
Abstract
Background
The role of type I interferon (IFN-I) signaling in systemic lupus erythematosus (SLE) has been well established. However, unanswered questions remain regarding the applicability of these findings to pediatric-onset SLE. The aim of this review is to provide an overview of the novel discoveries on IFN-I signaling in pediatric-onset SLE.
Data sources
A literature search was conducted in the PubMed database using the following keywords: “pediatric systemic lupus erythematosus” and “type I interferon”.
Results
IFN-I signaling is increased in pediatric SLE, largely due to the presence of plasmacytoid dendritic cells and pathways such as cyclic GMP-AMP synthase–stimulator of interferon genes–TANK-binding kinase 1 and Toll-like receptor (TLR)4/TLR9. Neutrophil extracellular traps and oxidative DNA damage further stimulate IFN-I production. Genetic variants in IFN-I-related genes, such as IFN-regulatory factor 5 and tyrosine kinase 2, are linked to SLE susceptibility in pediatric patients. In addition, type I interferonopathies, characterized by sustained IFN-I activation, can mimic SLE symptoms and are thus important to distinguish. Studies on interferonopathies also contribute to exploring the pathogenesis of SLE. Measuring IFN-I activation is crucial for SLE diagnosis and stratification. Both IFN-stimulated gene expression and serum IFN-α2 levels are common indicators. Flow cytometry markers such as CD169 and galectin-9 are promising alternatives. Anti-IFN therapies, such as sifalimumab and anifrolumab, show promise in adult patients with SLE, but their efficacy in pediatric patients requires further investigation. Janus kinase inhibitors are another treatment option for severe pediatric SLE patients.
Conclusions
This review presents an overview of the IFN-I pathway in pediatric SLE. Understanding the intricate relationship between IFN-I and pediatric SLE may help to identify potential diagnostic markers and targeted therapies, paving the way for improved patient care and outcomes.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interferons (IFNs) constitute a crucial class of cytokines with significant roles in the immune response. They can be categorized into three families based on their structural characteristics, immunomodulatory functions, and the cells responsible for their secretion. Type I interferon (IFN-I) represents one of these subfamilies and include 17 proteins: 13 for IFN-α, IFN-β, IFN-ω, IFN-ε, and IFN-κ [1]. Generally, the levels of IFN-I peak during the initial days following acute viral infections and return to normal once the virus has been cleared, indicating a transient and time-limited response [2]. However, elevated IFN-I was observed in systemic lupus erythematosus (SLE) patients [3]. Over the past few decades, research has shed light on the role of IFN-I in the context of SLE [4]. It has been well established that IFN-I signaling is activated in SLE, with elevated levels of IFN-I contributing to the progression of the disease through various mechanisms. These mechanisms include the induction of autoreactive T cells, the promotion of autoantibody production, and the upregulation of proinflammatory cytokine expression [5]. In addition, anomalies in other pathways in SLE, such as complement deficiency, could also cause secondary overexpression of IFN-I [6]. However, the majority of these studies have been conducted in adult populations, leaving uncertainties regarding whether similar conditions apply to pediatric-onset SLE. Therefore, this review aimed to explore the relationship between the IFN-I pathway and SLE, focusing specifically on investigations conducted in pediatric patients.
Type I interferon signaling in classic systemic lupus erythematosus
Multiple studies have established a robust association between elevated IFN-I levels and SLE. Aberrant IFN-I signaling is thought to contribute to the perpetuation of autoimmunity in SLE patients [7]. Several mechanisms underlying IFN-I dysregulation in SLE include increased IFN-I production by plasmacytoid dendritic cells, defective IFN-I receptor signaling, and a failure to negatively regulate IFN-I production [8]. Notable genes involved in IFN-I signaling, such as IFN-regulatory factors (IRFs) [9] and signal transducer and activator of transcription (STATs) [10], have been linked to SLE susceptibility. IFN-I plays a multifaceted role in SLE pathogenesis. It promotes B-cell activation, disrupts immune tolerance, and drives the differentiation of autoreactive T cells [11,12,13]. Additionally, IFN-I induces the expression of various proinflammatory cytokines, amplifying the inflammatory cascade in affected tissues [14]. These collective effects contribute to the production of autoantibodies and immune complex deposition, which are central to the immunopathology of SLE.
In pediatric SLE, the overproduction of IFN-I is also prominent. Most IFN-I is produced by plasmacytoid dendritic cells and usually depends on the cyclic GMP-AMP synthase–stimulator of interferon genes–TANK-binding kinase 1 (cGAS-STING-TBK1) pathway as well as the Toll-like receptor (TLR)4/TLR9 pathways [15]. The former represents an instinct nucleic acid-sensing pathway, while the latter is usually activated during bacterial infections [16, 17]. A recent study revealed that in pediatric SLE, the presence of undigested nucleic acids and bacterial stimulation can potentially collaborate in a positive feedback loop, ultimately leading to dysregulated IFN production [18].
The triggers of IFN-I overproduction in SLE could vary, and neutrophil extracellular traps (NETs) might be important. A study demonstrated that mature neutrophils from pediatric SLE patients were primed in vivo by IFN-I and underwent cell death when exposed to SLE-derived anti-ribonucleoprotein antibodies. This resulted in the release of NETs. Then, NETs activate plasmacytoid dendritic cells to enhance the uptake and recognition of mammalian DNA, prompting them to produce elevated levels of IFN-α [19]. Another possible trigger of IFN-I production is oxidative DNA damage. Experiments revealed that defects in OGG1, a DNA repair enzyme that repairs 8-OH-dG DNA lesions, which accumulates in oxidized DNA [20], enhanced IFN-driven gene expression and was associated with increased autoantibodies. Furthermore, the expression of OGG1 was notably lower in lesional skin than in non-lesional skin in patients diagnosed with discoid lupus [21].
In pediatric SLE, the overproduction of IFN-I could also be a result of impaired inhibition of related pathways. An Australian group elucidated the functional implications of these rare and infrequent missense variants in the interacting proteins B-lymphoid kinase and B-cell scaffold protein with ankyrin repeats 1, which were found either individually or in combination in a significant proportion of individuals with lupus. The rare variants identified in SLE patients, as opposed to those exclusively found in the control group, hinder the suppression of IRF5 and IFN in human B-cell lines and augment the presence of pathogenic lymphocytes in mice prone to lupus [22]. In addition, another group found de novo protein kinase C and casein kinase substrate in neurons 1 (PACSIN1) missense variants in a child with SLE [23]. Their study established that PACSIN1 formed a trimolecular complex involving tumor necrosis factor receptor-associated factor 4 (TRAF4) and TRAF6, which plays a crucial role in the regulation of IFN-I. The Q59K mutation in PACSIN1 reduced its interaction with TRAF4. Consequently, this alteration resulted in uncontrolled TRAF6-mediated activation of IFN-I.
In pediatric SLE patients, several variants of IFN-I-related genes were found to be associated with SLE susceptibility, similar to the findings in the adult population. IRF5 is one such gene that has been identified as an autoimmune susceptibility gene [24]. To date, at least four variants of IRF5 have been associated with SLE risk [25,26,27,28,29]. Li et al. demonstrated that individuals carrying the IRF5-SLE risk haplotype had increased IFN pathway enrichment and decreased reactive oxygen species pathway expression and thus had increased circulating plasmacytoid dendritic cells and plasma cells, as well as elevated spontaneous NETosis [30]. In a recent Mexican study, two tyrosine kinase 2 (TYK2) variants related to infection risk were shown to be protective against SLE [31].
