
Abstract
Background and objective
The long-QT syndrome (LQTS) is associated with premature sudden cardiac deaths affecting whole families and is caused by mutations in genes encoding for cardiac proteins. When the same mutation is found in different families (recurrent mutations), this may imply either a common ancestor (founder) or multiple de novo mutations. We aimed to review recurrent mutations in patients with LQTS.
Methods
By use of our databases, we investigated the number of mutations that were found recurrently (at least three times) in LQT type 1–3 patients in the Netherlands. We studied familial links in the apparently unrelated probands, and we visualised the geographical distribution of these probands. Our results were compared with published literature of founder effects in LQTS outside the Netherlands.
Results
We counted 14 recurrent LQT mutations in the Netherlands. There are 326 identified carriers of one of these mutations. For three of these mutations, familial links were found between apparently unrelated probands.
Conclusion
Whereas true LQT founder mutations are described elsewhere in the world, we cannot yet demonstrate a real founder effect of these recurrent mutations in the Netherlands. Further studies on the prevalence of these mutations are indicated, and haplotype-sharing of the mutation carriers is pertinent to provide more evidence for founder mutation-based LQTS pathology in our country.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The long-QT syndrome (LQTS) is an autosomal dominant, inherited, primary electrical cardiac disease. It is characterised by structurally normal hearts in which serious ventricular tachyarrhythmias occur, also known as torsades de pointes, which may lead to ventricular fibrillation and sudden cardiac death. The electrocardiographic signature of LQTS is prolongation of the rate-corrected QT interval on the electrocardiogram. The estimated prevalence is 1:2000 [1]. Patients with LQTS present, frequently in childhood, with typical symptoms as fainting, often triggered by specific circumstances, among which is physical or emotional stress. Since 1995, 12 different genes with several hundreds of causal mutations have been identified, resulting in 12 LQT subtypes (Table 1). Gene-specific triggers for symptoms are pertinent such as swimming and diving in LQT1 and arousal in LQT2, while other patients develop symptoms during rest (LQT3) [2–6]. The expression of symptoms of the disease is variable and not all carriers are symptomatic (known as incomplete penetrance). Indeed, most patients present with QT prolongation and repolarisation abnormalities on the baseline ECG, but not all carriers have clearly prolonged QT intervals [7]. Early treatment is required because of a high risk of severe arrhythmias when left untreated.
Molecular testing of LQTS patients is mandatory as there is evidence for gene-specific triggers of events and therapeutic efficacy [5, 8, 9]. In this manuscript, as part of a series on Dutch founder mutations and recurrent mutations in inherited diseases, we present an overview of the yield of the molecular genetic tests performed in index cases (probands) with a definite or suspected LQTS in the Netherlands. In a number of countries, including Finland, Sweden, Saudi Arabia, and South Africa, LQTS founder mutations have been described, i.e., single mutations occurring within several families which originate from common ancestors (the “founder”) [10–16]. However, in general, LQTS is regarded as a disease where each family has its own “private” mutation [17–19]. In the Netherlands, only one founder mutation is currently known, the SCN5A mutation p.Tyr1795_Glu1796insAsp (known as 1795insD) which gives rise to an SCN5A overlap syndrome reported earlier in this series [20, 21]. Nevertheless, some mutations within the most commonly involved LQT genes are identified repeatedly. In this overview, we analyse whether these mutations are possibly also founder mutations, instead of mutations which occurred multiple times on a de novo basis.
Methods
In the Netherlands, the first cardiogenetic outpatient clinics started in the Academic Medical Centres of Amsterdam (AMC), Utrecht and Maastricht in early 1996, followed by the other Academic Centres in later years. Molecular genetic testing for LQTS is only performed in the Molecular Genetic Departments of Maastricht and Amsterdam (AMC). From both centres, we collected the genetic results of all probands with a (suspected) diagnosis of LQTS for whom DNA was sent to the molecular genetics facilities. A proband is the first member of a family to be analysed for a familial mutation. We investigated the number of mutations which were found in the most common LQTS types 1–3, from 1996 to November 2009. All mutations were selected on recurrence in at least three probands who were originally assumed independent. We collected the pedigree data of these families and tried to connect families with an identical mutation to each other using genealogical information, such as place of birth and the names of parents or grandparents. Furthermore, with help of the postal codes of the probands, we tried to establish whether they originated from the same region, which would further support a common antecedent. This analysis was based on the PC2 code (the first two numbers of the postal code) of the probands. Data were visualised with MapInfo Professional (MapInfo, Toronto, Canada). In the present analysis, we excluded the previously mentioned SCN5A mutation p.Tyr1795_Glu1796insAsp.
