
Abstract
Calcific aortic valve disease (CAVD) results in aortic valve stenosis and is one of the most common cardiac diseases in both Western and developing countries. The burden of this disease is expected to increase rapidly in the future, but there are still no relevant pharmacological therapies available and aortic valve replacement remains the sole definite therapy. This review presents an overview of the most common causes of CAVD, followed by current debates and trials related to the onset and progression of this disease. Several differences and similarities between the different causes of CAVD are presented. Additionally, stages of CAVD are compared with stages in atherosclerosis. Finally, future directions for research on CAVD will be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Calcific aortic valve disease (CAVD) is the most frequent native valve disease in Europe (43.1%) followed by mitral regurgitation (31.5%), aortic regurgitation (13.3%) and mitral stenosis (12.1%) [1]. Aortic valve stenosis (AS) is thought to represent the late stage of the pathological process of CAVD, following aortic valve sclerosis, i.e. thickening of the aortic valve cusp without obstruction of the left ventricular outflow (Fig. 1). Sclerotic and stenotic aortic valves are characterised by a chronic inflammatory cell infiltrate, which consists mostly of macrophages and T-lymphocytes, accumulation of lipids, thickening and fibrosis and eventual mineralisation [2]. The prevalence of aortic valve sclerosis is 29% in the overall population, and up to 37% in those older than 75 years [3]. Estimates of patients in whom sclerosis develops into AS ranges from 15–30% within 6 to 8 years [4]. Approximately 2–3% of the population of 65 years and older have been estimated to have AS [3]. Life expectancy in patients with AS is severely reduced, as indicated by Otto et al., who found that the probability to be alive after two years for asymptomatic patients with a peak jet velocity of > 4 m/s and without aortic valve replacement was only 21 ± 18% [5].

Echographic images showing the aortic valve in different stages of the disease: normal (a), sclerotic (b, early stage, low transvalvular gradient) and stenotic (c, end stage). All images were obtained using short-axis TEE in the same angle. NC: Non-coronary cusp, L: Left coronary cusp, R: Right coronary cusp. Arrow marks improper closing of the severely stenotic valve causing a valvular leak
Grossly, the most common aetiologies for CAVD are degenerative, rheumatic and congenital (81.9%, 11.2% and 5.4% of the patients respectively) [1]. Despite several prospective clinical trials, there are no effective pharmacological therapies available for CAVD and the only effective treatment is valve replacement. Several procedures are available for aortic valve replacement, which include conventional replacement surgery with biological or synthetic prostheses and less invasive trans-apical or trans-femoral therapies. Surgical treatment options for end-stage aortic stenosis will not be discussed any further in this review.
In this review, we will provide an overview of the three most common aetiologies and pathogeneses of CAVD and present some of the latest concepts and results in clinical trials aiming to prevent CAVD.
Degenerative aortic valve disease
The most frequent cause of CAVD is degenerative valve disease and several risk factors have been correlated to the progression this condition. The prospective Cardiovascular Health Study correlated age, male gender, hypertension, elevated levels of lipoprotein (a) and low-density lipoprotein cholesterol (LDL), and smoking with the presence of aortic valve sclerosis and stenosis [3]. Others also identified these risk factors, in addition to diabetes and elevated body mass index, the metabolic syndrome and end-stage renal disease, amongst others [6]. Risk factors for degenerative CAVD are thus suggested to be similar to the traditional risk factors for atherosclerosis, which also include increasing age, male gender, hypertension, diabetes, triglycerides, and smoking [3, 7] and it has been hypothesised that acquired valve disease is a manifestation of atherosclerosis. However, an inconsistency has been found in the coexisting prevalence between CAVD and coronary artery disease (CAD) as only 50% of patients with severe CAVD have significant CAD, and the majority of patients with CAD do not have CAVD [8]. This shows that risk factors may be similar, but that there are also notable differences between atherosclerosis and CAVD.
