
Abstract
Arrhythmogenic cardiomyopathy, or its most well-known subform arrhythmogenic right ventricular cardiomyopathy (ARVC), is a cardiac disease mainly characterised by a gradual replacement of the myocardial mass by fibrous and fatty tissue, leading to dilatation of the ventricular wall, arrhythmias and progression towards heart failure. ARVC is commonly regarded as a disease of the intercalated disk in which mutations in desmosomal proteins are an important causative factor. Interestingly, the Dutch founder mutation PLN R14Del has been identified to play an additional, and major, role in ARVC patients within the Netherlands. This is remarkable since the phospholamban (PLN) protein plays a leading role in regulation of the sarcoplasmic reticulum calcium load rather than in the establishment of intercellular integrity. In this review we outline the intracellular cardiac calcium dynamics and relate pathophysiological signalling, induced by disturbed calcium handling, with activation of calmodulin dependent kinase II (CaMKII) and calcineurin A (CnA). We postulate a thus far unrecognised role for Ca2+ sensitive signalling proteins in maladaptive remodelling of the macromolecular protein complex that forms the intercalated disk, during pro-arrhythmic remodelling of the heart.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Genetic causes of cardiomyopathy
Over the last decades, multiple studies have identified over 100 genes and mutations involved in various forms of cardiac disease such as hypertrophic, dilated, and arrhythmogenic cardiomyopathy (HCM, DCM, and ACM, respectively) [1]. These genes mainly encode proteins of the contractile machinery, but also proteins that participate in calcium homeostasis and ones that reside in the intercalated disk (ID). As a consequence, mutations in these genes have pathogenic effects on several cardiac cellular functions, such as contractile force generation, force regulation, signal transduction, excitability and intracellular calcium handling. Since the discovery of the first causative mutations for cardiomyopathies, attention for such inherited genetic disorders has greatly increased. To uncover the consequences of these genetic mutations contributes importantly to knowledge of the underlying pathogenic mechanisms and has introduced a regimen of clinical genetic evaluation of affected individuals and their relatives. Early identification of asymptomatic mutation carriers, via genetic screening, is of major prognostic importance since cardiomyopathies, with an emphasis on ACM, can lead to sudden cardiac death as a result of malignant ventricular arrhythmias even before the onset of any symptoms.
Interestingly, there are multiple genes in which genetic aberrancies cause different types of cardiomyopathies and even opposite functional effects are present. For example, mutations in proteins involved in calcium handling, such as phospholamban (PLN), which mostly correlate to DCM also manifest in the setting of ACM [2]. On the other hand, mutations in the gene encoding the desmosomal protein plakophilin2 (PKP2) are classically correlated with ARVC (a subform of ACM), [3] but can additionally be identified in DCM patients [4]. Likewise, mutations in PKP2 are also present in patients suffering from Brugada syndrome, a disease commonly related to mutations in multiple ion channels instead of desmosomal proteins [5]. These studies suggest that distinct cardiac proteins should not be considered to be completely autonomous working entities, but rather that they function in different macromolecular protein complexes. In this review we will focus specifically on gene mutations that relate to ACM and the potential mechanisms that drive proarrhythmic cardiac remodelling in this disease. For a subset of these mutations, particularly those involving PLN, we postulate a potential role for calcium sensitive signalling pathways.
Pathophysiological remodelling in arrhythmogenic cardiomyopathy
During cardiomyopathy, not exclusively limited to ARVC, ventricular arrhythmias can arise out of electrical and structural cardiac remodelling, which disrupts cell-cell communication, excitability and the pump function of the heart [6]. Electrical remodelling is based on abnormal expression, distribution and regulation of ion channels and gap junctions, and a heterogeneous expansion of the extracellular matrix (fibrosis formation) which results in a disturbed electrical cell-cell signalling between cardiomyocytes or between cardiomyocytes and (myo)fibroblasts [6, 7]. Increased pathophysiological intracellular calcium concentrations, caused by dysfunction of calcium regulatory proteins, negatively affects the contraction and relaxation dynamics of the heart (reviewed by ‘Qu et al. 2015’) [6]. Structural cardiac remodelling involves myocyte death, fibre disorders, changes in cell size (hypertrophy), and collagen deposition (fibrosis). Structural remodelling disturbs the impulse propagation between cardiomyocytes and hampers pump function of the entire heart [6].
