
Abstract
Background
Progressive pseudorheumatoid dysplasia (PPRD) is a rare genetic disease with autosomal recessive inheritance. There was a lack of genotype–phenotype correlation data from the Chinese population. This study aimed to identify the genotype and phenotype characteristics of Chinese PPRD patients and to conduct a genotype–phenotype analysis of Chinese PPRD patients.
Methods
Genetic analysis was performed for suspected PPRD patients from Peking Union Medical College Hospital. Medical records were collected from the electronic medical record system and patient-held portable health records. Published Chinese PPRD cases were gathered from both international and Chinese local databases. We collected demographic information, genetic variants, clinical manifestations, and imaging characteristics for further analysis.
Results
We included 105 Chinese PPRD patients in the current study. Thirty-three variants, including nine novels and five hotspot variants, were identified, with 26/33 (79%) variants exclusively seen in the Chinese population. Chinese PPRD patients share a phenotype similar to that in international reports. Joint involvement may progress with age (R2 = 0.2541). Long bone shortening and severe deformities occur in three patients with biallelic null variants, of which at least one variant is located in exon 2. Among hotspot variants, c.624dupA (p.C209Mfs*21) were associated with later onset and more involved joints. Elbow joints were more likely to be affected in patients carrying c.624dupA (p.C209Mfs*21) and c.866dupA (p.S209Efs*13). Shoulder joints are more likely to be involved in patients with biallelic null variants (P = 0.027).
Conclusions
Chinese PPRD patients share a unique mutation spectrum. Among the five hotspot variants, c.624dupA is associated with later onset of disease, more extensive joint involvement, and a tendency to affect elbow joints. Biallelic null variants with at least one variant in exon 2 could be a likely cause of long bone shortening and severe deformities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Progressive pseudorheumatoid dysplasia (PPRD), initially described in 1982 [1], is a rare autosomal recessive disorder characterized by spondyloepiphyseal dysplasia. The causal gene CCN6 (cellular communication network factor 6 gene), previously known as WISP3 (WNT1-inducible signaling pathway protein-3 gene), was first cloned and sequenced in 1998 [2]. The first seven Chinese cases were reported in 1986 without genetic confirmation [3]. Due to the low incidence of PPRD, currently published case reports or series often consist of only a few patients [4,5,6]. Neither large cohort analyses nor epidemiological surveys of PPRD have been conducted in China. To summarize the genotypic and phenotypic features of PPRD in the Chinese population, we report a single-center cohort of 61 patients genetically diagnosed with PPRD from Peking Union Medical College Hospital (PUMCH) and conduct a retrospective analysis of 44 genetically confirmed Chinese PPRD cases by searching the literature before October 2021.
Methods
Patients
This retrospective study included all patients with suspected PPRD seen at the genetic clinics of the Pediatric Department, PUMCH between January 2007 and October 2021. The inclusion criteria were as follows: (1) progressive polyarticular deformities with a limited range of motion (involving bilateral interphalangeal joints, elbows, knees, or hips) or progressive bone malformations, with varying degrees of severity and sequences of progression; (2) with or without spinal deformities such as scoliosis and kyphosis; and (3) no fever and rashes. The exclusion criteria were as follows: (1) persistent elevation of inflammatory markers, such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP); and (2) other monogenic diseases presenting with spondyloepiphyseal dysplasia.
Inpatient and outpatient medical records were collected from patient-held portable health records and the electronic medical record system of PUMCH. Potential cases with WISP3 or CCN6 variants were collected, followed by genetic confirmation. This study was approved by the Ethics Committee of PUMCH (S-609).
Literature-derived patients
We searched PubMed, Wanfang Data, China Science and Technology Journal Database (CQVIP), China Knowledge Resource Integrated Database (CNKI), and Chinese Medical Journal Network by using combinations of the following keywords: “progressive pseudorheumatoid dysplasia”, “spondyloepiphyseal dysplasia”, “WISP3”, “CCN6”, “Chinese”, and “China”. We put no restrictions on the publication dates. Cases with no genetic confirmation were excluded. We also removed duplicate cases in different studies and the cases overlapping our PUMCH cohort. Cases with detailed clinical data were selected for further analysis of their demographic information, genetic mutations, clinical manifestations, and radiological characteristics.