Type I interferonopathies in monogenic systemic lupus erythematosus
Monogenic SLE represents a distinct subset of pediatric SLE patients, and the relationship between monogenic SLE and type I interferonopathies is intricate, showing overlapping features that blur the lines between these distinct entities. Monogenic SLE refers to a subset of SLE cases driven primarily by single-gene mutations, often affecting key components of immune pathways, while type I interferonopathies encompass a group of inborn errors of immunity characterized by sustained activation of the IFN-I signaling pathway (Fig. 1). Notably, a considerable number of type I interferonopathies, including Aicardi-Goutières syndrome (AGS), proteasome-associated autoinflammatory syndrome (PRAAS) and STING-associated vasculopathy with onset in infancy (SAVI), manifest clinical and laboratory features akin to SLE (Table 1). These conditions involve dysregulated IFN-I signaling, leading to a cascade of immune dysregulation and autoimmune-like phenotypes resembling SLE, such as the presence of autoantibodies, skin manifestations, and neurological abnormalities. Therefore, while monogenic SLE typically results from mutations in genes directly linked to lupus susceptibility, there is a notable convergence between certain type I interferonopathies and SLE, emphasizing the pivotal role of IFN-I dysregulation in the pathogenesis of both monogenic and multifactorial forms of SLE. Understanding these overlaps provides insights into shared immunopathogenic mechanisms and highlights the intricate interplay between genetic factors and aberrant interferon signaling in autoimmune diseases.

IFN-I pathway and known genes related to type I interferonopathies and monogenic SLE. Ribonuclease H2 (encoded by RNASEH2A, RNASEH2B and RNASEH2C), SAMHD1, TREX1 and DNases (encoded by DNASE1, DNASE2 and DNASE1L3, etc.) participate in RNA‒DNA hybrids, deoxynucleotide triphosphates (dNTPs) and single-stranded DNA as well as double-stranded DNA metabolism. Mutations in these genes cause excess accumulation of DNA, while mutations in LSM11 and RNU7-1 lead to a disorder of histone stoichiometry, resulting in the sensing of nuclear DNA. Together, these factors activate the interferon pathway through cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling via the activation of TANK-binding kinase 1 (TBK1)-interferon regulatory factor 3 (IRF3). Mutations in STING1, COPA and ARF1 result in prolonged presence of STING in the Golgi apparatus, leading to constitutive activation of IFN-I. Mutations in IFIH1 (encoding MDA5) and DDX58 (encoding RIG-I) cause abnormal sensing of exogenous viral double-stranded RNA (dsRNA), and loss-of-function mutations in ADAR1 lead to the generation of immunogenic dsRNA. These mutations induce interferon production by activating mitochondrial antiviral signaling protein (MAVS)-TBK1-IRF3. OTUD1 acts as a suppressor by deubiquitinating IRF3 and loss-of-function mutations in OTUD1, lead to overactivation of IRF3 and thus upregulation of IFN. Moreover, single-stranded RNA (ssRNA) from viruses and bacteria, detected by TLR8, can trigger the IFN-I response via NF-κB activation. After IFN-I binds to its receptor, JAK1 and TYK2 are phosphorylated and subsequently activate STAT1 to induce the expression of interferon-stimulated genes (ISGs). Gain-of-function mutations in JAK1 and STAT1 therefore result in overactivation of IFN-I. USP18, ISG15 and STAT2 participate in the regulation of the IFN pathway by inhibiting signaling downstream of IFNARs, while deficiency of USP18 and ISG15, as well as separation-of-function mutations in STAT2, promote the induction of interferon signaling. Currently, there are at least 9 genes related to the proteasome that causes interferonopathies, but the exact underlying mechanism remains unclear. In addition, the relationships of mutations in ACP5, RELA, and SAMD9L with disease remain unknown. IFN-I type I interferon, SLE systemic lupus erythematosus, RNASEH2A ribonuclease H2 subunit A, SAMHD1 SAM-domain- and HD-domain-containing protein 1, TREX1 3′ repair exonuclease 1, DNASE1L3 deoxyribonuclease 1 like 3, COPA coatomer protein complex subunit alpha, ARF1 ADP-ribosylation factor 1, IFIH1 interferon induced with helicase C domain 1, RIG-I retinoic acid-inducible I, ADAR1 adenosine deaminase RNA specific 1, OTUD1 ovarian tumor deubiquitinase 1, TLR Toll-like receptor, NF-κB nuclear factor-κB, JAK1 Janus kinase 1, TYK2 tyrosine kinase 2, STAT1 signal transducer and activator of transcription 1, USP18 ubiquitin-specific peptidase 18, IFNARs IFN-α receptors, SOCS1 suppressor of cytokine signaling 1, SAMD9L sterile alpha motif domain-containing 9 like, PSM proteasome
Aicardi–Goutières syndrome
AGS was first reported in 1984 by neurologists Jean Aicardi and Françoise Goutières. To date, seven related pathogenic genes have been discovered, including 3′ repair exonuclease 1 gene (TREX1; AGS1), ribonuclease H2 subunit B gene (RNASEH2B; AGS2), RNASEH2C (ASG3), RNASEH2A (AGS4), SAM-domain- and HD-domain-containing protein 1 gene (SAMHD1; AGS5), adenosine deaminase RNA specific 1 gene (ADAR1; AGS6), and interferon induced with helicase C domain 1 gene (IFIH1; AGS7) [32]. TREX1, also known as DNase III, is a 3′-5′ DNA exonuclease present in the cytoplasm, and it acts on single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA). TREX1 cleaves mismatched and modified nucleotides from the 3′ end of DNA, degrading DNA from retroviruses and reverse transcriptase [33]. SAMHD1 degrades deoxynucleoside triphosphates (dNTPs) into 2′-deoxynucleosides and triphosphate subunits, thereby stably consuming the cellular dNTP pool and preventing the accumulation of cytoplasmic ssDNA [34]. RNASEH2, ribonuclease H2, is an enzyme composed of three subunits, including the catalytic subunit RNASEH2A and the non-catalytic subunits RNASEH2B and RNASEH2C. RNASEH2 degrades the RNA portion of RNA/DNA hybrids and hydrolyzes the phosphodiester bonds of ribonucleotides embedded in DNA double strands, preventing genomic instability and the accumulation of abnormal nucleic acids [35]. Their deficiencies lead to the accumulation of ssDNA, dsDNA, and RNA/DNA hybrids in the body, which are recognized by cGAS. This triggers the activation of STING, which in turn recruits TBK1 to phosphorylate IRF3, leading to the production of IFN-β.
ADAR1 primarily acts on RNA as an adenosine deaminase, catalyzing the hydrolytic deamination of adenosine to inosine in double-stranded RNA (dsRNA) [36]. Loss of ADAR1 function leads to an increase in dsRNA levels. MDA5, also known as IFIH1, recognizes dsRNA, inducing the activation and oligomerization of mitochondrial antiviral signaling proteins [37]. This activates TBK1 and IκB kinase, ultimately resulting in the activation of the transcription factors nuclear factor (NF)-κB, IRF3, and IRF7. These transcription factors translocate to the cell nucleus, where they promote the production of IFN-I.
Initially, AGS was thought to manifest as progressively worsening neurological symptoms, including spasms, hypertonia, and microcephaly, with severe cases resulting in early death. Patients with AGS exhibit elevated levels of IFN-stimulated genes (ISGs) and IFN-α in their blood and cerebrospinal fluid. As more cases have been documented, a wider range of clinical phenotypes have emerged, including chilblain-like skin rashes, intermittent fever, cytopenia, hepatosplenomegaly, elevated transaminases, hypothyroidism, interstitial lung disease, intracranial calcifications, and white matter lesions in the brain, accompanied by positive autoantibodies, leading to a misdiagnosis of SLE [35].