Pathogenesis of degenerative CAVD
Injury and activation of the valve endothelium by mechanical forces, such as shear stress and transvalvular pressure, is thought to be causative for the onset of CAVD [9]. Similar to atherosclerosis, endothelial damage might initiate a number of events such as accumulation of lipoproteins and inflammation [2, 10]. Several adhesion molecules, which are normally not expressed by the valvular endothelium, are found in non-rheumatic aortic valve disease. Monocytes can adhere to these adhesion molecules and migrate into the subendothelial space [10], where they release cytokines, chemokines, growth factors and proteolytic enzymes. In addition, ApoA and ApoB have been found to accumulate in developing lesions of CAVD. Oxidative modifications make these lipoproteins highly cytotoxic for most cells and the products generated by lipid oxidation have shown pro-inflammatory properties. It is likely that inflammation and lipid oxidation cause activation and differentiation of the valve interstitial cells (VICs), which are responsible for the maintenance and repair of the valve matrix structure.
In pathological processes, inflammatory cells are the main source of matrix-metallo proteinases (MMPs). Tenascin-C (TN-C) is an extra-cellular matrix glycoprotein, which is often co-expressed with MMPs, and is overexpressed by interstitial myofibroblasts in aortic valve calcification. TN-C actively participates in physiological bone formation and stimulates osteoblastic differentiation [11]. During progression of aortic valve sclerosis to CAVD, positive feedback mechanisms between MMP-2 and TN-C lead to increased alkaline phosphatase expression, which is a crucial enzyme for bone formation and bone cell differentiation, and deposition of calcium phosphates [12]. MMP activity has also been identified as an activator of transforming growth factor-beta 1 (TGF-β1) [13], which is overexpressed in stenotic valves [14] and has been suggested to play an active role in progression of the disease [15].
Macrophages have also been found to express angiotensin-converting enzyme (ACE) in atherosclerosis. In valvular tissue, local production of ACE has been demonstrated [16]. Enzymatically active ACE was also found to be present in aortic sclerosis and CAVD lesions, where it might participate in disease progress with local production of angiotensin II [17]. Angiotensin II is an important mediator of inflammation and fibrosis and therefore a potential initiator of processes involved in CAVD progression [18].
Lastly, it has been suggested that VIC differentiation towards osteoblasts in valvular calcification is enhanced by the osteoprotegerin (OPG)/receptor activator of nuclear factor-κB (RANK)/RANK ligand (RANKL) system. RANKL expression was increased, while OPG was decreased in human calcified aortic valves [19]. In cultured VICs, RANKL stimulation leads to the induction of an osteogenic phenotype, a significant rise in matrix calcification, alkaline phosphatase activity, expression of osteocalcin, and increased functioning of the transcription factor Runx2, which is essential for onset of genes involved in osteogenic differentiation.
Together, these data provide a brief overview of the possible processes involved in risk factors, onset, progression and end-stage differentiation in degenerative CAVD, while additional pathways have been reviewed by others [6]. Risk factors similar to atherosclerosis, combined with high continuous mechanical stress, are thus most likely important for onset of CAVD, followed by inflammatory processes and gradual osteogenic differentiation with AS as the end-stage (Fig. 2a and b).

a Stenotic tricuspid valve, prior to replacement. b Excised cusps of stenotic tricuspid valve, showing large calcific nodules in the tissue, primarily located at the belly region of the valve. c Stenotic congenital bicuspid valve, prior to replacement. d Excised cusps of stenotic congenital bicsupid valve, also showing large calcified deposition in the belly region of the cusp. e Explanted aortic valve after rheumatic aortic valve disease, obtained from Robins and Cotran, Pathologic Basis of Disease, 7th edition, Elsevier Health Sciences. This shows thickening of the cusps and fusion of the commissures following healing and scarring after the acute phase of the disease
Congenital aortic valve disease
Congenital aortic valve disease includes unicuspid, bicuspid, quadricuspid and even pentacuspid valves. Bicuspid aortic valve (BAV) is the most common congenital cardiac abnormality, occurring in about 1–2% of the population, with a higher prevalence in males [20]. Unicuspid aortic valves, which is the second most common congenital valve disease, occurs in 0.02% of the population. In this review, the focus will be on BAVs.