Genetic analysis
Sanger sequencing was performed at the PUMCH laboratory for all 42 PUMCH patients with suspected PPRD between 2007 and 2013. Targeted next-generation sequencing (NGS) panel or Sanger sequencing was applied by third-party companies for the remaining PUMCH patients from 2014 to date.
Sanger sequencing
We obtained peripheral blood from PUMCH patients and/or their parents, followed by DNA extraction. Polymerase chain reaction (PCR) primers (Supplementary Table 1) were designed with Primer3.0 online software. We performed PCR amplification and product purification. Sanger sequencing was then applied to sequence the five exons of WISP3 as well as exon‒intron boundaries.
Targeted next-generation sequencing
Peripheral blood was obtained from PUMCH patients and/or their parents for targeted NGS by third-party companies. The general steps include DNA extraction, DNA library preparation, enrichment and sequencing of targeted genes, and bioinformatic analysis. The sequencing results were validated with Sanger sequencing using primers designed by third-party companies.
Statistical analysis
For descriptive statistics, median and range were used for numerical data that may not follow a normal distribution. The arithmetic mean and standard deviation were calculated for normally distributed numerical data. Categorical data were described with frequencies and proportions. Statistical inferences were made by SPSS 17.0 software. For hypothesis testing of normally distributed numerical data among multiple groups, we performed one-way ANOVA under the equal variance hypothesis. If the null hypothesis was rejected, the least significant difference t test approach was then applied to identify the significantly different pairs. If variances were heterogeneous across groups, Tamhane’s multiple comparison procedure was followed for intergroup analysis. For categorical data, Chi-square tests and Fisher’s exact test were applied. To compare multiple groups, we weighed the data by frequency and applied Fisher’s exact tests pair by pair. The threshold of statistical significance was set as a P value < 0.05.
Results
Genetic testing results
Genetic testing of PPRD patients from PUMCH
Genetic analysis of our PUMCH cohort was conducted on 55 Chinese patients from 47 pedigrees (Fig. 1). Briefly, we enrolled and conducted genetic tests on 79 patients with suspected PPRD from the Department of Pediatrics, PUMCH. We successfully diagnosed 61 patients from 52 pedigrees in our center, of which six patients from five pedigrees were excluded from genetic analysis due to a lack of variation details from their medical records.

Flowchart of patient collection. PUMCH Peking Union Medical College Hospital, PPRD progressive pseudorheumatoid dysplasia, NGS next-generation sequencing
Biallelic compound heterozygous or homozygous CCN6 variants were identified in 52 patients, including one patient who carried three CCN6 variants. Three patients were heterozygous for CCN6. In sum, twenty CCN6 variants were identified from the 55 patients, including nine novel variants (NM_198239.2): c.135dupA (p.Q46Tfs*31); c.166C>T (p.Q56*); c.238A>G (p.K80E); c.250delG (p.E84Kfs*21); c.419dupT (p.L140Ffs*30); c.556C>T (p.Q186*); c.625T>A (p.C209S); c.802T>C (p.C268R); c.1012C>T (p.Q338*).
CCN6 mutations in Chinese PPRD patients
After a literature search, forty-four literature-derived patients from 36 pedigrees were included in this study (Fig. 1). Twenty-four variants were collected from the literature, including 11 variants also observed in our PUMCH cohort. Biallelic CCN6 variants were found in 39 patients. The remaining five patients carried only one monoallelic variant.