DDX58 gain-of-function
DDX58 encodes retinoic acid-inducible I (RIG-I), an intracytoplasmic nucleic acid sensor. The hyperactivation of RIG-I impairs ATPase activity, leading to constitutive self-RNA recognition and IFN-I pathway activation [38]. Initially, DDX58 gain-of-function was thought to present as atypical Singleton–Merten syndrome, characterized by abnormal valvular and thoracic calcifications and osteoporosis [39]. Recently, a new DDX58 pathogenic variant, R109C, was found to be associated with lupus nephritis [40]. These patients manifested autoimmune symptoms, including nephritis, hemolytic anemia, positive antinuclear antibodies (ANAs), anti-dsDNA and anticardiolipin antibodies and decreased C3, accompanied by an elevated IFN signature. Further investigations indicated that compared with previous variants, the R109C variant leads to a loss of RIG-I autoinhibition, which is a distinct pattern.
Abnormalities in toll-like receptors
TLRs are single-spanning membrane proteins located on the surface or within endosomes of immune cells. Defects in TLR signaling pathways are linked to increased susceptibility to bacterial infections and compromised inflammatory responses during infections. Conversely, gain-of-function mutations in both TLR7 and TLR8, which detect both viral and bacterial single-stranded RNA [41], are related to SLE [42, 43]. TLR8 is found in monocytes/macrophages, myeloid dendritic cells, and granulocytes, while TLR7 is present in plasmacytoid dendritic cells, B cells, and monocytes/macrophages [44]. Upon stimulation, these receptors activate NF-κB, mitogen-activated protein kinase (MAPK), and IRF, leading to the transcription of inflammatory cytokines, chemokines, and costimulatory molecules. However, their downstream signaling pathways differ across cell types. In monocytes, TLR8 activation triggers robust NF-κB and IFN-I responses, promoting T helper (Th) 1 cytokine production. Conversely, TLR7 primarily activates MAPK signaling and induces Th17 cytokines. Notably, TLR7 inhibits the TLR8-mediated IFN-I response [45]. With respect to TLR8 gain-of-function, patients often exhibit early-onset severe cytopenia, hepatosplenomegaly, and lymphadenopathy accompanied by progressive autoinflammatory features, including fever, arthritis and central nervous system vasculitis. Laboratory examinations revealed elevated proinflammatory cytokines, including interleukin (IL)-18, IL-1β, and tumor necrosis factor (TNF)-α, as well as an increased IFN signature [42, 46], indicating the possibility of interferonopathies. However, this is still unclear since the IFN signature associated with TLR8 gain-of-function is lower than that associated with classical type I interferonopathies [46]. In addition, TLR7 gain-of-function did not increase the IFN signature [43].
Deoxyribonuclease deficiencies
Deoxyribonucleases (DNases) are a group of enzymes responsible for catalyzing the degradation of DNA molecules, thus preventing the recognition of self-DNA. DNase I is a prominent serum endonuclease primarily responsible for degrading extracellular dsDNA from dying cells. DNase II is a crucial lysosomal endonuclease that plays a pivotal role in breaking down exogenous DNA encountered through endocytosis. Deoxyribonuclease 1 like 3 (DNase1L3) shares homology with DNase I and is presumed to play a role in the clearance of NETs. Deficiencies in these enzymes cause the accumulation of nucleic acid, leading to the activation of DNA sensors and the IFN-I signaling pathway [47].
In terms of DNase I deficiency, mice deficient in DNASE1 develop ANA and glomerulonephritis [48]. There have been reports of several cases of SLE in which individuals had a mutation in DNASE1, resulting in markedly elevated levels of anti-nucleosome antibodies [49, 50].
Clinical manifestations of DNase II deficiency are similar to those of AGS and may present with symptoms in the neonatal period, including hepatosplenomegaly, cholestatic hepatitis, and, in severe cases, progression to cirrhosis. Additionally, patients may exhibit chilblain-like skin rashes, cytopenia, proteinuria, non-destructive deformative joint disease, neurological symptoms, and intracranial calcifications, and some may show features of immunodeficiency and autoimmune diseases [51].
The absence of DNASE1L3 can lead to early-onset and clinically severe SLE. In 2011, Al-Mayouf et al. reported 17 patients with SLE from six unrelated families attributed to DNASE1L3 deficiency. All of these patients developed childhood-onset SLE, with ages of onset ranging from 2 to 12 years. Notably, all patients tested positive for ANA and ds-DNA antibodies but also displayed decreased C3 and C4 levels. Among this group, 11 patients were positive for anti-neutrophil cytoplasmic antibodies (ANCAs), and 11 were diagnosed with lupus nephritis [52].
LSM11 deficiency and RNU7-1 deficiency
LSM11 and RNU7-1 are two components of the replication-dependent histone pre-mRNA processing complex. In 2020, Uggenti et al. discovered that biallelic mutations in either LSM11 or RNU7-1 led to a dysregulation of histone messenger RNA transcripts, as well as a disturbance of histone protein composition, which further activated interferon signaling through a cGAS–STING-dependent pathway [53]. According to previous reports, patients exhibit typical AGS phenotypes.
STING-associated vasculopathy with onset in infancy
STING is a transmembrane protein encoded by STING1 that is located primarily in the endoplasmic reticulum and serves as an essential sensor of self-DNA molecules. Generally, STING is activated by its ligand cGAS, triggering the activation of the IFN-I signaling pathway via TBK1 and IRF3. However, gain-of-function mutations in STING1 can result in ligand-independent activation of STING, leading to sustained activation of the IFN-I signaling pathway [54]. SAVI is characterized by interstitial lung disease, cutaneous involvement such as erythematous or purpuric plaques and nodules, livedo reticularis, and painful ulcerative lesions, as well as systemic symptoms such as failure to thrive, fever, malaise, and chronic anemia, sometimes accompanied by positive autoantibodies such as low-titer ANA or positive anti-phospholipid antibodies [55, 56].
COPA syndrome
Coatomer protein complex subunit alpha (COPA) syndrome is caused by a heterozygous mutation in the COPA gene, which encodes the α-subunit of coat protein complex I (COPI) and is involved in the trafficking of membranes from the Golgi apparatus to the endoplasmic reticulum [57]. Defects in the COPI complex lead to prolonged activation of STING in the Golgi apparatus, resulting in the overexpression of IFN-I. The clinical characteristic of COPA syndrome is lung disease (including cystic, hemorrhagic, and interstitial lung disease), with a smaller subset also experiencing arthritis and kidney disease. Laboratory studies may reveal ANA and ANCAs. In a recent report, a 10-year-old girl who was diagnosed with lupus nephritis and carried a mutation in the COPA gene indicated that COPA syndrome could also be a cause of lupus nephritis [58]. Another study suggested that alveolar hemorrhage and pulmonary vasculitis in both SLE patients and COPA syndrome patients might be initiated by endothelial injury, resulting in endoplasmic reticulum stress, lung endothelial cell apoptosis and myeloid cell recruitment [59].
ARF1 deficiency
ADP-ribosylation factor 1 (ARF1) is a negative regulator of cGAS-STING signaling. It was recently found to be related to interferonopathy. Heterozygous ARF1 missense mutations produce GTPase-defective ARF1, which leads to prominent mitochondrial DNA release and the accumulation of STING, resulting in increased IFN-I signaling [60]. All patients exhibited significant developmental delay, and 75% (three of four) of patients had skin lesions such as chilblain lupus. Notably, no patients had epilepsy, and images of the central nervous system were normal in all patients, without intracranial calcification or other manifestations observed in AGS patients.
OTUD1 deficiency
The ovarian tumor deubiquitinase 1 gene (OTUD1) encodes a deubiquitinase enzyme that plays a pivotal role in cellular processes by interacting with IRF3. Its primary function involves the deubiquitination of IRF3, leading to the removal of polyubiquitin chains from this transcription factor and subsequently resulting in the suppression of IFN gene transcription. However, when loss-of-function (LOF) missense mutations occur within the OTUD1 gene, this finely tuned regulatory mechanism is disrupted. Consequently, these mutations cause the overactivation of IRF3, leading to the aberrant upregulation of IFN genes. This dysregulation of the immune response has been closely associated with the development of a diverse array of autoimmune diseases, which notably include early-onset SLE [61].