ARVC, the most familiar form of ACM, is a cardiac disease mainly characterised by a gradual replacement of the myocardial mass by fibrous and fatty tissue, which causes dilatation of the ventricular wall and progression towards heart failure. As such, this is classically defined as structural remodelling, but it also includes a disruption of proper electrical signalling, with a high susceptibility for ventricular arrhythmias. As described in several studies, remodelling also results in a combination of reduced sodium channel (Nav1.5), gap junction channels (constituted of connexin43, Cx43) and plakoglobin localisation at the ID of patients [8]. ARVC is a familial disease with an estimated frequency of 1:5000 people all over the world, though in some regions prevalence is more pronounced [9]. In about 60% of the ARVC cases, a mutation is the underlying cause of the disease [2]. Mutations in genes encoding the desmosomal proteins PKP2, plakoglobin (gene name JUP), desmoplakin (DSP), desmocollin-2 (DSC2) and desmoglein-2 (DSG2), are highly related to development of ARVC [1]. Those mutations are predominantly found in the PKP-2 gene, but have also been discovered in genes encoding non-desmosomal proteins such as ryanodine receptor2 (RYR2), transforming growth factor β3 (TGFβ3), lamin A/C (LMNA), transmembrane protein 43 (TMEM43), desmin (DES), titin (TTN), αT-catenin (CTNNA3) and PLN [1].
The fact that mutations in genes encoding desmosomal and non-desmosomal proteins commonly lead to a comparable clinical phenotype suggests that more than one underlying mechanism could be involved in the process of cardiac remodelling in ARVC patients. This is strengthened by the identification of a role for mutations in the sarcoplasmic reticulum (SR) associated proteins PLN and RYR2, and the fact that in the other 40% of patients diagnosed with ARVC, no causative mutations, or mutations not yet linked to ARVC, have been detected thus far [1]. PLN is a transmembrane SR phosphoprotein and a key regulator of calcium homeostasis. As expected, mutations in PLN mostly lead to inhibition of the calcium re-uptake into the SR [10]. Interestingly, next to the classical desmosomal mutations that trigger instability of the ID (potentially due to trafficking defects), the pathogenic PLN-R14Del mutation is identified in patients diagnosed with ARVC. In such patients, a differential diagnosis of DCM or ARVC can be made [2]. In the Netherlands, the PLN-R14Del mutation is a founder mutation and has been identified in 10–15% of patients diagnosed with either DCM or ARVC [11]. This similarity in pathophysiological remodelling driven by dysfunction/mislocalisation of proteins in the ID and proteins related to the intracellular calcium handling may perhaps not be as long distance as it seems at first glance. Present studies provide an interesting new view into the possible interaction between structures classically defined as separate (ID and calcium homeostasis). Next to the well-known classical model, based on malfunctioning/mislocalised desmosomal proteins, also involvement of alternative mechanisms underlying ARVC phenotype should be mentioned. The canonical Wnt/β-catenin signalling pathway and hippo pathway illustrate pathways which are affected by ID proteins under pathogenic conditions, ending in recapitulation of the ARVC phenotype [12, 13]. In this regard, the common observation that plakoglobin signals are reduced or even absent at the ID is important [8, 14], while glycogen synthase kinase 3β (GSK3β) seems to accumulate at the ID during pathological remodelling [15].
Cross talk within the intercalated disk
To withstand the strong and frequent mechanical forces of the beating heart, individual cardiomyocytes tightly connect to each other via a subcellular domain named the ID [14]. IDs are of major physiological importance as they not only physically connect cardiomyocytes by means of desmosomes and adherence junctions, but also couple cardiomyocytes electrically to ensure fast propagation of electrical signals, through gap junctions [14]. Gap junctions are agglomerates of multiple intercellular channels that consist of two connexons (hemi channels), which are engaging structures composed of hexametric clusters of connexin proteins, delivered by cells on both sides of each junction. In the working myocardium, these channels are predominantly composed of connexin40 (Cx40, atria) and Cx43 (atria and ventricles). Electrical coupling depends on the organisation of gap junctions at the ID, whose abundance, resistivity and distribution defines the characteristics of electrical impulse propagation between cardiomyocytes. The classical outlook of the arrangement at the ID with its separated structures such as desmosomes, gap junctions, ion channels and connexons, is currently changing. Knowledge about the relation between different structures of the ID is expanding in detail and it is becoming more and more clear that the ID in fact is a complex and sophisticated protein network.
Accumulating experimental evidence supports the idea that the ID should be considered to be a singular arranged functional entity in which macromolecular complexes mechanically and electrically interact in synchrony [16]. This is exampled by the fact that the loss of desmosomal integrity, caused by PKP2 dysfunction, leads to a reduced sodium current (I Na), aiding an interaction between the desmosome and sodium channel complex [5]. This interaction could partly be related to disturbed trafficking of proteins to the ID. A study by Sato et al. presented colocalisation of Nav1.5 (the alpha-subunit of the sodium channel) with PKP2 [17], whereas a very recent study of the same group identified colocalisation of these sodium channels with N‑cadherin, a protein of the adherence junction [16]. When PKP2 expression was decreased through RNA silencing in neonatal rat ventricular myocytes, this also resulted in a decrease of the total Cx43 content and a significant redistribution of Cx43 to the intracellular space. Multiple experiments showed that Cx43 and PKP2 coexist in the same macromolecular complex. Besides this, ankyrin-G (encoded by ANK3) shows to be a key component of linkage within the ID as it is related to three complexes often considered independent: the voltage-gated sodium channel, gap junctions, and the cardiac desmosome [17, 18]. The intermolecular interactions between proteins relevant to mechanical junctions, and those that constitute gap junctions/sodium channels embeds important implications for the pathophysiology of inherited arrhythmias, such as in ACM [17]. This specifically accounts for patients in which the ACM is caused by mutations in desmosomal proteins. However, it is more complex to understand why patients show a similar clinical phenotype when causative mutations are present in non-desmosomal proteins such as PLN, a protein heavily involved in calcium homeostasis. Certainly if we bear in mind that also in these patients pro-arrhythmic remodelling of the ID is reported [11, 19].