BAV is the second most common cause of CAVD and is the result of abnormal aortic cusp formation during a complex developmental process. From a recently suggested classification system, it can be extracted that there are several types of BAVs, with different incidences [21]. It showed that only few patients congenitally have two commissures and no raphe (type 0: 7%) and that the majority have conjoined or fused cusps resulting in one (type 1: 88%) or more (type 2: 5%) raphes. Moreover, there seems to be a genetic predisposition for the location of the raphe in the type 1 BAVs, as suggested by the high incidence of fusion of the left and right coronary cusp (71%) and the low incidence of fusion of the left and non-coronary cusp (3%) [21]. Although the information on the effect of these different types of BAVs on calcification is not yet known, there are indications that CAVD tends to progress more rapidly if the cusps are asymmetrical and in an anteroposterior position [20]. Patients with BAV are generally 10 years younger than those with tricuspid CAVD when requiring aortic valve replacement. It is likely that the progression of CAVD in these patients is similar to that in patients with tricuspid valve degenerative calcification [22], but that the process starts earlier in life due to higher mechanical stresses on the cusps and more turbulent blood flow due to abnormal configuration of the cusps [23].
Pathogenesis of BAV
It has been suggested that onset of CAVD in BAVs is the result from less natural folding, incomplete opening and wrinkling of the bicuspid valve cusps during the cardiac cycle, making it more prone to tissue damage as compared with tricuspid valves [24]. The increased turbulence in the blood flow causes trembling and whipping of the cusps, predisposing them to fibrosis and calcification [24]. In addition, recent modelling studies noted that the orifice area of BAVs, functioning normally on echocardiography, may already resemble valves with some degree of stenosis [25]. The overloaded stresses revealed by the model may accelerate degeneration as described for degenerative valve disease [25]. The models do not explain why aortic valves become bicuspid in the first place, but this has been correlated with mutations in Notch signalling. Notch is a receptor which has been attributed several important roles in embryonic development, but that is also linked to endothelial functioning and suppression of osteogenic transcription factor Runx2. Hence, mutations in Notch could be causative for accelerated progression of valve calcification as well [6]. Lastly, recent discoveries have identified altered expression of microRNAs (miRs) 26a, 30b and 195 in calcific and non-calcific regurgitant BAVs [26]. MiRs are small sequences of nucleotides that can influence protein expression and they may provide targets for future therapies as has been shown in several in vivo studies [27].
In summary, progression of calcification in BAV appears to have many similarities with degenerative CAVD in tricuspid valves, although it most likely progresses faster due to higher mechanical stresses on the cusp. A severely stenotic congenital bicuspid valve is presented in Fig. 2c and d.
Rheumatic aortic valve disease
Although the incidence of rheumatic aortic valve disease has declined in industrialised countries, it is estimated that worldwide nearly 20 million people are affected by acute rheumatic fever and rheumatic heart disease, of which 95% in developing countries. The first step in the pathogenesis of rheumatic valve calcification is pharyngitis due to Streptococcus pyogenes, or group A streptococci (GAS), but only 0.3–3% of individuals with acute GAS pharyngitis go on to develop rheumatic fever [28]. Despite a significant decline in the incidence of acute rheumatic fever in developed countries there has been no apparent decline in GAS pharyngitis [28]. This may be explained by the rapid and appropriate diagnosis, and antibiotic management of even uncomplicated GAS infections, or the occurrence of less virulent serotypes becoming more dominant with time.