JAK1 gain-of-function
Janus kinase 1 (JAK1) is one of the most important kinases downstream of IFN-I, and the JAK1 gain-of-function variant A634D was reported to cause a syndrome characterized by hepatosplenomegaly, eosinophilia, enteritis, thyroid disease, growth retardation, and susceptibility to viral infection [65, 66]. Membranous nephropathy has also been reported [66]. Tofacitinib, a pan-JAK inhibitor, has been reported to ameliorate clinical and biological immune dysfunction [66].
USP18 deficiency and ISG15 deficiency
Ubiquitin-specific peptidase 18 (USP18) downregulates IFN-I signaling by inhibiting the interaction between JAK1 and IFN-α receptors (IFNARs), while interferon-stimulated gene 15 (ISG15) is a ubiquitin-like protein induced by IFN-I that can bind to USP18, thereby protecting USP18 from proteasome degradation and enhancing its suppressive function. Defects in either ISG15 or USP18 can ultimately cause sustained activation of the IFN-I signaling pathway [67, 68]. Patients typically exhibit a broad range of neurological and immunological manifestations. These can include basal ganglia calcification, seizures, and susceptibility to mycobacterial infections. Additionally, these patients often display higher levels of autoantibodies than healthy individuals [69].
STAT2 deficiency
In 2019 and 2020, two reports suggested that homozygous mutations, specifically the p.Arg148Trp or p.Arg148Gln separation-of-function mutations in the STAT2, resulted in the inability of the protein to interact with USP18. This interaction is pivotal for the recruitment of USP18 to IFNAR2. As a consequence of these mutations, patients exhibit severe neuroinflammatory diseases characterized by progressive intracranial calcification, white matter disease, and intracranial hemorrhage. In addition to these neurological symptoms, patients also exhibit systemic inflammation and multisystemic dysfunction, which includes recurrent fever, hepatosplenomegaly, cytopenia with pronounced thrombocytopenia, elevated ferritin levels, heightened liver enzyme activity, and nephrotic range proteinuria [70, 71].
SOCS1 haploinsufficiency
Suppressor of cytokine signaling 1 (SOCS1) haploinsufficiency was initially reported in 2020, and it is primarily attributed to monoallelic LOF variants (large deletions, frameshift mutations, and nonsense variants) [72,73,74,75]. SOCS1 regulates the JAK-STAT pathway by inhibiting JAK1/2 phosphorylation and suppressing the activity of TYK2 [76]. Thus, individuals with SOCS1 dysfunction exhibit an enhanced IFN signature. Based on limited published reports to date, disease onset typically occurs within the first decade of life. Patients may present with various rheumatologic manifestations, including polyarthritis, recurrent mucosal ulcerations, fever, glomerulonephritis, alopecia, autoimmune endocrinopathies, psoriasis, and features resembling SLE. There are also other manifestations, including gastrointestinal tract abnormalities (hepatosplenomegaly, hepatitis, and colitis), recurrent infections, variable T-cell lymphopenia, and natural killer cell dysfunction. Furthermore, secondary granulomatous interstitial lung disease related to immune dysregulation has been documented. Notably, recent findings have revealed benign variants of SOCS1 in cis, leading to early-onset SLE-like symptoms. This finding underscores the significance of SOCS1 in cytokine signaling, as even a partial disruption of one allele by approximately 20%–30% may suffice to drive aberrant IFN-I signaling [77].
STAT1 gain-of-function
Individuals with STAT1 gain-of-function mutations exhibit increased expression of ISGs in whole blood. However, it is essential to note that STAT1 mutations involve the activation of multiple cytokine pathways, in addition to the IFN-I signaling pathway. This multifaceted cytokine activation likely contributes to aspects of their clinical phenotypes that are not typically observed in other type I interferonopathies. While some patients with STAT1 gain-of-function mutations present with intracranial calcification and aortic calcification, the core clinical phenotype primarily includes chronic mucocutaneous candidiasis and autoimmune hypothyroidism [78,79,80,81].
Proteasome-associated autoinflammatory syndrome
Dysfunction of immune proteasomes, whose primary role is the hydrolysis of intracellular senescent and exogenous proteins, is characterized by this process. This dysfunction is attributed to mutations in immunoproteasome subunits and constituent subunits [proteasome subunit alpha type 3 (PSMA3), PSMA5, PSMB4, PSMB8, PSMB9, PSMB10], regulatory particles (PSMC5, PSMD12), and proteasome assembly units [POMP (proteasome maturation protein), PSMG2]. These mutations result in diminished enzymatic hydrolysis activity and contribute to proteasome-related diseases [82,83,84,85,86]. Notably, in 2023, Papendorf et al. reported the discovery of a novel variant in PSMC5, a gene previously unassociated with the proteasome. However, it is essential to note that this finding cannot be definitively attributed to the condition, as the patient also possessed a pathogenic maternally inherited PSMB8 variant and a de novo PSMA5 mutation [86]. Common clinical features of this disorder include the presence of pernio-like purplish nodular lesions (neutrophilic dermatosis), panniculitis accompanied by progressive lipodystrophy and muscle atrophy, and joint contractures leading to extremity deformities. Hepatosplenomegaly and hypochromic or hemolytic anemia have also been reported. Early metabolic syndrome, characterized by systemic hypertension and dyslipidemia, affects 40%–80% of patients [84, 87,88,89]. Furthermore, proteasome defects have been associated with neurological diseases, such as the development of microcephaly and cognitive delay [90]. Clinical studies have noted elevated levels of IFN-I in PRAAS patients. Nevertheless, the precise molecular mechanisms underlying the enhanced IFN levels and their connection to proteasome dysfunction remain to be elucidated.
Spondyloenchondrodysplasia with immune dysregulation
The clinical manifestations of spondyloenchondrodysplasia with immune dysregulation (SPENCDI) associated with ACP5 mutations are similar to those of AGS in terms of neurological manifestations (spasticity, intracranial calcifications) as well as manifestations of autoimmunity (mimicking SLE) [62, 63]. Autosomal recessive mutations in the ACP5 gene lead to a deficiency of tartrate-resistant acid phosphatase (TRAP), which is normally expressed in osteoclasts and myeloid cells. TRAP plays a crucial role in the processing and degradation of osteopontin (OPN) within plasmacytoid dendritic cells by catalyzing the dephosphorylation of OPN. Defects in TRAP cause prolonged phosphorylation of OPN, which leads to sustained activation of the TLR9 pathway, inducing the nuclear translocation of the IRF7 and NF-κB transcription factors and ultimately resulting in increased expression of IFN-I, IL-6, and TNF [64].
RELA dominant-negative mutation
RELA encodes the p65 protein, a member of the NF-κB family of transcription factors. Earlier research indicated that RELA haploinsufficiency leads to chronic mucocutaneous ulceration and autoimmune hematological disorders, largely dependent on TNF [91]. However, a recent study showed that patients with RELA dominant-negative (DN) mutations exhibit clinical features similar to those of patients with RELA haploinsufficiency plus inflammatory symptoms such as periodic fever, inflammatory bowel diseases, juvenile idiopathic arthritis, and skin involvement such as erythema nodosum or pustulosis [92]. Basic research has shown that myeloid cells with RELA DN mutations have increased TLR7 and myeloid differentiation factor 88 expression and thus produce more IFN-I in response to TLR7 activation [92].
SAMD9L-associated autoinflammatory disease
It is caused by truncating mutations within the nucleotide-binding oligomerization domain of SAMD9L, which plays a role as an autonomous brake to block the translation of several proteins to inhibit growth factor signaling [93] and cell cycle progression and stimulate inflammation [94]. The truncating mutations resulted in the absence of the winged helix domain/helical domain 2 and tetracopeptide repeat domain, obscuring the nucleotide-binding domain oligomerization interface. Consequently, the truncated protein tends to multimerize into its active form, inhibiting mRNA translation even in the absence of viral RNA triggering [95]. These patients often exhibit early-onset multisystemic inflammation, including panniculitis (characterized by neutrophil inflammation), interstitial pneumonia, intracranial calcification, pancytopenia, and highly elevated inflammatory markers (C-reactive protein and erythrocyte sedimentation rate) [94]. These immunophenotypes suggested a progressive decrease in B cells and natural killer cells.