Calcium dynamics and pathophysiological signalling
PLN plays a leading role in cardiac excitation contraction coupling which relies on tightly regulated calcium fluxes that are triggered when an action potential enters from adjacent cells. Voltage gated L‑type calcium channels (LTCC) on the cell membrane of striated muscle cells open and calcium enters the cell. This influx of calcium into the cell subsequently triggers opening of RYR2, leading to a coordinated efflux of calcium out of the SR. Local calcium peaks result in binding of calcium ions to the troponin complex, which consists of three regulatory proteins that form a contractile complex. If calcium ions unbind from troponin the myofilaments are able to relax again. Calcium is removed out of the cardiomyocytes by Na+/Ca2+ exchange channels (NCX) at the cell membrane or stored back in the SR by sarco/endoplasmic reticulum Ca2+-ATPase channels (SERCA, reviewed by ‘Luo et al., 2013’) [20]. The open and closed state of gap junction/ion channels and channels involved in calcium handling is tightly controlled by phosphorylation and dephosphorylation, through kinases and phosphatases. Under physiological conditions their activation is mainly regulated by protein kinase A (PKA), protein kinase C (PKC), and mitogen-activated protein kinase signalling. Downstream activity of protein kinases such as PKA and PKC regulates Ca2+ homeostasis by phosphorylation of Ca2+ channels, such as LTCC, SERCA, and RYR2. This regulation is important to adapt to changing demands as, for example, an elevated cardiac output. Dephosphorylated PLN interacts with SERCA and inhibits Ca2+ pump activity into the SR [10]. By phosphorylation of PLN upon β‑adrenergic stimulation and enhanced cyclic AMP-dependent protein kinase A (PKA) activity, the inhibitory effect of PLN on SERCA is relieved. This leads to increased initial rates of SR Ca2+ uptake, accelerated relaxation, enhanced SR Ca2+ load and increased Ca2+ release by RYR2 during systole [21].
Under pathophysiological conditions, functional control of cellular excitability and excitation-contraction coupling shifts from kinases such as PKA and PKC towards calmodulin dependent kinase II (CaMKII) and calcineurin A (CnA) [22, 23]. Under pathological conditions the diastolic calcium levels can become persistently higher, which in the end compromises pump function through insufficient relaxation and a reduced force of contraction due to lower calcium availability during systole [20]. Diastolic calcium concentrations do not solely rely on LTCC and SR Ca2+ regulation, since alterations in the kinetics of sodium channels (triggering late sodium current, I Na-L), NCX and PLN activity are also of importance [10, 24]. An increase in the amplitude of I Na-L prolongs the action potential duration, increases the transmural dispersion of repolarisation which triggers arrhythmogenesis [25]. An increase in I Na-L also increases the intracellular sodium concentration and subsequently raises [Ca2+]i via the reverse-mode NCX [26].
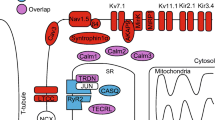
Besides affecting kinases and phosphatases such as CaMKII and CnA, the intracellular calcium concentration is also capable of affecting the ID structure directly. Although it is known that elevating intracellular calcium conditions from very low towards the physiological state causes targeting and maturation of desmosomes from the endoplasmic reticulum to the cellular membrane [27], currently it is still unknown whether such influences can also be triggered by a persistently elevated diastolic calcium level above the physiological concentration. Importantly, the signal transduction pathways induced under persistently elevated calcium levels further trigger cardiac remodelling and electrical and contractile modifications [24]. In the following we propose a link between increased diastolic Ca2+ concentrations, activation of CaMKII and CnA, and electrical and structural cardiac remodelling in the setting of ACM (Fig. 1).

