standards. Front Microbiol 10:109. https://doi.org/10.3389/fmicb.2019.00109
Grabarkiewicz-Szczesna J, Chełkowski J (1992) Metabolites produced by Alternaria species and their natural occurence in Poland. In: Chełkowski J (ed) Alternaria: Biology, plant diseases and metabolites. Elsevier, Amsterdam, pp 363–380
Habler K, Hofer K, Geißinger C, Schüler J, Hückelhoven R, Hess M, Gastl M, Rychlik M (2016) Fate of Fusarium toxins during the malting process. J Agric Food Chem 64:1377–1384. https://doi.org/10.1021/acs.jafc.5b05998
Jacob F, Mitteleuropäische Brautechnische Analysenkommission (2011) Raw materials: barley, adjuncts, malt, hops and hop products: collection of brewing analysis methods of the Mitteleuropäische Brautechnische Analysenkommission (MEBAK). Chairman Dr. Fritz Jacob, self-published by MEBAK, Freising-Weihenstephan 46, 127–128
Lacey J (1992) Effects of environment on growth and mycotoxin production by Alternaria species. In: Chełkowski J (ed) Alternaria: Biology, plant diseases and metabolites. Elsevier, Amsterdam, pp 381–407
Lee HB, Patriarca A, Magan N (2015) Alternaria in food: ecophysiology, mycotoxin production and toxicology. Mycobiology 43:93–106. https://doi.org/10.5941/MYCO.2015.43.2.93
Liu Y, Rychlik M (2015) Biosynthesis of seven carbon-13 labeled Alternaria toxins including altertoxins, alternariol, and alternariol methyl ether, and their application to a multiple stable isotope dilution assay. Anal Bioanal Chem 407:1357–1369. https://doi.org/10.1007/s00216-014-8307-5
Logrieco A, Moretti A, Solfrizzo M (2009) Alternaria toxins and plant diseases: an overview of origin, occurrence and risks. World Mycotoxin Journal 2:129–140. https://doi.org/10.3920/WMJ2009.1145
Magan N, Lacey J (1985) The effect of water activity and temperature on mycotoxin production by Alternaria alternata in culture and wheat grain. In: Lacey J (ed) Trichothecenes and other mycotoxins: Proceedings. Wiley, Chichester, p 243
Mandel J, Mansfield JE (1964) The statistical analysis of experimental data. Phys Today 18:66–68. https://doi.org/10.1063/1.3047724
Nguyen TTT, Kim J, Jeon SJ, Lee CW, Magan N, Lee HB (2018) Mycotoxin production of Alternaria strains isolated from Korean barley grains determined by LC-MS/MS. Int J Food Microbiol 268:44–52. https://doi.org/10.1016/j.ijfoodmicro.2018.01.003
Noots I, Delcour JA, Michiels CW (1999) From field barley to malt: detection and specification of microbial activity for quality aspects. In: Critical reviews in microbiology 25(2):121–153. https://doi.org/10.1080/10408419991299257
Prusova N, Dzuman Z, Jelinek L, Karabin M, Hajslova J, Rychlik M, Stranska M (2021) Free and conjugated Alternaria and Fusarium mycotoxins during pilsner malt production and double-mash brewing. Food Chem 369:130926. https://doi.org/10.1016/j.foodchem.2021.130926
Puntscher H, Kütt M-L, Skrinjar P, Mikula H, Podlech J, Fröhlich J, Marko D, Warth B (2018) Tracking emerging mycotoxins in food: development of an LC-MS/MS method for free and modified Alternaria toxins. Anal Bioanal Chem 410:4481–4494. https://doi.org/10.1007/s00216-018-1105-8
Scheibenzuber S, Hoffmann T, Effenberger I, Schwab W, Asam S, Rychlik M (2020) Enzymatic synthesis of modified Alternaria mycotoxins using a whole-cell biotransformation system. Toxins (basel). https://doi.org/10.3390/toxins12040264
Scheibenzuber S, Dick F, Asam S, Rychlik M (2021) Analysis of 13 Alternaria mycotoxins including modified forms in beer. Mycotoxin Res 37:149–159. https://doi.org/10.1007/s12550-021-00424-0
Scott PM (2001) Analysis of agricultural commodities and foods for Alternaria mycotoxins. J AOAC Int 84:1809–1817. https://doi.org/10.1093/jaoac/84.6.1809
Strandberg JO (1992) Alternaria species that attack vegetable crops: biology and options for disease management. In: Chełkowski J (ed) Alternaria: Biology, plant diseases and metabolites. Elsevier, Amsterdam, pp 175–208
Vogelgesang J, Hädrich J (1998) Limits of detection, identification and determination: a statistical approach for practitioners. Accred Qual Assur 3:242–255. https://doi.org/10.1007/s007690050234
Walravens J, Mikula H, Rychlik M, Asam S, Ediage EN, Di Mavungu JD, van Landschoot A, Vanhaecke L, Saeger SD (2014) Development and validation of an ultra-high-performance liquid chromatography tandem mass spectrometric method for the simultaneous determination of free and conjugated Alternaria toxins in cereal-based foodstuffs. J Chromatogr A 1372C:91–101. https://doi.org/10.1016/j.chroma.2014.10.083
Walravens J, Mikula H, Rychlik M, Asam S, Devos T, Njumbe Ediage E, Di Diana MJ, Jacxsens L, van Landschoot A, Vanhaecke L, Saeger SD (2016) Validated UPLC-MS/MS methods to quantitate free and conjugated Alternaria toxins in commercially available tomato products and fruit and vegetable juices in Belgium. J Agric Food Chem 64:5101–5109. https://doi.org/10.1021/acs.jafc.6b01029
Wang M, Jiang N, Xian H, Wei D, Shi L, Feng X (2016) A single-step solid phase extraction for the simultaneous determination of 8 mycotoxins in fruits by ultra-high performance liquid chromatography tandem mass spectrometry. J Chromatogr A 1429:22–29. https://doi.org/10.1016/j.chroma.2015.12.004
Woudenberg JHC, Groenewald JZ, Binder M, Crous PW (2013) Alternaria redefined. Stud Mycol 75:171–212. https://doi.org/10.3114/sim0015
Funding
Open Access funding enabled and organized by Projekt DEAL. This IGF project (AiF 19766 N) was supported via AiF within the program for promoting the Industrial collective Research (IGF) of the German Ministry of Economic Affairs and Energy (BMWi), based on a resolution of the German Parliament, the Forschungskreis der Ernährungsindustrie e.V., the Wissenschaftliche Station für Brauerei in München e.V., and the Wissenschaftsförderung der Deutschen Brauwirtschaft e.V.
Author information
Authors and Affiliations
Contributions
Sophie Scheibenzuber, Marina Bretträger, Martina Gastl, and Michael Rychlik contributed to the study conception. Marina Bretträger and Martina Gastl were responsible for sampling and malting. Method development was performed by Sophie Scheibenzuber and Fabian Dick; method validation and sample analysis were conducted by Sophie Scheibenzuber. The first draft of the manuscript was written by Sophie Scheibenzuber and was revised by Stefan Asam and Michael Rychlik. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scheibenzuber, S., Dick, F., Bretträger, M. et al. Development of analytical methods to study the effect of malting on levels of free and modified forms of Alternaria mycotoxins in barley. Mycotoxin Res 38, 137–146 (2022). https://doi.org/10.1007/s12550-022-00455-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12550-022-00455-1