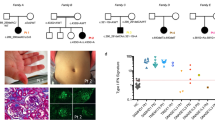
All interferonopathies discussed above could present as SLE-like symptoms, and thus, it is important to identify them from among all patients with suspected SLE. Importantly, compared to patients with classical SLE, patients with type I interferonopathies usually exhibit several specific clinical manifestations, including early onset, skin vasculopathy (chilblains, livedo reticularis and panniculitis, as shown in Fig. 2), central nervous system involvement (intracranial calcification, as shown in Fig. 2; seizures and psychomotor development delay), interstitial lung disease (as shown in Fig. 2), elevated transaminases and hypothyroidism. In addition, since patients with these interferonopathies could present with SLE, the study of type I interferonopathies could further reveal the pathogenesis of SLE and provide potential therapeutic targets in the future.

Images showing typical manifestations of type I interferonopathies (permissions were obtained by parents for publication). a Chilblains; b livedo reticularis; c intracranial calcification; d interstitial lung disease
Measurement of the activity of IFN-I signaling in SLE
Efforts have been made to establish assays for detecting biomarkers associated with the IFN-I pathway since they have the potential to diagnose and stratify patients. In 2023, Burska et al. conducted a literature review including 276 papers on IFN-I assays, and those assays included immunoassays for IFN-I proteins (n = 58), single-molecule assays (SimoA; majorly measuring IFN-α, n = 8), immunoassays for IFN-inducible proteins (n = 42), flowcytometry for IFN-I-inducible markers (n = 12), RNA microarrays for IFN signatures, scores or modules of ISGs (n = 70), RNA sequencing (n = 9), NanoString for the expression of ISGs (n = 5), IFN-I scoring by quantitative polymerase chain reaction (n = 122) and others [96].
The widely used assay for assessing interferon pathway activation is to analyze the expression of ISGs. Usually, a set of the most strongly and consistently expressed ISGs is selected, and one of the commonly used sets consists of six genes: IFI27, IFI44L, IFIT1, ISG15, RASD2, and SIGLEC1 [97]. It is the most developed technique, and many centers have used it routinely [98]. Nevertheless, studies indicate that different clinical conditions may have distinct subsets of ISGs expressed, which means that a given subset of ISGs might not be sensitive for detecting activation [99]. In addition, there is still some overlap between the IFN-I and IFN-II pathways, and thus, an increase in the ISG could sometimes be a consequence of IFN-II pathway activation instead of IFN-I [100,101,102].
In early studies, detecting IFN-α, whose half-life is 2–3 hours [103], was difficult due to its relatively low concentration in serum [104]. With the help of sensitive SimoA methods, several studies have attempted to measure the concentrations of IFN-I [105]. A study consisting of 48 pediatric SLE patients and 67 healthy controls demonstrated that serum IFN-α2 levels are strongly positively correlated with the IFN-I gene signature and that serum IFN-α2 levels are significantly associated with both the safety of estrogens according to the Lupus Erythematosus National Assessment-SLE disease activity index (SLEDAI) and the British Isles Lupus Assessment Group 2004, while the IFN-I gene signature did not show this association [106]. Nevertheless, soluble IFN-α2 is only a member of the IFN-I family and might not be able to represent the total activity of IFN-I. In addition, this assay is only able to measure the level of IFN-α2 in serum and is not sufficient to reflect specific local conditions, such as those in the kidney or joints.
There are many other attempts to find more assays that are convenient for detecting IFN-I activation in clinical practice. The detection of surface molecules by flow cytometry seems to be promising. CD169, also known as Siglec-1, is specifically expressed on monocytes, dendritic cells and tissue macrophages. It is induced by stimulation with IFN-I, including IFN-α, IFN-β and IFN-ω, but not IFN-γ. Sakumura et al. demonstrated that CD169 expression on CD14+ monocytes, as detected by flow cytometry, is elevated in pediatric SLE patients and parallels the concentration of IFN-α [107]. Additionally, Hoogen’s group showed that Galectin-9 is correlated with disease activity and serves as a reliable and easily measurable serum biomarker for detecting the interferon signature in patients with SLE [108]. Their conclusion is further supported by Yuksel’s study, which showed that the serum level of Galectin-9 is correlated with the SLEDAI [109]. The expression of SAMHD1 was also suggested to be associated with the IFN-I signaling pathway. By examining 98 pediatric SLE patients and 44 gender- and age-matched healthy donors, SAMHD1 was found to be positively correlated with several ISG, including myxovirus resistance protein A, IRF3 and IRF7 [110].
As concluded by the 2023 European Alliance of Associations for Rheumatology points to consider, current measurement assays measure various aspects of the IFN pathway, but they do not provide a comprehensive assessment of the entire pathway, and some lack specificity for IFN-I. The selection of the most appropriate assay depends on the specific research or clinical question [111].
Clinical implications of IFN-I in SLE
Since the IFN-I pathway plays an essential role in SLE pathogenesis, the detection of IFN-I is widely used in the diagnosis, stratification, and prognosis of patients with SLE. In addition, anti-IFN treatments are assumed to be a potential method for treating some specific SLE patients. In terms of the application of IFN-I in the diagnosis of SLE. In 2018, Wahadat et al. reported that 57% of pediatric SLE patients had a positive IFN-I signature [112]. Another study performed by Zorn-Pauly et al. suggested that in newly diagnosed SLE patients, almost all patients had IFN-I pathway activation, and a negative flow cytometry result for CD169 is efficient for excluding SLE, whose negative predictive value is > 99% [113].
Assays testing IFN-I pathway activation also have an important role in the stratification of patients with SLE. By using blood genomics, researchers identified three subtypes of SLE: “IFN-high”, “NE-high” and “mixed” [114]. They assumed that high levels of interferons (IFN-high) may arise due to the excessive production of IFNs triggered by viral infections, subsequently leading to the development of autoantibodies. On the other hand, elevated levels of NETs (NE-high) may predominantly result from infections caused by bacteria and fungi, which stimulate neutrophils to generate NETs and induce individual autoimmune responses. They also showed that the “mixed” group had the highest SLEDAI scores, while the “NE-high” group tended to cluster with healthy patients. Another group discovered a distinct clinical subset of pediatric SLE patients using interferon scores and complement levels [115]. These studies suggested that these patients had lower anti-dsDNA levels and might represent a predominant autoinflammatory subset of pediatric SLE patients. However, in their cohort, the IFN score was not correlated with disease activity, which contradicts the results of other groups. Several attempts have been made to identify differences between SLE patients with a high IFN signature and those with a low IFN signature, but the results were not satisfactory. There was a study suggesting significant associations between a high IFN signature and higher titers of anti-dsDNA, elevated serum B-cell activating factor and hypocomplementemia but no correlations with clinical disease activity [116].
Additionally, some markers of IFN-I are related to specific phenotypes. It was suggested that IFN-α and neopterin in cerebrospinal fluid (CSF) constitute promising biomarkers of neuropsychiatric SLE in a 5-year retrospective monocentric pediatric SLE cohort [117]. CSF IFN-α levels are significantly greater in patients with active neuropsychiatric SLE than in patients with inactive SLE, while the serum IFN-α levels are similar. In addition, correlating with the improvement in symptoms of neuropsychiatric SLE, the CSF concentration of IFN-α decreased. SAMDH1, as mentioned above, was also indicated to be related to vasculitis. Its levels were significantly increased (P < 0.05) in pediatric SLE patients with butterfly erythema, alopecia, and photosensitivity [110].