Results
Since 1996, the genetic laboratories in Amsterdam (n = 630) and Maastricht (n = 242) have received 872 Dutch samples of unrelated probands for LQTS diagnostics. All cases are included, patients with a definite diagnosis and those in which LQTS were suspected. We combined the data of both centres. All together, a potential disease-causing mutation was found in 266 cases (31%). Most were “unique” familial mutations. However, in KCNQ1 (LQT1), eight mutations were detected in at least three families (different probands). In KCNH2 and SCN5A (LQT2 and LQT3, respectively), five and one recurrent mutations were found in at least three families (Table 2). In three out of these 14 recurrent mutations, we were able to indicate familial links between probands, although we have not yet succeeded in connecting all probands with these mutations. Pedigrees are provided of these initially apparently unrelated families who could subsequently be connected by genealogy after they appeared to carry an identical disease-causing mutation (Fig. 1a–c). Geographic characteristics are provided for probands that have the most common recurrent mutations (Fig. 2a–c). The subsequent paragraphs will discuss these mutations.

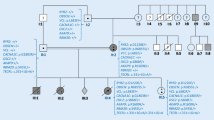
a Pedigree LQT1: p.Tyr184Ser. b Pedigree LQT2: p.Arg582Cys. c Pedigree LQT3: p. Ile1768Val

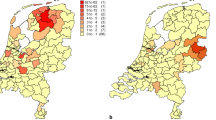
a Disparity of the p.Tyr184Ser mutation in KCNQ1. b Disparity of the p.Arg582Cys mutation in KCNH2. c Disparity of the p.Ile1768Val mutation in SCN5A
KCNQ1
The p.Tyr184Ser mutation in KCNQ1 (which means that, on amino acid position 184 in the protein, the amino acid tyrosine is replaced by the amino acid serine) is caused by a missense mutation in the intracellular loop between the second and third transmembrane segment of this potassium ion channel. It has been found in eight LQT probands in the Netherlands so far. The mutation is a frequently reported mutation also outside the Netherlands (USA/Japan) [22]. The mutation clearly co-segregates with the disease, indicating that there is little doubt that the mutation is pathogenic. Three of the eight Dutch probands carrying this mutation were found to be genealogically connected to each other. This family has a shared male ancestor, born in 1721 (i.e., six generations from the probands (Fig. 1a). Although the number of patients is small and this mutation was found outside the Netherlands as well, the postal codes of the known probands are closely clustered in the region surrounding Utrecht, which strengthens the idea of a founder effect rather than a recurrent mutation (Fig. 2a).
KCNH2
The p.Arg582Cys mutation in KCNH2 (on position 582 in the protein the amino acid arginine was changed into cysteine) is caused by a missense mutation between the fifth transmembrane segment and the pore (S5-pore) region of the KCNH2-ion channel. The mutation is reported to be present in at least 25 patients in the USA and The Netherlands. In our population, it was detected in five probands; two of these probands were genealogically linked over six to seven generations through a male born in 1747 and his wife born in 1748 (Fig. 1b). The geographic disparity of the five probands is quite large, as presented in Fig. 2b.
SCN5A
The p.Ile1768Val (isoleucine is changed by the amino acid valine on position 1768) mutation in SCN5A is caused by a missense mutation located in the sixth transmembrane spanning the segment in domain 4 of the sodium ion channel protein. Patients carrying this mutation often present with severe arrhythmias or (aborted) sudden cardiac death. This mutation was found in nine unrelated probands, two of whom are connected. The ancestral couple was born in 1758 (male) and 1774 (female), seven generations ago (Fig. 1c). Most probands are living in one region (again close to Utrecht), also strengthening the idea of a founder mutation although there is some geographic disparity (Fig. 2c).
Finally, it could be demonstrated that a recurrent mutation is not necessarily a founder mutation. Whereas the p.Arg259Cys and the p.Gly314Ser mutation in KCNQ1 were both identified in four probands, it occurred de novo in at least one of them. Neither of the pairs of parents of the proband carried the causal mutation and the family history was negative for sudden cardiac death or LQTS (Table 2). Hence, the recurrent identification of these mutations might also be based on the de novo mutation of a nucleotide sensitive to spontaneous changes. Indeed, both mutations occur at CpG dinucleotides known to be mutation hotspots [23].