Each year there are 470,000 incident cases of rheumatic fever worldwide and 233,000 deaths due to acute rheumatic fever and rheumatic heart disease [29]. The mean annual incidence rates are lowest in American and Western European countries and relatively a bit higher in Eastern Europe, Asia, Australasia, and the Middle East. The highest reported mean annular incidences were found in India and in a Maori community in New Zealand [30]. However, the prevalence in African countries such as Kenya and Congo has been estimated to be a lot higher [28]. Even these estimates have been suggested to be underestimations, since more systematic screening with echocardiography resulted in even higher numbers in other developing countries, such as Cambodia and Mozambique [31]. (Fig. 3)

Graphic presentation of measured or estimated incidence of rheumatic aortic valve disease in Western and developing countries
Pathogenesis of rheumatic valve disease
The cytoplasmic membrane of GAS contains several proteins and groups that are highly similar to proteins found in the human host. This results in molecular mimicry, or the sharing of epitopes between antigens of the host and the bacteria, and may stimulate existing B and T cells to respond to self proteins [32]. Rheumatic heart disease can be a serious autoimmune consequence of acute rheumatic fever. For example, the sites where anti-streptococcal/anti-myosin antibodies bind to valvular endothelium may serve as infiltration sites for activated T cells and macrophages [32, 33]. Inside the inflammatory infiltrate, cells produce valve inflammation, scarring and destruction of valvular tissue. The valve turns into a local microenvironment for continuous cytokine production, lymphocyte infiltration, inflammation, and scarring [33]. Infiltrated lymphocytes and macrophages allow for the formation of so-called Aschoff bodies in the valve. Aschoff bodies are characteristic lesions of rheumatic heart disease, consisting of granulomatous structures with fibrinoid change and chronic inflammation of the valve interstitium. Scarring and eventual neovascularisation of the previously avascular valve tissue will lead to irreversible deformation of the valve (Fig. 2e) [32, 33]. Concomitant identification of osteopontin, osteocalcin, periostin and increased bone matrix protein synthesis suggests that a similar osteoblast-like phenotype is involved in rheumatic valves as well as in degenerative CAVD [34].
Pharmacological treatment
In search for effective pharmacological treatment of CAVD, the discussion has often focused on similarities between CAVD and atherosclerosis [35], although contradictory studies have also been published [6, 36]. Currently, the debate seems to shift back to focusing on the differences between the two conditions. Atherosclerotic lesions generally consist of foam cells that fuse to form lipid accumulations surrounded by fibrous caps, while early valvular lesions have been suggested to lack foam cells and progress to mineralised matrix and in some cases bone structures [6]. However, lipid accumulations are also observed in minimally sclerotic valves derived from explanted hearts for transplantation [2] and mature lamellar bone formation has also occasionally been described in atherosclerotic lesions (Fig. 4) [37]. These data show that both atherosclerosis and CAVD may occur in several manifestations, but that most manifestations of atherosclerotic lesions also occur in valves and vice versa. Thus, in many cases, risk factors and causes of atherosclerotic and valvular lesions may be similar, but different mechanic environments and cells involved may cause slightly different progressions and end-stages of the diseases. These small differences might explain why therapies used to slow progression of atherosclerosis are not always successful for inhibition of CAVD progression.

Atherosclerosis versus degenerative aortic valve disease. a, c, e and g. Atherosclerosis. a Coronary artery vessel wall with an early atherosclerotic lesion (fatty streak). Adv = adventitia; Med = media and Fs = fatty streak. Bar = 150 μm. c Higher magnification of A showing foamy macrophages in the intima. Bar = 50 μm. e Advanced atherosclerotic lesion in the aorta. Bar = 300 μm. g higher magnification of E showing bone formation in the atherosclerotic lesion. Foamy macrophages are present in between the bone fragments. Bar = 100 μm. b, d, f, h. Degenerative aortic valve disease. b Cross section through aortic valve leaflet showing the three layer structure with early lesions. Bar = 250 μm. d higher magnification of B showing foamy macrophages and cholesterol clefts in the lamina fibrosa of the valve leaflet. Bar = 100 μm. f Advanced degenerative lesion with bone formation in aortic valve. Bar = 400 μm. h Higher magnification of F showing blood-forming bone marrow next to the bone fragments in the aortic valve leaflet. Bar = 125 μm
One of the key factors in the pathogenesis of CAVD appears to be lipoproteins. The lipid-lowering agents HMG-CoA reductase inhibitors (statins) are used to lower cholesterol levels, exert significant anti-inflammatory effects and also preserve endothelial function in atherosclerotic disease. Several studies have been published in which serum cholesterol levels were lowered and calcium accumulation or inflammation in the valve were reduced, resulting in inhibited progression of CAVD [38]. However, these studies were predominantly retrospective or had small sample sizes. Three recent prospective randomised double-blind studies revealed that statin treatment did not have any effect on the progression of CAVD. According to the Scottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression (SALTIRE) study [39] high-dose statin therapy does not halt the progression of CAVD over a 2-year follow-up period, despite reducing the LDL cholesterol by more than 50%. Similar results were found in the Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial [40], during a median follow-up of at least 4 years. An average reduction in LDL cholesterol of at least 50% was found in addition to reduced incidence of ischaemic cardiovascular events, but no changes in CAVD progression. The third prospective trial, Aortic Stenosis Progression Observation: Measuring Effect of Rosuvastatin (ASTRONOMER) [41], included patients who were nearly 10 years younger than in the SALTIRE and SEAS trials, to better represent the population suffering from CAVD (58 years on average). There was a 54.5% reduction in LDL concentration, but no reduction of progression of CAVD. The results of studies are consistent in demonstrating that lipid lowering does not affect CAVD progression in patients with no clinical indications for lipid lowering.