The prominent role of IFN-I in SLE pathogenesis has also spurred the development of targeted therapies. Anti-IFN therapies, including sifalimumab (an anti-IFN-α monoclonal antibody) and anifrolumab (a monoclonal antibody targeting the IFN-I receptor), have demonstrated favorable outcomes in phase II randomized controlled trials involving adult SLE patients with moderate-to-severe disease [118,119,120]. These therapies, when used in conjunction with standard-of-care medications, have shown efficacy in the absence of severe nephritis and neuropsychiatric involvement. However, pediatric patients were not included in those trials, and thus, the efficacy and efficacy of these biologics in pediatric populations still need further investigation. Additionally, JAK inhibitor treatment is an alternative therapeutic approach for pediatric patients with severe pediatric SLE. It has shown promise in gradually reducing inflammatory markers and IFN scores and promoting the upregulation of the DNA repair pathway [121]. Many molecules, such as inhibitors of TBK1 and IRF5, are also able to partially inhibit the IFN pathway, showing promising results in in vitro experiments [112, 122].
Conclusions
The role of IFN-I in pediatric-onset SLE is complex and multifaceted, as highlighted in this comprehensive review. While much of the existing research on IFN-I has focused on adult SLE patients, it has become increasingly evident that IFN-I signaling plays a pivotal role in the pathogenesis of pediatric SLE. Several key insights and implications emerge from this review: (1) IFN-I signaling in pediatric SLE: robust evidence demonstrating that IFN-I signaling is significantly activated in pediatric SLE patients; (2) genetic variants and susceptibility: genetic factors, such as variants in IFN-I-related genes such as IRF5 and TYK2, have been identified as risk factors for pediatric SLE. These genetic associations shed light on the genetic underpinnings of the disease and provide potential targets for future research and therapeutic interventions; (3) type I interferonopathies: this review highlights the overlap between type I interferonopathies and pediatric SLE. These conditions, characterized by sustained IFN-I activation, can closely mimic SLE symptoms, underscoring the complex interplay between genetic factors and immune dysregulation in autoimmune diseases. It is important to be aware of the possibility of type I interferonopathies in patients with SLE. On the other hand, the study of type I interferonopathies provides a way to discover the pathogenesis of SLE and potential therapeutic targets in the future; and (4) diagnostic and therapeutic implications: the detection of IFN-I activation has significant diagnostic and therapeutic implications for pediatric SLE patients. Biomarkers such as ISG signatures, CD169 expression, and Galectin-9 levels hold promise for improved diagnosis, stratification, and monitoring of disease activity in pediatric patients. Additionally, the emergence of anti-IFN therapies and JAK inhibitors presents potential treatment options, although their efficacy and safety in pediatric populations require further investigation.
In conclusion, this review underscores the pivotal role of IFN-I signaling in the pathogenesis of pediatric SLE and its potential as a diagnostic marker and therapeutic target. Additionally, this review describes the manifestations of interferonopathies with monogenic SLE presentations as well as the pathogenic mechanism of related genes, highlighting the importance of identifying those diseases, especially for patients with early-onset clinical IFN signatures, skin vasculopathy with chilblains, livedo reticularis and panniculitis, involvement of the central nervous system, and interstitial lung disease. Further research, particularly in the pediatric population, is needed to refine our understanding of the precise contributions of IFN-I to disease progression and to translate this knowledge into more effective and personalized management strategies for pediatric SLE patients. The complex interplay between genetic, immunological, and environmental factors in pediatric SLE patients requires continued investigation to improve patient outcomes and quality of life.
Data availability
All data generated or analysed during this study are included in this published article.
References
Psarras A, Emery P, Vital EM. Type I interferon-mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. Rheumatology (Oxford). 2017;56:1662–75.
Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. 2015;15:231–42.
Bezalel S, Guri KM, Elbirt D, Asher I, Sthoeger ZM. Type I interferon signature in systemic lupus erythematosus. Isr Med Assoc J. 2014;16:246–9.
Ronnblom L, Alm GV. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol. 2001;22:427–31.
Jiang J, Zhao M, Chang C, Wu H, Lu Q. Type I interferons in the pathogenesis and treatment of autoimmune diseases. Clin Rev Allergy Immunol. 2020;59:248–72.
Finke D, Randers K, Hoerster R, Hennig H, Zawatzky R, Marion T, et al. Elevated levels of endogenous apoptotic DNA and IFN-alpha in complement C4-deficient mice: implications for induction of systemic lupus erythematosus. Eur J Immunol. 2007;37:1702–9.
Cetin Gedik K, Lamot L, Romano M, Demirkaya E, Piskin D, Torreggiani S, et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology points to consider for diagnosis and management of autoinflammatory type I interferonopathies: CANDLE/PRAAS SAVI and AGS. Ann Rheum Dis. 2022;81:601–13.
Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthr Rheum. 2004;50:1861–72.
Feng D, Stone RC, Eloranta ML, Sangster-Guity N, Nordmark G, Sigurdsson S, et al. Genetic variants and disease-associated factors contribute to enhanced interferon regulatory factor 5 expression in blood cells of patients with systemic lupus erythematosus. Arthr Rheum. 2010;62:562–73.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86.
Liu M, Guo Q, Wu C, Sterlin D, Goswami S, Zhang Y, et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol Immunol. 2019;16:367–79.
Dong X, Antao OQ, Song W, Sanchez GM, Zembrzuski K, Koumpouras F, et al. Type I interferon-activated STAT4 regulation of follicular helper T cell-dependent cytokine and immunoglobulin production in lupus. Arthr Rheumatol. 2021;73:478–89.
Buang N, Tapeng L, Gray V, Sardini A, Whilding C, Lightstone L, et al. Type I interferons affect the metabolic fitness of CD8+ T cells from patients with systemic lupus erythematosus. Nat Commun. 2021;12:1980.
Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol. 2014;192:5459–68.
Eloranta ML, Alm GV, Ronnblom L. Disease mechanisms in rheumatology–tools and pathways: plasmacytoid dendritic cells and their role in autoimmune rheumatic diseases. Arthr Rheum. 2013;65:853–63.
Vance RE. Cytosolic DNA sensing: the field narrows. Immunity. 2016;45:227–8.
Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, et al. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–6.
Tesser A, Piperno GM, Pin A, Piscianz E, Boz V, Benvenuti F, et al. Priming of the cGAS-STING-TBK1 pathway enhances LPS-induced release of type I interferons. Cells. 2021;10:785.
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011. https://doi.org/10.1126/scitranslmed.3001201.
Boiteux S, Radicella JP. The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch Biochem Biophys. 2000;377:1–8.
Tumurkhuu G, Chen S, Montano EN, Ercan Laguna D, De Los SG, Yu JM, et al. Oxidative DNA damage accelerates skin inflammation in pristane-induced lupus model. Front Immunol. 2020;11:554725.
Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A, et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat Commun. 2019;10:2201.
Xie C, Zhou H, Athanasopoulos V, Shen Q, Zhang Y, Meng X, et al. De novo PACSIN1 gene variant found in childhood lupus and a role for PACSIN1/TRAF4 complex in toll-like receptor 7 activation. Arthr Rheumatol. 2023;75:1058–71.
Eames HL, Corbin AL, Udalova IA. Interferon regulatory factor 5 in human autoimmunity and murine models of autoimmune disease. Transl Res. 2016;167:167–82.
Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–37.
Sigurdsson S, Goring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, et al. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Hum Mol Genet. 2008;17:872–81.
Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–5.
Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104:6758–63.
Lofgren SE, Yin H, Delgado-Vega AM, Sanchez E, Lewen S, Pons-Estel BA, et al. Promoter insertion/deletion in the IRF5 gene is highly associated with susceptibility to systemic lupus erythematosus in distinct populations, but exerts a modest effect on gene expression in peripheral blood mononuclear cells. J Rheumatol. 2010;37:574–8.