Discussion
Most patients with LQTS have mutations in genes, which encode for cardiac ion channels or their subunits. Worldwide, most families carry a KCNQ1 mutation (type 1 LQTS) or a KCNH2 mutation (type 2 LQTS). Both genes encode for potassium channels (IKs and IKr, respectively) in which a reduced outward current leads to prolonged repolarisation and, hence, a prolonged QT interval. Additional to classical risk factors as QTc interval length, sex, age, and history of prior syncope, the risk stratification of LQT1 and LQT2 patients significantly depends of the location of the mutation within the gene (transmembrane or C terminus in KCNQ1 and the transmembrane pore region vs. N terminus, transmembrane nonpore, or C terminus regions in KCNH2) [22, 24]. Furthermore, the biophysical function of a mutation in KCNQ1 patients (haploinsufficiency or dominant-negative effects) and the topology (α-helical domain vs. the β-sheet domain or other uncategorised locations) of the mutation in KCNH2 patients are independent risk stratifiers for cardiac events [22, 24]. Finally, the type of mutation (nonmissense vs. missense) in the C terminus region of KCNH2 determines the risks of cardiac events significantly [22, 24, 25]. Patients with LQT3 have a defect in the sodium channel gene SCN5A (type 3 LQTS). Here, persistent inward sodium current underlies the prolonged action potential duration.
Approximately 65% of patients with a definite diagnosis have a disease-causing mutation in one of these three genes (LQT1-3). In general, the yield of genetic testing strongly depends on the case: in 60% to 70% of cases with a definite diagnosis and/or a disease-related family history, a mutation is identified [18, 19, 26–28]. When all referrals are counted, including asymptomatic patients with slightly prolonged QT interval durations, the yield significantly drops to about 35% [18, 19, 26–28]. If the molecular screening is limited to familial cases (>1 clinically affected individual or a family history of unexplained sudden death at age ≤ 40 years), the yield can be as high as 90% [29].
In the series on Dutch inherited cardiac diseases caused by a founder mutation, we analysed and described which mutations were found at least three times in apparently unrelated Dutch LQT probands. In the Netherlands, so far, we do not seem to have clear LQTS founder mutations, with the exception of the p.Tyr1795_Glu1796insAsp in the SCN5A gene [20, 21]. However, LQTS-causing founder mutations have been discovered in other parts of the world, i.e., South Africa, Sweden, Saudi Arabia, and Finland [10–16]. An example is the p.Ala341Val mutation, identified in at least 22 LQT1 families in South Africa. The disease allele in all of these families descends from a common ancestor who migrated from the Netherlands to South Africa more than 300 years ago [11–13]. Interestingly, as far as we know, this mutation is not a recurrent mutation in the Dutch population. Although these patients carry the same underlying gene defect, there are significant phenotypical differences. In general, the associated phenotype seems to be highly malignant and even more malignant than other mutations in the same region of the KCNQ1 gene [26]. A recent study by Winbo et al. presents the dominant-negative p.Tyr111Cys mutation in KCNQ1 in 80 carriers from 15 families of which eight are connected with a common ancestor born 11 generations previously in the inland of northern Sweden. Contrary to the p.Ala341Val mutation, this mutation appears to be very benign, with no cases of sudden cardiac death in its population and only a single reported case of aborted cardiac arrest [14].
In Finland, four mutations are frequently detected: the p.Gly589Asp and the c.1129-2A > G (originally annotated as IVS7-2A > G) in KCNQ1 and the p.Leu552Ser and p.Arg176Trp mutations in KCNH2, accounting for up to 70% of the known genetic causes for Finnish LQTS [10, 15]. These Finnish founder mutations cause a slightly prolonged QTc interval, and the phenotype seems to be rather benign, similar to the reported Swedish founder mutation. The intron mutation c.387-5 > A in KCNQ1 and the p.Gln1070X mutation in KCNH2, both found homozygously in consanguineous, but apparently unrelated, Saudi Arabian families, appeared to be founders as well, since both families had identical haplotypes [16]. In these families, there are several severely affected individuals due to homozygous mutation carriership.
We have not yet performed haplotype analysis in the families with recurrent LQT mutations. However, the described links in the three pedigrees (Fig. 1a, c) make it likely that these recurrent mutations are indeed true founder mutations.
Recognising founder mutations is important for several reasons. First, it could significantly improve the turnaround time of DNA diagnostics if this could start with searching for frequent founder mutations. The results of DNA diagnostics would be available much faster, followed by fast available cascade screening of the family. Secondly, the genotype–phenotype correlation of the particular mutation can be studied extensively, which will improve risk stratification and optimal treatment strategies in these families. Furthermore, a population carrying a founder mutation is particularly of interest for research purposes, i.e., for the identification of other genetic variants that may influence the phenotype (known as genetic modifiers). The first evidence for such a genetic modifier in a LQTS founder population was recently described by Crotti et al. [30]. This study in the South African founder population (the Ala341Val mutation in KCNQ1, see above) revealed that common NOS1AP variants are associated with the clinical severity of congenital LQTS. The QT duration of carriers of the founder mutations and the NOS1AP variant is significantly prolonged compared with the QT duration of those patients who carry the founder mutation alone [30]. Concerning LQTS, the NOS1AP is the first important study of a series of modifiers, based on data of founder populations.