Overview illustrating the pro-arrhythmic interactions between Ca2+-sensitive proteins, intercalated disk proteins and ion channels. SR sarcoplasmatic reticulum, RyR ryanodine receptor, SERCA SR Ca2+ ATPase, PLN phospholamban, LTCC L-type Ca2+ channel, NaL late sodium current, NCX Na+/Ca2+ exchanger, NKA Na+/K+-ATPase, AnkG ankyrin-G, DSC2/DSG2 desmocolin-2/desmoglein-2, PKP2 plakophilin-2, Cx43 connexin43, N-Cad N-Cadherin, CaMKII Ca2+/calmodulin-dependent protein kinase II, CaM calmodulin, CnA calcineurin A, EB-1 end-binding protein 1, NFAT nuclear factor of activated T‑cells, CACNA1D voltage-dependent L‑type calcium channel subunit alpha-1D, ATP2A2 sarcoplasmic/endoplasmic reticulum calcium ATPase 2, SCN5A sodium channel protein type 5 subunit alpha, Col1A1/1A2/3A1 alpha-1 type I/1 type 2/3 type 1 collagen, TGFβ1 transforming growth factor beta 1, CTGF connective tissue growth factor, TIMP1 metallopeptidase inhibitor 1
Phospholamban dysfunction
Deregulated intracellular Ca2+ handling in cardiomyocytes is frequently attributed to impaired SR functioning, leading to heart failure, in which dysfunction of the different key proteins involved can each be causative. Dysfunction of PLN has been reported to play a key role in cardiac pathophysiology and up to now several different mutations have been identified in patients [21]. Characterisation of those PLN mutations generally revealed disruption of intracellular calcium handling, precipitating clinical manifestations of ACM, DCM and heart failure (see Table 1).
Of special attention in this review is the PLN-R14Del mutation since this mutation is known to trigger development of ACM in patients [19]. PLN is a phosphoprotein which, when dephosphorylated, inhibits the Ca2+ affinity of SERCA. PLN phosphorylation can take place at the Ser16 by PKA (during sympathetic stimulation) or at Thr17 by CaMKII (predominantly during pathophysiological conditions), which reverse the inhibitory effect on SERCA [28]. The PLN-R14Del mutation is associated with the deletion of the highly conserved basic amino acid Arg-14 in the coding region. As a result, phosphorylation is still possible but the inhibitory effect is no longer relieved. Coexpression of wild-type PLN and mutant PLN-R14Del, in human embryonic kidney (HEK) cells confirmed the super inhibition of the SERCA channel [10]. This super inhibitory effect is likely caused by partial destabilisation of the PLN pentamer, leading to production of highly inhibitory monomers and a persistent PLN-SERCA association. The dominant inhibitory effect of this mutant was not alleviated by phosphorylation through PKA, leading to ventricular dilatation, contractile dysfunction and ventricular arrhythmias. Transgenic mice, overexpressing the PLN-R14Del protein, recapitulated the human cardiomyopathy and resulted in premature death. Next to the disturbance in Ca2+ transients, PLN-R14Del patients also show classical ARVC-like features as fibrofatty replacement and myocardial disarrangement [10]. A study in endomyocardial samples taken from PLN-R14Del patients showed depressed or absent plakoglobin levels indicative of remodelling of the ID in 71% of the cases [11].
In addition to abnormalities in Ca2+ transients and electrical instability, in cardiomyocytes derived from human induced pluripotent stem (iPS) cells that were generated from patients bearing the PLN-R14Del mutation, an abnormal distribution of the mutated protein towards the cytoplasm was reported [29]. Knocking down the mutated PLN gene and simultaneous overexpression of a normal PLN gene (genetic repair) appeared able to reverse this pathological phenotype in vitro. In a different experimental mouse model, the mutation was introduced into PLN null mice, to generate a homozygous PLN-R14Del knockout model [21]. Comparable with the situation in PLN-L39stop mutants, colocalisation of PLN with SERCA was lost and no inhibition on SR Ca2+ transport and contractility in these PLN-R14Del mice was present. In contrast, PLN-R14Del mutated protein misrouted towards the sarcolemmal Na+/K+-ATPase (NKA) in the plasma membrane and stimulated its activity. Eventually these PLN-R14Del mice showed cardiac remodelling in terms of ventricular dilation and interstitial fibrosis [21].
The missense mutation at PLN residue 9 (PLN-R9C) has been reported to cause inherited DCM and heart failure in patients [30]. Studies in transgenic PLN-R9C mice showed that the mutated protein did not impair Ca2+ transients by direct inhibition of SERCA, rather than it depended on trapping of PKA by the mutant protein that resulted in a blocked PKA-mediated phosphorylation of wild-type PLN. This finally delayed intracellular calcium transients through super inhibition of SERCA [30]. A different T116G point mutation resulting in PLN-Leu-39-stop (PLN-L39stop) was also discovered in familial cases of heart failure [31]. Whereas the heterozygous mutation caused cardiac hypertrophy, homozygous patients developed lethal DCM and heart failure. Homozygous hearts showed a 50% reduction of PLN mRNA while PLN protein diminished to a negligible level suggesting that disturbed calcium handling could not be causative for the disease. Surprisingly, homozygous patients demonstrated extensive fibrofatty replacement and myocardial disarrangements. In vitro, adenoviral transfection of recombinant PLN-L39stop in adult rat cardiomyocytes indeed confirmed the absence of a maladaptive effect on SERCA and contractility. Viral transfection of PLN-L39stop in HEK cells revealed destabilised PLN expression and redirected minor amounts of PLN towards the cytosol or plasma membranes. This study emphasised a clear contrast between the disease phenotypes in human and rodents since ablation of PLN in mice seemed to be beneficial in the setting of heart failure, whereas human carriers of PLN-L39stop develop severe and lethal cardiomyopathies [31].