Li D, Matta B, Song S, Nelson V, Diggins K, Simpfendorfer KR, et al. IRF5 genetic risk variants drive myeloid-specific IRF5 hyperactivation and presymptomatic SLE. JCI Insight. 2020;5:e124020.
Contreras-Cubas C, Garcia-Ortiz H, Velazquez-Cruz R, Barajas-Olmos F, Baca P, Martinez-Hernandez A, et al. Catalytically impaired TYK2 variants are protective against childhood- and adult-onset systemic lupus erythematosus in Mexicans. Sci Rep. 2019;9:12165.
Dell’Isola GB, Dini G, Culpepper KL, Portwood KE, Ferrara P, Di Cara G, et al. Clinical spectrum and currently available treatment of type I interferonopathy Aicardi-Goutières syndrome. World J Pediatr. 2023;19:635–43.
Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38:917–20.
Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–32.
Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A:296–312.
Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJ, et al. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. 2014;45:386–93.
Rice GI, Del Toro DY, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–9.
Lässig C, Matheisl S, Sparrer KMJ, Mann CCD, Moldt M, Patel JR, et al. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. Elife. 2015;4:e10859.
d’Angelo DM, Di Filippo P, Breda L, Chiarelli F. Type I Interferonopathies in children: an overview. Front Pediatr. 2021;9:631329.
Peng JH, Wang YS, Han X, Zhang CM, Chen X, Jin Y, et al. Clinical implications of a new pathogenic variant that causes lupus nephritis due to RIG-I hyperactivation. J Am Soc Nephrol. 2023;34:258–72.
Farrugia M, Baron B. The role of toll-like receptors in autoimmune diseases through failure of the self-recognition mechanism. Int J Inflam. 2017;2017:8391230.
Fejtkova M, Sukova M, Hlozkova K, Kramarzova KS, Rackova M, Jakubec D, et al. TLR8/TLR7 dysregulation due to a novel TLR8 mutation causes severe autoimmune hemolytic anemia and autoinflammation in identical twins. Am J Hematol. 2022;97:338–51.
Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605:349–56.
Bender AT, Tzvetkov E, Pereira A, Wu Y, Kasar S, Przetak MM, et al. TLR7 and TLR8 differentially activate the IRF and NF-κB pathways in specific cell types to promote inflammation. Immunohorizons. 2020;4:93–107.
de Marcken M, Dhaliwal K, Danielsen AC, Gautron AS, Dominguez-Villar M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci Signal. 2019;12:eaaw1347.
Aluri J, Bach A, Kaviany S, Paracatu LC, Kitcharoensakkul M, Walkiewicz MA, et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood. 2021;137:2450–62.
Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New horizons in the genetic etiology of systemic lupus erythematosus and lupus-like disease: monogenic lupus and beyond. J Clin Med. 2020;9:712.
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in dnase1-deficient mice. Nat Genet. 2000;25:177–81.
Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28:313–4.
Bodano A, Amarelo J, Gonzalez A, Gomez-Reino JJ, Conde C. Novel DNASE I mutations related to systemic lupus erythematosus. Arthr Rheum. 2004;50:4070–1.
Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun. 2017;8:2176.
Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43:1186–8.
Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. 2020;52:1364–72.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–18.
Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124:5516–20.
Vece TJ, Watkin LB, Nicholas S, Canter D, Braun MC, Guillerman RP, et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol. 2016;36:377–87.
Boulisfane-El Khalifi S, Viel S, Lahoche A, Fremond ML, Lopez J, Lombard C, et al. COPA syndrome as a cause of lupus nephritis. Kidney Int Rep. 2019;4:1187–9.
Zhuang H, Hudson E, Han S, Arja RD, Hui W, Lu L, et al. Microvascular lung injury and endoplasmic reticulum stress in systemic lupus erythematosus-associated alveolar hemorrhage and pulmonary vasculitis. Am J Physiol Lung Cell Mol Physiol. 2022;323:L715–29.
Hirschenberger M, Lepelley A, Rupp U, Klute S, Hunszinger V, Koepke L, et al. ARF1 prevents aberrant type I interferon induction by regulating STING activation and recycling. Nat Commun. 2023;14:6770.
Lu D, Song J, Sun Y, Qi F, Liu L, Jin Y, et al. Mutations of deubiquitinase OTUD1 are associated with autoimmune disorders. J Autoimmun. 2018;94:156–65.
Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. 2011;43:127–31.
Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. 2011;43:132–7.
An J, Briggs TA, Dumax-Vorzet A, Alarcon-Riquelme ME, Belot A, Beresford M, et al. Tartrate-resistant acid phosphatase deficiency in the predisposition to systemic lupus erythematosus. Arthr Rheumatol. 2017;69:131–42.
Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immun. 2017;139:2016–2020.e5.
Gruber CN, Calis JJA, Buta S, Evrony G, Martin JC, Uhl SA, et al. Complex autoinflammatory syndrome unveils fundamental principles of JAK1 kinase transcriptional and biochemical function. Immunity. 2020;53:672–84.e11.
Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med. 2016;213:1163–74.
Martin-Fernandez M, Bravo Garcia-Morato M, Gruber C, Murias Loza S, Malik MNH, Alsohime F, et al. Systemic type I IFN inflammation in human ISG15 deficiency leads to necrotizing skin lesions. Cell Rep. 2020;31:107633.
Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517:89–93.
Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol. 2019;4:eaav7501.
Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med. 2020;217:e20192319.
Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020;583:90–5.
Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. 2020;146:1194–200.
Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. 2020;11:5341.
Michniacki TF, Walkovich K, DeMeyer L, Saad N, Hannibal M, Basiaga ML, et al. SOCS1 haploinsufficiency presenting as severe enthesitis, bone marrow hypocellularity, and refractory thrombocytopenia in a pediatric patient with subsequent response to JAK inhibition. J Clin Immunol. 2022;42:1766–77.
Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. 2018;9:1558.
Du Y, Brodeur KE, Hsu E, Chen L, Chen Q, Liu M, et al. In cis “benign” SOCS1 variants linked to enhanced interferon signaling and autoimmunity. J Autoimmun. 2023;140:103119.
Smyth AE, Kaleviste E, Snow A, Kisand K, McMahon CJ, Cant AJ, et al. Aortic calcification in a patient with a gain-of-function STAT1 mutation. J Clin Immunol. 2018;38:468–70.
Kaleviste E, Saare M, Leahy TR, Bondet V, Duffy D, Mogensen TH, et al. Interferon signature in patients with STAT1 gain-of-function mutation is epigenetically determined. Eur J Immunol. 2019;49:790–800.
Stellacci E, Moneta GM, Bruselles A, Barresi S, Pizzi S, Torre G, et al. The activating p.Ser466Arg change in STAT1 causes a peculiar phenotype with features of interferonopathies. Clin Genet. 2019;96:585–9.
Okada S, Asano T, Moriya K, Boisson-Dupuis S, Kobayashi M, Casanova JL, et al. Human STAT1 gain-of-function heterozygous mutations: chronic mucocutaneous candidiasis and type I interferonopathy. J Clin Immunol. 2020;40:1065–81.
Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, nakajo-nishimura syndrome. Proc Natl Acad Sci U S A. 2011;108:14914–9.
Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. 2015;125:4196–211.
de Jesus AA, Brehm A, VanTries R, Pillet P, Parentelli AS, Montealegre Sanchez GA, et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J Allergy Clin Immunol. 2019;143:1939–43.e8.
Yan K, Zhang J, Lee PY, Tao P, Wang J, Wang S, et al. Haploinsufficiency of PSMD12 causes proteasome dysfunction and subclinical autoinflammation. Arthr Rheumatol. 2022;74:1083–90.
Papendorf JJ, Ebstein F, Alehashemi S, Piotto DGP, Kozlova A, Terreri MT, et al. Identification of eight novel proteasome variants in five unrelated cases of proteasome-associated autoinflammatory syndromes (PRAAS). Front Immunol. 2023;14:1190104.
Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendinez F, Hernandez A, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. 2010;62:489–95.
Garg A, Hernandez MD, Sousa AB, Subramanyam L, de Martinez Villarreal L, dos Santos HG, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab. 2010;95:E58–63.
Ebstein F, Poli Harlowe MC, Studencka-Turski M, Kruger E. Contribution of the unfolded protein response (UPR) to the pathogenesis of proteasome-associated autoinflammatory syndromes (PRAAS). Front Immunol. 2019;10:2756.
Kury S, Besnard T, Ebstein F, Khan TN, Gambin T, Douglas J, et al. De novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am J Hum Genet. 2017;100:689.
An JW, Pimpale-Chavan P, Stone DL, Bandeira M, Dedeoglu F, Lo J, et al. Case report: novel variants in RELA associated with familial Behcet’s-like disease. Front Immunol. 2023;14:1127085.
Moriya K, Nakano T, Honda Y, Tsumura M, Ogishi M, Sonoda M, et al. Human RELA dominant-negative mutations underlie type I interferonopathy with autoinflammation and autoimmunity. J Exp Med. 2023;220:e20212276.
Wang Q, Zhai YY, Dai JH, Li KY, Deng Q, Han ZG. SAMD9L inactivation promotes cell proliferation via facilitating GI-S transition in hepatitis B virus-associated hepatocellular carcinoma. Int J Biol Sci. 2014;10:807–16.
de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. 2020;130:1669–82.
Russell AJ, Gray PE, Ziegler JB, Kim YJ, Smith S, Sewell WA, et al. SAMD9L autoinflammatory or ataxia pancytopenia disease mutations activate cell-autonomous translational. Proc Natl Acad Sci U S A. 2021;118:e2110190118.
Burska A, Rodriguez-Carrio J, Biesen R, Dik WA, Eloranta ML, Cavalli G, et al. Type I interferon pathway assays in studies of rheumatic and musculoskeletal diseases: a systematic literature review informing EULAR points to consider. RMD Open. 2023;9:e002876.
Rice GI, Melki I, Fremond ML, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol. 2017;37:123–32.
Nombel A, Foray AP, Garnier L, Lombard C, Hachulla E, Bader-Meunier B, et al. Assessment of type I interferon response in routine practice in France in 2022. RMD Open. 2023;9:e003211.
Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. 2016;165:551–65.
Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111–22.
Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, et al. Synergistic activation of inflammatory cytokine genes by interferon-gamma-induced chromatin remodeling and toll-like receptor signaling. Immunity. 2013;39:454–69.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49.
Ramos TI, Villacis-Aguirre CA, Santiago Vispo N, Santiago Padilla L, Pedroso Santana S, Parra NC, et al. Forms and methods for interferon’s encapsulation. Pharmaceutics. 2021;13:1533.
Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthr Rheum. 2006;54:1906–16.
Mathian A, Mouries-Martin S, Dorgham K, Devilliers H, Barnabei L, Ben Salah E, et al. Monitoring disease activity in systemic lupus erythematosus with single-molecule array digital enzyme-linked immunosorbent assay quantification of serum interferon-alpha. Arthr Rheumatol. 2019;71:756–65.
Wahadat MJ, Qi H, van Helden-Meeuwsen CG, Huijser E, van den Berg L, van Dijk-Hummelman A, et al. Serum IFNα2 levels are associated with disease activity and outperform IFN-I gene signature in a longitudinal childhood-onset SLE cohort. Rheumatology (Oxford). 2023;62:2872–9.
Sakumura N, Yokoyama T, Usami M, Hosono Y, Inoue N, Matsuda Y, et al. CD169 expression on monocytes as a marker for assessing type I interferon status in pediatric inflammatory diseases. Clin Immunol. 2023;250:109329.
van den Hoogen LL, van Roon JAG, Mertens JS, Wienke J, Lopes AP, de Jager W, et al. Galectin-9 is an easy to measure biomarker for the interferon signature in systemic lupus erythematosus and antiphospholipid syndrome. Ann Rheum Dis. 2018;77:1810–4.
Yuksel K, Sag E, Demir S, Ozdel S, Kaya UA, Atalay E, et al. Plasma checkpoint protein levels and galectin-9 in juvenile systemic lupus erythematosus. Lupus. 2021;30:998–1004.
Zhao X, Li C, Li S, Zhang J, Kuang W, Deng J, et al. SAMHD1 associates with inflammation and vasculitis in paediatric-onset systemic lupus erythematosus. Clin Exp Rheumatol. 2022;40:1801–7.
Rodriguez-Carrio J, Burska A, Conaghan PG, Dik WA, Biesen R, Eloranta ML, et al. 2022 EULAR points to consider for the measurement, reporting and application of IFN-I pathway activation assays in clinical research and practice. Ann Rheum Dis. 2023;82:754–62.
Wahadat MJ, Bodewes ILA, Maria NI, van Helden-Meeuwsen CG, van Dijk-Hummelman A, Steenwijk EC, et al. Type I IFN signature in childhood-onset systemic lupus erythematosus: a conspiracy of DNA- and RNA-sensing receptors? Arthr Res Ther. 2018;20:4.
Zorn-Pauly L, von Stuckrad ASL, Klotsche J, Rose T, Kallinich T, Enghard P, et al. Evaluation of SIGLEC1 in the diagnosis of suspected systemic lupus erythematosus. Rheumatology (Oxford). 2022;61:3396–400.
Cui M, Li T, Yan X, Wang C, Shen Q, Ren H, et al. Blood genomics identifies three subtypes of systemic lupus erythematosus: “IFN-high,” “NE-high,” and “mixed”. Mediators Inflamm. 2021;2021:6660164.
Tesser A, de Carvalho LM, Sandrin-Garcia P, Pin A, Pastore S, Taddio A, et al. Higher interferon score and normal complement levels may identify a distinct clinical subset in children with systemic lupus erythematosus. Arthr Res Ther. 2020;22:91.
Kennedy WP, Maciuca R, Wolslegel K, Tew W, Abbas AR, Chaivorapol C, et al. Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci Med. 2015;2:e000080.
Labouret M, Costi S, Bondet V, Trebossen V, Le Roux E, Ntorkou A, et al. Juvenile neuropsychiatric systemic lupus erythematosus: identification of novel central neuroinflammation biomarkers. J Clin Immunol. 2023;43:615–24.
Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75:1909–16.
Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthr Rheumatol. 2017;69:376–86.
Wu LX, Xu DC, Sun K, Huang H, Jiang WW, Li WF. Systemic lupus erythematosus in a male teenager manifested with cardiogenic shock and extremities infarction. World J Emerg Med. 2022;13:406–8.
Pin A, Tesser A, Pastore S, Moressa V, Valencic E, Arbo A, et al. Biological and clinical changes in a pediatric series treated with off-label JAK inhibitors. Int J Mol Sci. 2020;21:7767.
Song S, De S, Nelson V, Chopra S, LaPan M, Kampta K, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest. 2020;130:6700–17.
Funding
This study was supported by National Key Research and Development Program of China (2021YFC2702005) and National High level Hospital Clinical Research Foundation (2022-PUMCH-B-079).
Author information
Authors and Affiliations
Contributions
ZY contributed to data curation, investigation, methodology, writing of the original draft, reviewing and editing. SHM contributed to conceptualization, funding acquisition, writing of the original draft, reviewing and editing. All the authors approved the final version to be published.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. For images of patients, permissions were obtained by parents for publication.
Conflict of interest
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article. Author Hong-Mei Song is a member of the Editorial Board for World Journal of Pediatrics. The paper was handled by the other editor and has undergone rigorous peer review process. Author Hong-Mei Song was not involved in the journal's review of, or decisions related to this manuscript. The authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, Y., Song, HM. Type I interferon pathway in pediatric systemic lupus erythematosus. World J Pediatr (2024). https://doi.org/10.1007/s12519-024-00811-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12519-024-00811-4