The recurrent mutations that are “possible” founder mutations in the Netherlands are listed in Table 2. Care should be taken not to jump too prematurely to this conclusion as there is evidence that both mutations p.Arg259Cys and p.Gly314Ser can occur de novo. Spontaneous mutagenesis at hotspots may result in recurrent mutations in unrelated probands.
The LQTS1 and LQTS2 recurrent mutations p.Tyr184Ser and p.Arg582Cys, respectively, have a phenotype that is not particularly benign or particularly malignant. However, in the SCN5A p.Ile1768Val families, premature sudden death is prevalent, and carriers of this particular mutation should have close follow-up and timely treatment, which unfortunately in this subtype may have to be the implantation of a device (PM or ICD?).
Conclusions
We have identified recurrent mutations in all different LQTS subtypes. In three of these families, a genealogical link was found between (far-related) independent probands. This supports the concept that some recurrent mutations are indeed founder mutations. Haplotype analysis in these and other families with a recurrent mutation is pertinent to perform further research on the founder aspects of these mutations. Eventually, this may result in a better understanding of mutation–disease relations and in better tailoring of potentially life-saving treatment.
References
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7.
Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, et al. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. 1999;15:876–9.
Wilde AAM, Jongbloed RJE, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS) patients from KVLQT1-related patients (LQTS). J Am Coll Cardiol. 1999;33:327–32.
Ackerman MJ, Tester DJ, Porter CJ. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. 1999;74:1088–94.
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype–phenotype correlation in the long QT syndrome. Specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95.
Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson J, Priori SG, et al. Influence of genotype on the clinical course of the long-QT syndrome. N Engl J Med. 1998;339:960–5.
Hofman N, Wilde AA, Kääb S, van Langen IM, Tanck MW, Mannens MM, et al. Diagnostic criteria for congenital long QT syndrome in the era of molecular genetics: do we need a scoring system? Eur Heart J. 2007;28:575–80.
Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. New Engl J Med. 2003;348:1866–74.
Tan HL, Bardai A, Shimizu W, Moss AH, Schulze-Bahr E, Noda T, et al. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: possible therapy implications. Circulation. 2006;114:2096–103.
Marjamaa A, Salomaa V, Newton-Cheh C, Porthan K, Reunanen A, Karanko H, et al. High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann Med. 2009;41:234–40.
Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–10.
Brink PA, Schwart PJ. Of founder populations, long QT syndrome, and destiny. Heart Rhythm. 2009;6:S25–33.
Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, et al. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 2007;116:2366–75.
Winbo A, Diamant UB, Stattin EL, Jensen SM, Rydberg A. Low incidence of sudden cardiac death in a Swedish Y111C type 1 long-QT syndrome population. Circ Cardiovasc Genet. 2009;2:558–64.
Fodstad H, Swan H, Laitinen P, Piipo K, Paavonen K, Viitasalo M, et al. Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann Med. 2004;36:53–63.
Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Khalaf K, Mannens MMAM, et al. Clinical and genetic analysis of long QT syndrome in children from six families in Saudi Arabia: are they different? Pediatr Cardiol. 2009;30:490–501.
Wilde AA, Bezzina CR. Genetics of cardiac arrhythmias. Heart. 2005;91:1352–8.
Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–60.
Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, et al. Spectrum and prevalence of mutations from the first 2500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–303.
Bezzina C, Veldkamp MW, van den Berg MP, Postma AV, Rook MB, Viersma JW, et al. A single Na (+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13.
Postema PG, van den Berg MP, van Tintelen JP, van den Heuvel F, Grundeken N, Hofman N, et al. SCN5A 1795insD, the first described arrhythmia overlap syndrome and one of the largest and best characterized families worldwide. Neth Heart J. 2009;17:422–8.
Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–9.
Antonarakis SE, Krawczak M, Cooper DN. Disease-causing mutations in the human genome. Eur J Pediatr. 2000;159:S173–8. Supplement 3.
Shimizu W, Moss AJ, Wilde AAM, Towbin JA, Ackerman MJ, January CT. Genotype–phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol. 2009;54:2052–62.
Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient mechanism. Circulation. 2006;113:365–73.
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85.
Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol. 2006;47:764–8.
Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975–80.
Hofman N, Tan HL, van Langen IM, Birnie E, Mannens MMAM, et al. Primary inherited electrical heart disease: a ten year experience in the Netherlands. Circulation. 2006;114:II–723. abstract.
Crotti L, Cristina M, Insolia R, Peljto A, Goosen A, Brink PA, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–63.
Acknowledgement
We gratefully acknowledge Y. Blauw (Boston Scientific, Guidant) for providing the MapInfo software.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hofman, N., Jongbloed, R., Postema, P.G. et al. Recurrent and Founder Mutations in the Netherlands: the Long-QT Syndrome. Neth Heart J 19, 10–16 (2011). https://doi.org/10.1007/s12471-010-0046-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-010-0046-4