Very recently it was shown that overexpression of PLN-R25C in adult rat cardiomyocytes extremely suppressed the affinity of SERCA for Ca2+, resulting in a decreased SR Ca2+ concentration and Ca2+ transients [32]. Interestingly, this super inhibition of SERCA caused elevated diastolic Ca2+ concentrations and CaMKII activation. CaMKII activation subsequently enhanced RYR2 phosphorylation and leakage of Ca2+ out of the SR, ending in an impaired cardiac contractility and ventricular arrhythmias. To further stress the pivotal role for CaMKII in this pathophysiological remodelling, KN93 inhibition of CaMKII abolished the PLN-R25C triggered Ca2+ sparks [32].
In general, dysfunctional PLN highly disrupts the cardiac intracellular calcium handling, finally ending in hypertrophy, DCM, ventricular arrhythmias, heart failure and premature death. Mainly SERCA, RYR2 and the SR load are affected, leading to increased diastolic calcium concentrations and decreased Ca2+ transients. In spite of the disturbed calcium homeostasis a subgroup of PLN-R14Del patients present an ARVC-like phenotype, resembled by fibro-fatty replacement and ID disarrangement. Comparable alterations were also reported in patients with a different PLN mutation, such as PLN-L39stop. The most recent study also presented an increased CaMKII activation by PLN-R25C. Several studies in cardiomyocytes have already proven that increased diastolic Ca2+ concentrations cause CaMKII auto-phosphorylation and CaMKII activation [22, 33, 34]. Importantly, next to CaMKII, also the Ca2+/CaM activated phosphatase CnA is sensitive to increased diastolic calcium concentrations [23]. In that regard it is of interest to uncover if a pathophysiological activation of CaMKII and CnA in ACM patients potentially contributes to the adverse remodelling in ACM. Because CaMKII and CnA play a key role in electrical and structural remodelling of the heart, as postulated in the following paragraphs, it is not unlikely that also in this particular disease they modulate aspects of electrical signalling, excitation-contraction coupling and tissue architecture.
CaMKII in cardiomyopathy
CaMKII and CnA are routinely present in the heart, but transcription, expression and activation of these proteins is enhanced during cardiomyopathy [35]. This phenomenon suggests a pathophysiological role for these proteins in the development of heart failure. Persistent activation of beta-adrenergic signalling pathways will activate CaMKII, but most important, phosphorylation of Ca2+ channels increases the diastolic intracellular Ca2+ concentration, which facilitates permanent activation of CaMKII via calmodulin (CaM). Moreover, CaMKII is able to maintain its activated state independent of calcium-activated CaM via autophosphorylation and via the influence of reactive oxygen species (reviewed by ‘Heineke et al. 2006’) [36]. Amongst many others, activated CaMKII phosphorylates cardiac proteins that increase inflammatory signalling and apoptosis [37]. A study in pneumocytes showed that activated CaMKII drives apoptosis which can lead to pulmonary fibrosis [38]. Chronic pressure overload in a murine knockout model of CaMKII resulted in a tempered increase in ventricular wall thickness and no significant levels of fibrosis [22]. Illustrative for its well-studied and maladaptive role in disturbance of calcium handling, a study in patients with heart failure suggested that pathophysiologically enhanced CaMKII levels in cardiomyocytes increased SR Ca2+ leak, which consequently depleted the SR as Ca2+ store, and decreased contractility [33]. A link of activated CaMKII to fatty replacement, one of the striking hallmarks of remodelling in ARVC hearts, currently is lacking.
CaMKII remodelling of ID proteins
Beyond its detrimental role in SR calcium handling, CaMKII can also influence electrical signalling between cardiomyocytes at the level of the ID, by phosphorylation of ion channels such as the sodium channel. The CaMKII-δ isoform stably interacts with two looping domains of the main cardiac voltage-dependent sodium channel (constituted from Nav1.5 proteins) and facilitates phosphorylation at the Ser-516, Ser571 and Thr-594 site [39]. The pathological regulation of cardiac sodium channels by CaMKII is not well studied in human disease, but animal models of triggered heart failure presented independent upregulation of CaMKII levels and enhanced I Na during cardiomyopathy [34]. In dogs with chronic heart failure the I Na-L and total sodium influx levels where enhanced by Ca2+/CaMKII signalling under physiological and pathophysiological conditions, which was manifested by slowing down of the inactivation kinetics of the channel [34]. In this model, the excess of sodium entry was counteracted by activation of the NCX, which in turn even further enhanced the already elevated diastolic calcium level. A recent study of Glynn et al. in mice revealed that phosphorylation of Nav1.5 at Ser571 regulates the increase in I Na-L, but not other channel properties previously linked to CaMKII [39]. Inhibition of CaMKII in mice resulted in down regulation of I Na, which negatively affected the sodium channels ability to depolarise the cell whereas phosphorylation of sodium channels in rabbit cardiomyocytes delayed the I Na recovery after inactivation and enhance the I Na-L [40]. The observations made in these different models indicate that enhanced activation of CaMKII increases the depolarising current, elongating the action potential and thereby increases the susceptibility to arrhythmias.