By combining PUMCH-identified variants with those of 44 genetically confirmed PPRD patients from the literature, we gathered 33 variants in 99 PPRD patients from 83 Chinese pedigrees (Fig. 2). All variants were located in exons 2–5 of CCN6 (NM_198239.2). Among the 33 variants, there were 15 frameshift, 10 missense, and seven nonsense variants, as well as one splicing variant (Table 1). The hotspot variants were c.667T>G (p.C223G), c.624dupA (p.C209Mfs*21), c.342T>G (p.C114W), c.866dupA (p.S290Efs*13), and c.589+2T>C. Nine of the 33 variants were novel variants. Among the previously reported variants, twenty variants have been recorded in the Human Gene Mutation Database (HGMD), with only seven reported in patients outside China. In sum, there are 26 variants exclusively seen in Chinese patients.

Frequency and distribution of CCN6 mutations. a Comparison of Chinese CCN6 variants (right panel) with non-Chinese variants from HGMD (left panel); b exon distribution of Chinese and non-Chinese variations; c hotspot variants in Chinese cohort as revealed by allele, patient, and pedigree counts. CCN6 cellular communication network factor 6 gene, PUMCH Peking Union Medical College Hospital, PPRD progressive pseudorheumatoid dysplasia, HGMD Human Gene Mutation Database
Clinical manifestations
After excluding poorly documented cases, duplicate ones, and those without genetic confirmation, ninety-six patients from 79 pedigrees were included in the clinical analysis, including 52 cases diagnosed by PUMCH and 44 cases retrieved from the literature (Fig. 1).
PPRD patients from PUMCH
The clinical analysis was conducted on 52 of the 61 genetically diagnosed patients, as nine patient-held portable medical records could not be retrieved. The 52 PUMCH-diagnosed PPRD patients, including 32 males and 20 females, came from 48 pedigrees. The median age of initial consultation at PUMCH was 12 years (range: 3–34), with 79% (41/52) under 18. The median age of onset was 6 years (range: 0.5–29). The dates of genetic diagnosis were documented in 35 patients. The median gap between disease onset and the genetic diagnosis was 6 years (range: 1–29). Thirteen patients were documented to have PPRD siblings. Consanguineous marriage was documented in four patients from three pedigrees. Ethnic information was recorded in 27 patients, all of whom were Han Chinese.
All patients were intellectually normal. Most patients were characterized by short stature, progressive deformities in multiple joints with limited range of motion and tenderness. The laboratory investigations returned all normal for complete blood count, biochemistry profile, ESR, CRP, and autoantibodies.
Chinese PPRD patients combined
Demographic information Combining PUMCH and literature-identified PPRD cases, the median age of onset was 6 years (range: 0.5–29). Of the 93 patients with documented initial symptoms, 72% (67/93) experienced their initial symptoms before seven. Only two patients had disease onset in adulthood, at 24 and 29 years old. The median age of initial consultation (at PUMCH or as reported from the literature) was 14 years (range: 2–59). Height was documented in 43 patients below 18 years old, with 70% (30/43) below the 3rd percentile. Weight was documented in 33 patients below 18 years old, with 33% (11/33) below the 3rd percentile. Seventy-one percent (12/17) of the adult patients with documented height were below the 3rd percentile (references were taken from the same gender 18-year-old healthy Chinese population). Forty-two percent (5/12) of adults were below the 3rd percentile in weight (reference as above). The proportions of short stature (height < 3rd percentile) were not significantly different in adult and pediatric patients (P = 1.000).
General signs Collectively, the 96 Chinese PPRD patients presented with short stature, short neck, thoracic deformities, short trunk, polyarticular joint enlargement and flexion deformities, spinal curvature, pronounced pelvic tilt in the standing position, and progressive gait changes ranging from normal gait to limping gait to waddling gait. In severe cases, patients were wheelchair-dependent and unable to walk at all. Muscular atrophy was observed in wheelchair-dependent patients. Seven patients were documented to use wheelchairs to enter the clinic, with ages ranging from 11 to 17 years.