Preliminary observational studies with ACE inhibitors (ACEi) have produced conflicting results. Rosenhek et al. [42] found that progression of CAVD was not significantly delayed in patients using ACEi. In contrast, a small retrospective study found an association between ACEi use and lower rate of aortic valve calcium accumulation [43]. More recent investigations showed that angiotensin receptor 1 blockers inhibited atherosclerotic changes and endothelial disruption in aortic valves [44]. More research may reveal new targets for therapies in this pathway.
Recent observational studies observed that patients receiving treatment for osteoporosis, especially bisphosphonates, had significantly less calcium deposition in the aortic valve and a slower progression of degenerative CAVD [41, 45]. In addition, members from our group have shown that valvular calcification correlates strongly to osteoporotic remodelling [46]. Lastly, a recent trial has shown that bisphosphonates can reduce cardiovascular calcification in patients >65 years, but that it had no effect in younger patients [47]. This may indicate that there are different stages in the progression of CAVD, in which recruitment of calcium is the last stage, and that bisphosphonates may be beneficial in inhibiting the progression of this last stage only.
For all the therapies described, both successful and unsuccessful results on progression of CAVD have been reported, but better matching of different stages of CAVD with specifically applied therapies may shift the balance towards more effective treatment.
Future directions
The previous sections show that there are no therapies available directed specifically against pathological processes in aortic valve disease. Instead, several experiments have been conducted with pharmacological agents that were successful against other cardiovascular diseases that resemble aspects of CAVD and were therefore thought to be promising for inhibition of CAVD as well. Limitations preventing successful development of pharmacological therapies for CAVD are the lack of suitable animal models, insufficient understanding of processes behind progression of the disease and most important: lack of knowledge on normal valve regeneration. VICs are able to actively maintain and repair the matrix structure of the valve, but it remains unknown which triggers are responsible for the activation of these regenerative capacities and more importantly, what causes it to get out of control and thereby initiate fibrosis and sclerosis in the cusps. For this purpose, it is important that in vitro and in vivo models are developed in which the processes that are specific for CAVD can be identified and targeted with newly developed therapeutic agents.
An example of these therapeutic agents could be modified small nucleotide chains called antagomirs, which can target specific miRs in vivo [48]. MiRs are known to be involved in processes such as angiogenesis and fibrosis and the first pre-clinical trials indicate that specific miRs can be successfully targeted to prevent or stimulate these events [49, 50]. The first investigations in BAVs reveal that there is also altered miR expression in calcified and non-calcified valves [21] and further investigations may reveal additional miRs, proteins and pathways involved in CAVD.
However, it should be noticed that successful development of pharmacological therapies that slow down progression of CAVD will not suffice to prevent surgery in most patients. Currently, most patients are diagnosed with CAVD when they have become symptomatic and at this stage, it may be too late for pharmacological intervention. Therefore, it is important that the search for medication is combined with additional genetic research in humans in order to identify patients at risk before CAVD is initiated. As described above, researchers have been able to identify several potentially interesting genes that may cause BAVs in mice, but these models cannot be translated to humans yet [6] and there is not much known about genetic predisposition for CAVD.