Conditional knockout of ankyrin-G in a mouse model showed that ankyrin-G targets Nav1.5 proteins and its regulatory protein CaMKII to the ID for which βIV-spectrin is necessary [18, 41]. Knockout of ankyrin-G resulted in a decreased Nav1.5 expression and membrane trafficking, and a concomitantly decreased I Na and I Na-L, highlighting the dependence of the sodium channel on other protein partners in the ID that serve as a regulatory platform [18, 42]. Moreover, ankyrin-G links sodium channels with broader ID signalling nodes, as loss of ankyrin-G also resulted in reorganisation of PKP2 and lethal arrhythmias in response to β‑adrenergic stimulation [18]. A study by Xu et al. clarified that enhanced cellular calcium entry totally diminished the open probability of Cx43 gap junction channels which suggests that gating was affected by a Ca2+/CaM-dependent mechanism [43]. These results reveal that CaM already links towards gap junctions by itself and additionally suggest a relation between CaMKII and Cx43. This was recently confirmed in an in vitro study with cultured cardiomyocytes, where one novel serine residue in the Cx43 carboxyl terminus was identified that can be phosphorylated by CaMKII, which suggests that CaMKII may phosphorylate, and thereby regulates the activity of Cx43 channels in normal and diseased hearts [44].
Role for calcineurin in fibrosis formation and conductional remodelling
Calcineurin is a serine-threonine dimeric phosphatase, consisting of the catalytic subunit CnA and the regulatory subunit calcineurin B (CnB). Next to the above described regulation of CaMKII, Ca2+ saturated CaM directly binds and activates CnA. Coherent to the fact that CaM needs to be activated by Ca2+, the amount of CaM needed for activation of CnA decreases by increasing intracellular calcium concentrations. Binding and dephosphorylation of nuclear factor of activated T‑cells (NFAT) transcription factors by CnA in the cytoplasm allows translocation of NFAT into the nucleus, which induces gene expression of adverse remodelling genes. Other kinases such as JNK, p38, GSK3β and PKA phosphorylate specific NFAT family members and block the nuclear translocation. (Reviewed by ‘Heineke et al. 2006’) [36].
Activity of CnA was significantly increased in the compensated and decompensated hypertrophic myocardium of patients with coronary artery disease, and it is suggested to play a major role in fibrosis formation [45]. Transgenic mice with a constitutively active form of CnA present in the myocardium display tremendous deposits of collagen in the myocardium and show trans-differentiation of ventricular fibroblasts into myofibroblasts, a phenotype known for its high profibrotic activity [23]. Mice overexpressing a constitutively active form of CnA presented pathological remodelling and impaired Cx43 protein levels. Fontes et al. studied the postnatal development of mice overexpressing this constitutively active form of CnA [46]. After one week these mice develop cardiac hypertrophy with reduced protein and RNA levels of Cx43 and Nav1.5. After three weeks the protein levels of Cx43 were still reduced and the connexin proteins were less phosphorylated, potentially as a result of dephosphorylation by CnA. Four weeks after birth these hearts display substantial levels of fibrosis, so increased levels of active CnA coincide with reduced expression of the sodium channel Nav1.5, Cx43 and eventually end in cardiac hypertrophy and fibrosis formation [46]. In addition, a different transgenic mouse model harbouring an inducible constitutively active CnA gene confirmed the presented adverse cardiac remodelling, but also indicated reversibility upon block of CnA [47]. CnA inhibition in a murine CaMKII knockout model confirmed the main role of CnA in cardiac hypertrophy, associated with fibrosis formation, apoptosis and systolic dysfunctions.