Joint involvement PPRD showed a distinct pattern of joint involvement (Fig. 3a). Bilateral interphalangeal joints were involved in 92% (88/96) of cases, including 25 cases of flexion deformities or claw hands. Knees were affected in 77% (74/96) of cases, including 72 cases involving bilateral knees and two cases unilaterally affecting the right knee. Hip joints were involved in 73% (70/96) of cases, including 68 bilaterally involved, one unilaterally right-sided, and one unilaterally left-sided. Elbows were affected in 67% (64/96) of patients, including 61 bilaterally involved patients and three unilaterally right-sided patients. Bilateral wrists were involved in 39% (37/96) of patients. The shoulder joint was involved in 14% (13/96) of patients (ten bilaterally and three unilaterally right-sided). Sacroiliac joints were involved in 5% (5/96) of patients. Interphalangeal joint involvement of the foot was extremely rare, as there were only four documented cases (three bilaterally and one unilaterally right-sided). For the axial skeleton, lumbar vertebrae were involved in 43% (41/96) of patients. The cervical spine was affected in 28% (27/96) of patients. Thirty-two (33%) patients had spinal curvature. Limited range of motion was documented in 95% (91/96) of patients, and 62% (60/96) of patients experienced joint tenderness (Fig. 3b).

Joint involvement of progressive pseudorheumatoid dysplasia (PPRD). a, b Frequencies of the joints involved in Chinese PPRD cohort; c positive correlation between affected joints and age. ROM range of motion
A 3-year-old boy with no joint involvement had bilateral symmetrical curvature of the tibia and fibula. An 11-year-old boy had only enlargement of bilateral interphalangeal joints of the hand and mild pectus carinatum. The number of involved joints (excluding interphalangeal joints) seemed positively correlated with age (Fig. 3c).
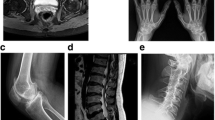
Imaging findings As revealed by X-ray or computed tomography scans, PPRD patients typically showed osteoporosis, polyarticular joint enlargement and degeneration, joint space narrowing, irregular articular surfaces, and dystrophy of the epiphyses or metaphyses (Fig. 4). Imaging findings of bilateral hands include thickening and enlargements of metaphyses, as well as narrowing, enlargement, and flexion deformities of interphalangeal joints. Spinal findings include narrowed anterior borders and abnormally flattened vertebral bodies, in addition to scoliosis, kyphosis, or lordosis. Severe long bone deformities occurred in three patients from two pedigrees. Case 4 is an adult male with bilateral humeral shortening and flexion deformities. He carries biallelic compound heterozygous variants at c.135dupA (p.Q46Tfs*31) and c.866dupA (p.S290Efs*3). Another two cases are siblings with homozygous variants at c.166C>T (p.Q56*). The older sister is an adult female with humeral head enlargement, ahumeral neck narrowing, humeral bone shortening, and localized flexion deformities. The 3-year-old brother had an early onset of the disease 6 months after birth. He developed progressive varus deformities of the bilateral lower extremities, with symmetrical curvature of the tibia and fibula.

Clinical and Imaging findings of progressive pseudorheumatoid dysplasia (PPRD). Typical imaging findings of PPRD (a–f) and imaging findings of PPRD cases with atypical severe long bone involvement (g–l). a, b Flexion deformity and claw shape of bilateral hands. X-ray showed osteoporosis of bilateral hands, epiphyseal enlargement of the metacarpophalangeal, proximal and distal interphalangeal joints, narrowing of the metacarpophalangeal joint spaces, irregularity of articular surfaces, and joint deformities (case 1); c widening of the pubic symphysis and bilateral sacroiliac joint spaces. Osteoporosis and degeneration of the bilteral hip joints, narrowed hip joints spaces, and ill-defined articular surfaces can be observed. Cystic changes were under the articular surfaces of bilateral femoral heads. The femoral necks were also shortened (case 2); d–f scoliosis or kyphosis of the spine (d and e from case 1; f from case 3). The vertebral bodies were flattened. There was a lack of regularity on the superior and inferior surfaces; g, j shortening and flexion deformities of the bilateral humeri with metaphyseal or epiphyseal enlargement of the upper and lower ends. The elbow joints were enlarged with narrowed joint spaces. There might be a rotational deformity of the humerus on the right shoulder (case 4); h, k enlargement of the humeral head. The humeral neck was narrowed. There might be a flexion deformity (case 1); i, l varus deformities of the bilateral lower extremities. The tibia and fibula on the bilateral sides were symmetrically curved. There were metaphyseal enlargements of the knees and ankles, and a widening of the joint spaces (case 5, the younger brother of case 1)
Association analysis between genotypes and clinical phenotypes
The association between mutational hotspots and phenotypes For genotype–phenotype analysis, we compared the age of onset, age at an initial consultation, gender ratio, number and patterns of joint involvement (excluding interphalangeal joints), percentage of spinal curvature, and joint symptoms (including a limited range of motion and arthralgia) in PPRD patients carrying the five hotspot variants (Table 2). Patients with c.624dupA (p.C209Mfs*21) variant had more involved joints than patients with the c.342T>G (p.C114W) and c.667T>G (p.C223G) variants (P = 0.013 and 0.016, respectively). Furthermore, elbow joints are more likely to be involved in patients carrying c.624dupA (p.C209Mfs*21) and c.866dupA (p.S290Efs*13) variants (P = 0.028).