Summary
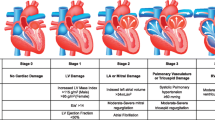
The pathogenesis of non-rheumatic aortic valve calcification is probably initiated by damage to the endothelial layer overlying the valve, followed by accumulation of low-density lipoprotein and infiltration of inflammatory cells. Oxidative stress, release of inflammatory cytokines and growth factors, and extracellular matrix remodelling create an environment that facilitates the proliferation and osteoblastic differentiation of VICs, which ultimately leads to calcification and occasionally ossification of the aortic valve. In BAV, calcification is likely to occur earlier than in patients with a tricuspid aortic valve, as altered mechanical stress accelerates valvular trauma. The processes following valvular trauma are probably similar in tricuspid and bicuspid valves. Rheumatic valve disease is initiated by inflammation, caused by pharyngitis due to GAS followed by the release of cytokines, activation of VICs, matrix remodelling and eventually osteogenic differentiation of VICs. These processes and the discovery of osteopontin, osteocalcin, and increased bone matrix protein synthesis in rheumatic aortic valves suggest that a similar osteoblast-like phenotype is involved in rheumatic valve disease as that in non-rheumatic CAVD. The three forms of CAVD, including currently debated pharmacological treatments, are depicted in Fig. 5. In conclusion, current evidence suggests that there are differences in the onset of several types of CAVD, but that there are similarities in the progression of the disease, which is interesting for development of pharmacological treatments in the future.

Overview of possible disease pathways as discussed and presented in this review. Degenerative CAVD and bicuspid CAVD are likely to follow a similar pathway, but the higher mechanical stress may induce the process early on. Genetical causes for bicuspid CAVD have not yet been identified, but cannot be ruled out yet. Rheumatic CAVD is caused by specific bacterial infections, but following healing and scarring (dashed/solid arrows), these valves also calcify and interstitial cells have been found to undergo osteogenic differentiation. The dashed arrow between matrix remodelling and calcification shows that this process may occur, but for severely stenotic valves, the path through osteogenic differentiation is probably more likely. The suggested therapies so far have not been successful in stopping progression of CAVD. Statins and inhibitors of ACE-related processes have been suggested to be primarily involved in prevention of onset of CAVD, while bisphosphonates were reported to be beneficial at later stages of the disease. For all therapies, both successful and unsuccessful results on progression of CAVD have been reported. Better matching of different stages of CAVD with specific applied therapies may shift the balance towards more effective treatment
References
Iung B, Baron G, Butchart EG, et al. A prospective survey of patients with valvular heart disease in Europe: the Euro Heart Survey on Valvular Heart Disease. Eur Heart J. 2003;24:1231–43.
Otto CM, Kuusisto J, Reichenbach DD, et al. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–53.
Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29:630–4.
Faggiano P, Antonini-Canterin F, Erlicher A, et al. Progression of aortic valve sclerosis to aortic stenosis. Am J Cardiol. 2003;91:99–101.
Otto CM, Burwash IG, Legget ME, et al. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation. 1997;95:2262–70.
Butcher JT, Mahler GJ, Hockaday LA. Aortic valve disease and treatment: the need for naturally engineered solutions. Adv Drug Deliv Rev 2011.
Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. 2001;88:693–5.
Otto CM, O’Brien KD. Why is there discordance between calcific aortic stenosis and coronary artery disease? Heart. 2001;85:601–2.
Sucosky P, Balachandran K, Elhammali A, et al. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler Thromb Vasc Biol. 2009;29:254–60.
New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–91.
Mackie EJ, Ramsey S. Modulation of osteoblast behaviour by tenascin. J Cell Sci. 1996;109(Pt 6):1597–604.
Mathieu P, Voisine P, Pepin A, et al. Calcification of human valve interstitial cells is dependent on alkaline phosphatase activity. J Heart Valve Dis. 2005;14:353–7.
Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24.
Jian B, Narula N, Li QY, et al. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–65. discussion 65–6.
Mohler 3rd ER, Chawla MK, Chang AW, et al. Identification and characterization of calcifying valve cells from human and canine aortic valves. J Heart Valve Dis. 1999;8:254–60.
Helske S, Lindstedt KA, Laine M, et al. Induction of local angiotensin II-producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004;44:1859–66.
O’Brien KD, Shavelle DM, Caulfield MT, et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106:2224–30.
Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–18.
Kaden JJ, Bickelhaupt S, Grobholz R, et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004;36:57–66.
Ward C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83:81–5.
Sievers HH, Schmidtke C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J Thorac Cardiovasc Surg. 2007;133:1226–33.