Conclusion and future perspective
ACM, or its most prominent subform ARVC, is commonly regarded as a disease of the ID in which mutations in desmosomal proteins are an important causative factor. Originally described as a composition of separate units, recent data indicate that the ID should be considered a single functional unit in which Nav1.5, Cx43, ankyrin-G and components of the adherence junction and desmosome (including PKP2 and plakoglobin) interact. As such, accumulating evidence in the last decade, suggests that destabilisation of the ID (through disturbed protein-protein interactions) and trafficking problems of its composing proteins trigger the remodelling. The latter aspect was recently illustrated through deficits in EB-1 ID protein trafficking causing a disturbed ID morphology resulting from a disrupted connection between the tubulin network (which directs the new proteins to the ID) and the existing macromolecular complex at the ID [48]. Other contributing mechanisms have recently also acquired more attention as detrimental roles are anticipated for intracellular accumulation of misfolded or mislocalised protein aggregates, and alterations in the hippo/Wnt signalling pathways. For the latter the specific contribution of GSK3β that accumulates at the ID, and plakoglobin that dispatches from the ID are topics of investigation (Fig. 2).

Figure illustrating five pathways putatively causing pathological remodelling in ACM: 1) Deficits in EB-1 ID protein trafficking provoke disturbance of the ID morphology. 2) Pathological activation of the hippo pathway and suppression of Wnt/β-catenin signalling, caused by molecular remodelling of ID proteins, Plakoglobin nuclear translocation and GSK3β-ID anchoring, recapitulates the ARVC phenotype. 3) Remodelling of ID proteins by Ca2+-activated CaMKII. 4) Possible direct effect of increased intracellular Ca2+ concentrations on the ID structure. 5) Intracellular accumulation and degradation of misfolded PLN proteins in PLN-R14Del mutation carriers. GSK3β glycogen synthase kinase 3 beta
The Dutch founder mutation PLN R14Del has been identified to play a major role in ACM (at least in the Netherlands) which seems, as explained, remarkable since PLN is an important protein involved in regulation of SR calcium load. Despite of that, patients with this mutation show, beyond clinical signs that fit with ARVC, also remodelling of proteins in the ID and fibrofatty replacement. The mutation results in dysfunction of PLN and thereby accumulation of diastolic calcium, which in turn would be able to activate Ca2+ sensitive proteins as CaMKII and CnA. Patients with a different PLN mutation, PLN-R25C, indeed show activation of CaMKII which in the past has been linked to DCM, heart failure and disturbed calcium handling. As has been reported for patients with desmosomal mutations, dysfunction of the cardiac sodium channels is a rather recent observation. Although the experimental proof is still lacking, if this dysfunction results in enhanced I Na-L, also under these conditions enhanced diastolic calcium levels can be expected and as such activation of the described Ca2+-sensitive pathways. Activated CaMKII likely modifies ID proteins, SR structures and ion channels in a direct fashion, where CnA rectifies their transcriptional regulation and stimulates formation of fibrosis. In addition, enhanced cytosolic Ca2+ concentrations can induce autophagy by activating CaMKII, a manifestation also seen in PLN-R14Del patients [49]. Obviously, further experimental investigation is needed to unravel the exact role of CaMKII and CnA in ACM.
Step by step, important accomplishments have been made in our knowledge and understanding. Progression, however, is sometimes compromised by the fact that the applied experimental models do not always completely recapitulate the situation seen in patients. Obviously patient material is extremely scarce and often reflects the situation in the end stage only. Examples of the hurdles that adhere to experimental models are the fact that calcium handling in mouse cardiomyocytes, and in cardiomyocytes derived from iPS cells, is significantly different from that in humans [50]. Also the important aspect of fibro-fatty replacement, one of the hallmarks of the disease, is often only partially recapitulated in mouse models (mostly only the deposition of fibrosis). Ongoing improvements in experimental approaches and implementation of new techniques (e. g. focusing on genetic repair and application of super-resolution microscopy) provide, however, enough fuel and hope for a fruitful progression of our aim to control this cardiac disease.
References
Van Tintelen JP, Pieper PG, Van Spaendonck-Zwarts KY, et al. Pregnancy, cardiomyopathies, and genetics. Cardiovasc Res. 2014;101:571–8.
Groeneweg JA, Bhonsale A, James CA, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015;8:437–46.
van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–8.
Cuenca S, Ruiz-Cano MJ, Gimeno-Blanes JR, et al. Genetic Basis of Familial Dilated Cardiomyopathy Undergoing Heart Transplantation. J Heart Lung Transplant. 2016;35:625–35.
Cerrone M, Noorman M, Lin X, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012;95:460–8.
Qu Z, Weiss JN. Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol. 2015;77:29–55.
de Jong S, van Veen TAB, van Rijen HVM, et al. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011;57:630–8.
Noorman M, Hakim S, Kessler E, et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–9.
Basso C, Bauce B, Corrado D, et al. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012;9:223–33.
Haghighi K, Kolokathis F. Gramolini, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci. U S A. 2006;103:1388–93.
van der Zwaag PA, van Rijsingen IAW, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–207.
Chen SN, Gurha P, Lombardi R, et al. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. 2014;114:454–68.
Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–21.
Noorman M, van der Heyden MAG, van Veen TAB, et al. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23–31.