Genotype–phenotype associations Of the 90 patients with complete clinical and genetic variant data, thirty-three patients carried biallelic CCN6 null variants. Eight biallelic null variant carriers had at least one variant in exon 2. We compared the clinical features of patients with different genotypes and found that biallelic null variants are more likely to involve shoulder joints (P = 0.027) and are associated with long bone shortening with severe deformities (P = 0.049). Further analysis revealed that long bone shortening with severe deformities was associated with a higher proportion of patients with biallelic null variants, including at least one variant located in exon 2 than other genotypes (P < 0.001) (Table 3).
Discussion
Progressive pseudorheumatoid arthritis (PPRD) is a rare progressive disease with spondyloepiphyseal dysplasia and an estimated incidence of 1/1,000,000 in the UK. No incidence rate data have been reported in China. The typical age of onset is 3–8 years [26, 27]. In this study, we conducted a large retrospective analysis of Chinese PPRD patients and showed for the first time that Chinese PPRD patients share a unique CCN6 mutation spectrum with five hotspots. Typical and atypical PPRD phenotypes were characterized, and potential genotype–phenotype associations were identified.
Chinese patients shared a unique CCN6 mutation spectrum (Fig. 2). As of April 2021, 78 PPRD-associated variants have been recorded by HGMD, including 13 Chinese-exclusive variants. Collectively, we identified a total of 33 variants widely distributed on CCN6 exons 2–5 among the Chinese patients. Only seven variants have been reported outside China. Seventy-nine percent (26/33) of variants were exclusively seen in the Chinese population, indicating a unique variant spectrum in the Chinese ethnic group. Nine novel CCN6 variants were also discovered. Among the novel variants, six were ACMG (American College of Medical Genetics) pathogenic null variants, including three frameshift and three nonsense variants that may trigger premature termination of protein translation. The remaining three novel variants are likely pathogenic missense variants. Five hotspot variants were identified.
We identified typical and atypical presentations as well as top joints involved in PPRD (Figs. 3, 4). Clinical features include abnormal gaits, fatigue, polyarticular joint deformities, arthralgia, and limited range of motion. The most commonly involved joints were bilateral interphalangeal joints of the hand, especially the distal joints, which can be considered a hallmark of this disease. The interphalangeal joints were followed by knees, hips, and elbows. Notably, almost all the cases showed symmetrical joint involvement, with only a few cases unilaterally affected. The unilaterally involved joints were more commonly positioned on the right side, probably associated with right-handedness. We observed a positive correlation between increased joint involvement with age, which was compatible with the progressive nature of this disease. Chinese PPRD patients presented with those from other countries. About 70% of Chinese PPRD patients’ heights were below the 3rd percentile, showing no significant differences between adult and pediatric patients.