Falcone MW, Roberts WC, Morrow AG, et al. Congenital aortic stenosis resulting from a unicommisssural valve. Clinical and anatomic features in twenty-one adult patients. Circulation. 1971;44:272–80.
Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol. 2010;55:2789–800.
Robicsek F, Thubrikar MJ, Cook JW, et al. The congenitally bicuspid aortic valve: how does it function? Why does it fail? Ann Thorac Surg. 2004;77:177–85.
Conti CA, Della Corte A, Votta E, et al. Biomechanical implications of the congenital bicuspid aortic valve: a finite element study of aortic root function from in vivo data. J Thorac Cardiovasc Surg. 2010;140:890–6. 6 e1–2.
Nigam V, Sievers HH, Jensen BC, et al. Altered microRNAs in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis. 2010;19:459–65.
da Costa Martins PA, Salic K, Gladka MM, et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. 2010;12:1220–7.
WHO. Rheumatic fever and rheumatic heart disease: Report of a WHO expert panel. WHO Technical Report Series. 2004;923.
Carapetis JR. Rheumatic heart disease in developing countries. N Engl J Med. 2007;357:439–41.
Tibazarwa KB, Volmink JA, Mayosi BM. Incidence of acute rheumatic fever in the world: a systematic review of population-based studies. Heart. 2008;94:1534–40.
Marijon E, Ou P, Celermajer DS, et al. Prevalence of rheumatic heart disease detected by echocardiographic screening. N Engl J Med. 2007;357:470–6.
Guilherme L, Kalil J, Cunningham M. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity. 2006;39:31–9.
Roberts S, Kosanke S, Terrence Dunn S, et al. Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis. 2001;183:507–11.
Rajamannan NM, Nealis TB, Subramaniam M, et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation. 2005;111:3296–301.
Helske S, Kupari M, Lindstedt KA, et al. Aortic valve stenosis: an active atheroinflammatory process. Curr Opin Lipidol. 2007;18:483–91.
Hoagland PM, Cook EF, Flatley M, et al. Case–control analysis of risk factors for presence of aortic stenosis in adults (age 50 years or older). Am J Cardiol. 1985;55:744–7.
Hunt JL, Fairman R, Mitchell ME, et al. Bone formation in carotid plaques: a clinicopathological study. Stroke. 2002;33:1214–9.
Shavelle DM, Takasu J, Budoff MJ, et al. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet. 2002;359:1125–6.
Cowell SJ, Newby DE, Prescott RJ, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–97.
Rossebo AB, Pedersen TR, Allen C, et al. Design and baseline characteristics of the simvastatin and ezetimibe in aortic stenosis (SEAS) study. Am J Cardiol. 2007;99:970–3.
Chan KL, Teo K, Dumesnil JG, et al. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–14.
Rosenhek R, Rader F, Loho N, et al. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–5.
O’Brien KD, Probstfield JL, Caulfield MT, et al. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005;165:858–62.
Arishiro K, Hoshiga M, Negoro N, et al. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. J Am Coll Cardiol. 2007;49:1482–9.
Skolnick AH, Osranek M, Formica P, et al. Osteoporosis treatment and progression of aortic stenosis. Am J Cardiol. 2009;104:122–4.
Hjortnaes J, Butcher J, Figueiredo JL, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J. 2010;31:1975–84.
Elmariah S, Delaney JA, O’Brien KD, et al. Bisphosphonate use and prevalence of valvular and vascular calcification in women MESA (The Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2010;56:1752–9.
van Mil A, Doevendans PA, Sluijter JP. The potential of modulating small RNA activity in vivo. Mini Rev Med Chem. 2009;9:235–48.
Bonauer A, Carmona G, Iwasaki M, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–3.
Elmen J, Lindow M, Schutz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9.
Acknowledgment
The authors gratefully acknowledge L.M. de Heer for providing the pictures A, B, C and D used in Fig. 2.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Riem Vis, P.W., van Rijswijk, JW., Chamuleau, S.A.J. et al. The pathophysiological basis of pharmacological interventions in CAVD. Neth Heart J 20, 270–278 (2012). https://doi.org/10.1007/s12471-011-0230-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-011-0230-1