Chelko SP, Asimaki A, Andersen P, et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;pii: e85923
Leo-Macias A, Agullo-Pascual E, Sanchez-Alonso JL, et al. Erratum: Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat Commun. 2016;7:10919.
Sato PY, Coombs W, Lin X, et al. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201.
Makara MA, Curran J, Little SC, et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. 2014;115:929–38.
Groeneweg JA, van der Zwaag PA, Jongbloed JDH, et al. Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Heart Rhythm. 2013;10:548–59.
Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 2013;113:690–708.
Haghighi K, Pritchard T, Bossuyt J, et al. The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase. J Mol Cell Cardiol. 2012;52:773–82.
Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009;106:2342–7.
Molkentin JD, Lu JR, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–28.
Luo A, Cao Z, Xiang Y, et al. Ketamine attenuates the Na+-dependent Ca2+ overload in rabbit ventricular myocytes in vitro by inhibiting late Na+ and L‑type Ca2+ currents. Acta Pharmacol Sin. 2015;36:1327–36.
Wang H‑G, Zhu W, Kanter RJ, et al. A novel NaV1.5 voltage sensor mutation associated with severe atrial and ventricular arrhythmias. J Mol Cell Cardiol. 2016;92:52–62.
Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92(Suppl 4):iv1–5.
Rötzer V, Hartlieb E, Vielmuth F, et al. E‑cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell Mol Life Sci. 2015;72:4885–97.
Mattiazzi A, Kranias EG. The role of CaMKII regulation of phospholamban activity in heart disease. Front Pharmacol. 2014;5:5.
Karakikes I, Stillitano F, Nonnenmacher M, et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun. 2015;6:6955.
Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a. Mutat Phospholamban Sci. 2003;299:1410–3.
Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–76.
Liu G‑S, Morales A, Vafiadaki E, et al. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res. 2015;107:164–74.
Sossalla S, Fluschnik N, Schotola H, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–61.
Maltsev VA, Reznikov V, Undrovinas NA, et al. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–608.
Zhang R, Khoo MSC, Wu Y, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17.
Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600.
Singh MV, Kapoun A, Higgins L, et al. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest. 2009;119:986–96.
Winters CJ, Koval O, Murthy S, et al. CaMKII inhibition in type II pneumocytes protects from bleomycin-induced pulmonary fibrosis by preventing Ca2+-dependent apoptosis. Am J Physiol Lung Cell Mol Physiol. 2016;310:L86–94.
Glynn P, Musa H, Wu X, et al. Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function In Vivo. Circulation. 2015;132:567–77.
Wagner S, Dybkova N, Rasenack ECL, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–38.
Hund TJ, Koval OM, Li J, et al. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–19.
Mohler PJ, Rivolta I, Napolitano C, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–8.
Xu Q, Kopp RF, Chen Y, et al. Gating of connexin 43 gap junctions by a cytoplasmic loop calmodulin binding domain. Am J Physiol Cell Physiol. 2012;302:C1548–56.
Huang RY-C, Laing JG, Kanter EM, et al. Identification of CaMKII phosphorylation sites in Connexin43 by high-resolution mass spectrometry. J Proteome Res. 2011;10:1098–109.
Diedrichs H, Hagemeister J, Chi M, et al. Activation of the calcineurin/NFAT signalling cascade starts early in human hypertrophic myocardium. J Int Med Res. 2007;35:803–18.
Fontes MSC, Raaijmakers AJA, van Doorn T, et al. Changes in Cx43 and NaV1.5 expression precede the occurrence of substantial fibrosis in calcineurin-induced murine cardiac hypertrophy. PLOS ONE. 2014;9:e87226.
Berry JM, Le V, Rotter D, et al. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ Res. 2011;109:407–17.
Agullo-Pascual E, Lin X, Leo-Macias A, et al. Super-resolution imaging reveals that loss of the C‑terminus of connexin43 limits microtubule plus-end capture and NaV1.5 localization at the intercalated disc. Cardiovasc Res. 2014;104:371–81.
Te Rijdt WP, van Tintelen JP, Vink A, et al. Phospholamban p.Arg14del cardiomyopathy is characterized by phospholamban aggregates, aggresomes and autophagic degradation. Histopathology. 2016;69:542–50.
Sabir IN, Killeen MJ, Grace AA, et al. Ventricular arrhythmogenesis: insights from murine models. Prog Biophys Mol Biol. 2008;98:208–18.
Acknowledgements
We acknowledge the support from the Netherlands CardioVascular Research Initiative: the Netherlands Heart Foundation, Netherlands Federation of University Medical Centers, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences (CVON2012-10-PREDICT, to TABvV).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C.J.M. van Opbergen, M. Delmar, T.A.B van Veen declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van Opbergen, C.J.M., Delmar, M. & van Veen, T.A.B. Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: focus on calcium sensitive pathways. Neth Heart J 25, 157–169 (2017). https://doi.org/10.1007/s12471-017-0946-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-017-0946-7