A later age of onset may be associated with c.725_726del and c.624dupA. In our cohort, only two patients had their disease onset in adulthood, all carrying c.725_726del. One patient [21] was 24 years old at the onset with a single maternal-derived monoallelic variant c.725_726del (p.K242Rfs*36). Another case was a 29-year-old male who carried biallelic compound heterozygous mutations of c.625T>A (p.C209S) and c.725_726del (p.K242Rfs*36). In addition, c.624dupA was found to be associated with later onset by comparison among hotspot variants.
Patients with c.624dupA and biallelic null variants are likely to present special phenotypes. After comparing the phenotypes of patients carrying the five hotspot variants, we revealed that patients carrying c.624dupA (p.C209Mfs*21) not only were associated with later onset of disease and more involved joints but also showed a higher likelihood of elbow involvement. By comparing clinical manifestations among PPRD patients with different genotypes, patients carrying biallelic null variants were more likely to have shoulder joints involved and present long bone shortening with severe deformities. All three patients with long bone shortening and severe deformities carried biallelic null variants, with at least one variant located in exon 2. These null variants may lead to premature termination of CCN6 translation and protein degradation, thereby contributing to shortening and severe long deformities.
Notably, we observed variable expressivity in PPRD patients. Patients with the same genotype from the same pedigree may show variable phenotypes. As the disease name suggests, the severity of this disease increases as patients age. Younger patients may have fewer involved joints and milder presentations, as exemplified in siblings with long bone deformities. The 3-year-old younger brother presented only deformities in the lower extremities, while the sister showed classic PPRD phenotypes involving multiple joints. The variability in phenotypes may be explained by the presence of modifier genes, as well as environmental triggers.
Sanger sequencing should be prioritized for PPRD screening. During 2007 and 2013, our laboratory at PUMCH performed Sanger sequencing for 42 patients suspected of PPRD and returned positive results for 34 cases and negative results for six cases. Two genetic testing results could not be retrieved. The diagnostic rate was 85% (34/40). We propose that by raising awareness of this disease among clinicians, PPRD can be diagnosed with a high diagnostic rate with Sanger sequencing.
One limitation of this study is the briefness of medical records and the accidental loss of genetic results and medical records. As most PPRD patients at PUMCH were seen in outpatient scenarios, their medical records were often briefly documented. The clinical data from the literature also vary widely in detail, and data from some sources may be too brief to analyze, leading to inevitable bias. Due to the progressive nature of this disease, it would be more advisable to perform stratified analysis by disease-burden years. However, the rarity of PPRD and the briefness of outpatient records limited our analysis within the general patient group.
In eight patients, only a single monoallelic CCN6 variant was found. As the PCR primers we designed were specifically designed for the five exons and intron‒exon junctions, our Sanger sequencing may fail to capture intronic variants and those on the 5′-UTR. Note that PPRD is typically characterized by autosomal recessive inheritance, and the patients showed classic PPRD phenotypes with a pathogenic variant identified on one allele. We speculate the presence of another pathogenic variant on the other allele that is located in the 5′-UTR or intronic regions.
In summary, through a retrospective analysis of genotypes and phenotypes in 105 Chinese PPRD patients, we characterized a unique genotype spectrum and identified nine novel variants and five hotspot variations among Chinese PPRD patients. We proposed genotype–phenotype associations in variants c.725_726del (p.K242Rfs*36), c.624dupA (p.C209Mfs*21), c.667T>G (p.C223G) and null variants on exon 2, for modifying onset age, joint involvement, and atypical deformities. For suspected PPRD patients with typical clinical features, Sanger sequencing, rather than NGS, should be prioritized.
Data availability
Data are available from the corresponding author upon reasonable request.
References
Wynne-Davies R, Hall C, Ansell BM. Spondylo-epiphysial dysplasia tarda with progressive arthropathy. A “new” disorder of autosomal recessive inheritance. J Bone Jt Surg Br. 1982;64:442–5.
Pennica D, Swanson TA, Welsh JW, Roy MA, Lawrence DA, Lee J, et al. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci U S A. 1998;95:14717–22.
Cao L, Xu A, Xu D, Fang M, Zhou E, Gao S, et al. Spondylo-epiphysial dysplasia tarda with progressive arthropathy (report of 7 cases). Zhonghua Fang She Xian Yi Xue Za Zhi. 1986;20:97–9 (in Chinese).
Liu Z, Chen X. Progressive pseudorheumatoid dysplasia: a case series report. Transl Pediatr. 2021;10:1932–9.
Yin L, Mao Y, Zhou Y, Shen Y, Chen H, Zhou W, et al. A retrospective study of nine patients with progressive pseudorheumatoid dysplasia: to explore early diagnosis and further treatment. Clin Rheumatol. 2022;41:877–88.
Uludağ Alkaya D, Kasapçopur Ö, Bursalı A, Adrovic A, Demir B, Aykut A, et al. Specific early signs and long-term follow-up findings of progressive pseudorheumatoid dysplasia (PPRD) in the Turkish cohort. Rheumatology (Oxford). 2022;61:3693–703.
Chen W, Mo S, Luo G, Wang Y, Deng X, Zhu J, et al. Progressive pseudorheumatoid dysplasia with new-found gene mutation of Wntl inducible signaling pathway protein 3. Pediatr Rheumatol Online J. 2018;16:55.
Liu L, Li N, Zhao Z, Li W, Xia W. Novel WISP3 mutations causing spondyloepiphyseal dysplasia tarda with progressive arthropathy in two unrelated Chinese families. Jt Bone Spine. 2015;82:125–8.
Ye J, Zhang HW, Qiu WJ, Han LS, Zhang YF, Gong ZW, et al. Patients with progressive pseudorheumatoid dysplasia: from clinical diagnosis to molecular studies. Mol Med Rep. 2012;5:190–5.
Yue H, Zhang ZL, He JW. Identification of novel mutations in WISP3 gene in two unrelated Chinese families with progressive pseudorheumatoid dysplasia. Bone. 2009;44:547–54.
Yu Y, Hu M, Xing X, Li F, Song Y, Luo Y, et al. Identification of a mutation in the WISP3 gene in three unrelated families with progressive pseudorheumatoid dysplasia. Mol Med Rep. 2015;12:419–25.
Li Y, Zeng Y, Chen Z, Xin H, Li X. Progressive pseudorheumatoid dysplasia confirmed by whole-exon sequencing in a Chinese adult before corrective surgery. J Orthop Surg Res. 2019;14:16.
Hu W, Zhang Z. Identification of WISP3 gene mutation in four families with progressive pseudorheumatoid dysplasia. Chin J Osteoporos Bone Miner Res. 2018;11:224–32 (in Chinese).
Yan W, Dai J, Xu Z, Shi D, Chen D, Xu X, et al. Novel WISP3 mutations causing progressive pseudorheumatoid dysplasia in two Chinese families. Hum Genome Var. 2016;3:16041.
Sun J, Xia W, He S, Zhao Z, Nie M, Li M, et al. Novel and recurrent mutations of WISP3 in two Chinese families with progressive pseudorheumatoid dysplasia. PLoS One. 2012;7:e38643.
Mao Y, Zhou W, Jin Y, Wang J, Shen Y, Zhou Y, et al. Clinical diagnosis of progressive pseudorheumatoid dysplasia in 5 cases and a report of novel WISP3 gene mutation. J Clin Pediatr. 2018;36:537–40.
Ye J, Zhang HW, Wang T, Cao LF, Qiu WJ, Han LS, et al. Clinical diagnosis and WISP3 gene mutation analysis for progressive pseudorheumatoid dysplasia. Zhonghua Er Ke Za Zhi. 2010;48:194–8 (in Chinese).
Hu Q, Liu J, Wang Y, Wang J, Shi H, Sun Y, et al. Delayed-onset of progressive pseudorheumatoid dysplasia in a Chinese adult with a novel compound WISP3 mutation: a case report. BMC Med Genet. 2017;18:149.
Wang Y, Xiao K, Yang Y, Wu Z, Jin J, Qiu G, et al. CCN6 mutation detection in Chinese patients with progressive pseudo-rheumatoid dysplasia and identification of four novel mutations. Mol Genet Genom Med. 2020;8:e1261.
Luo H, Shi C, Mao C, Jiang C, Bao D, Guo J, et al. A novel compound WISP3 mutation in a Chinese family with progressive pseudorheumatoid dysplasia. Gene. 2015;564:35–8.
Zhang Y, Yang C, Feng G, Chen X, Yin P. Spondyloepiphyseal dysplasia tarda with progressive arthropathy: a case report. J Pract Orthop. 2019;25:378–80 (in Chinese).
Yue H, He J, Gu J, Zhang H, Wang C, Ke Y, et al. Identification of a novel mutation in WISP3 gene in a Chinese family with spondyloepiphyseal dysplasia tarda associated with progressive pseudorheumatoid dysplasia. J Diagn Concepts Pract. 2011;10:418–22 (in Chinese).
Liu R, Zhai JY, Liu XY, Yao ZQ. Progressive pseudorheumatoid dysplasia misdiagnosed asankylosing spondylitis: a case report. Beijing Da Xue Xue Bao Yi Xue Ban. 2018;50:1112–6 (in Chinese).
Peng YQ, Liao EY, Gu HM, Wei QY, Zhou H, Li J, et al. Pathology and molecular pathogenesis of spondyloepiphyseal dysplasia tarda with progressive arthropathy caused by compound CCN6 heterogeneous gene mutations. Zhonghua Yi Xue Za Zhi. 2004;84:1796–803 (in Chinese).
Yang X, Song Y, Kong Q. Diagnosis and surgical treatment of progressive pseudorheumatoid dysplasia in an adult with severe spinal disorders and polyarthropathy. Jt Bone Spine. 2013;80:650–2.
Garcia Segarra N, Mittaz L, Campos-Xavier AB, Bartels CF, Tuysuz B, Alanay Y, et al. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): a review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. Am J Med Genet C Semin Med Genet. 2012;160C:217–29.
Torreggiani S, Torcoletti M, Campos-Xavier B, Baldo F, Agostoni C, Superti-Furga A, et al. Progressive pseudorheumatoid dysplasia: a rare childhood disease. Rheumatol Int. 2019;39:441–52.
Acknowledgements
We gratefully thank the patients and their families for their involvement in the study. We thank Jing-Ya Zhou (Department of Medical Records, Peking Union Medical College Hospital) and Lian Ma (Information Center, Peking Union Medical College Hospital) for their generous assistance in the data collection.
Funding
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-003), the National Key R&D Program of China (2021YFC2702001, 2016YFC0901500), and National High Level Hospital Clinical Research Funding (2022-PUMCH-B-079).
Author information
Authors and Affiliations
Contributions
WW contributed to the conceptualization, methodology, investigation, formal analysis, writing of the original draft, reviewing and editing. GSH contributed to data curation, writing of the original draft, reviewing and editing, and visualization. WM, ZLQ, JS, XJ, ZCH, ZJG, ZXF, XWB, and LW managed patients and contributed to the investigation and data curation. QZQ contributed to the management of the patients, investigation, data curation, conceptualization, methodology, resources, funding acquisition, reviewing and editing, supervision of the study, and project administration. SHM contributed to the management of the patients, investigation, data curation, conceptualization, methodology, resources, funding acquisition, reviewing and editing. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This study was approved by the Ethics Committee of Peking Union Medical College Hospital (S-609). Informed consent for this retrospective analysis was waived.
Conflict of interest
All authors have declared no conflict of interest. Author Hong-Mei Song is an Editorial Board member of the World Journal of Pediatrics. The paper was handled by the other Editors and has undergone a rigorous peer review process. Author Hong-Mei Song was not involved in the journal’s review or decisions making of this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, W., Gao, SH., Wei, M. et al. Unique mutation spectrum of progressive pseudorheumatoid dysplasia in the Chinese population: a retrospective genotype–phenotype analysis of 105 patients. World J Pediatr 19, 674–686 (2023). https://doi.org/10.1007/s12519-022-00674-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-022-00674-7