
Abstract
Thermoplastic polyurethane (PU) polymers with different chemical compositions were synthesized and casted to films, and their water vapor barrier properties at different range of relative humidity (RH) were characterized. The water vapor permeability (WVP) of packaging films is one of their most important properties to identify their suitability for use as packaging materials and is rather a complicated phenomena if the polymer has polar nature. The WVPs of PU films are determined both by permeation measurements which are a steady-state method and water vapor sorption measurements which are a non-steady-state method. Effective permeability (Peff), solubility (Seff), and diffusion (Deff) coefficients of PU films were determined at 23 °C within the RH range of 0–97%. It was found that Peff, Seff, and Deff increased with increasing RH gradient due to water vapor and polymer interactions. Microscopic images showed that 1,4-butanediol (BDO) helped to improve porous structure. Castor oil (CO) caused a decrease in the intensity of active absorption sites, namely, the C=O···H-N hydrogen bonds between chains. Results of two methods were yielded in the same magnitude of order. In most cases, the non-steady-state (sorption) method yields higher WVP values than steady state. At 0➔85% RH, the difference was up to 8-fold. Conditioning and equilibrating of films at 50% RH helped to reach sorption data approximate to permeation data. It was suitable to use sorption measurements to estimate the WVP which is a considerable simplification for polar polymers, e.g., developed PU film.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Respiring food products such as fresh produces (fruits and vegetable) tend to deteriorate quickly since they have an ongoing metabolism, i.e., respiration and transpiration. They are also prone to contamination by bacteria, yeasts, and/or molds [23]. Fresh produce needs to be packaged in films with a high oxygen and water vapor permeability. However, most polymeric films used in current modified atmosphere packaging (MAP) have too low water vapor permeability relative to the moisture loss of fresh produce. Therefore, a high relative humidity (RH) forms in headspace causing water vapor condensation leading to microbial growth and decay of produce [7, 30]. The condensation problem is aggravated also by temperature fluctuations during storage, distribution, and marketing [30].
An interesting polymer for fresh produce packaging can be thermoplastic polyurethane (PU) with controllable gas permeability with respect to temperature [33, 38]. Polyurethanes are one of the most important classes of polymers which are produced by the polyaddition process of diisocyanates and polyols and chain extender and make generally polar polymers with segmented (hard and soft) polymeric character [12]. The performance of polyurethanes can be controlled by chain structures and, in particular, by polar interactions and hydrogen bonds between their soft and hard segments [12, 14, 34]. Many studies have found a relationship between the structure and the barrier properties of polyurethanes [15, 22].
It is very important to calculate the water vapor permeability (WVP) of the packaging system. There are numerous standards to experimentally obtain WVP or water vapor transmission rate (WVTR). The most important international standards are American Standards ASTM F 1249, ASTM E 398 or ISO 15106, and ASTM E 96; the German standard, DIN 53122; the European standard, EN 1015-19:1998/A1:2004; and the Italian standard, Normal-21/85 [31]. However, all those methods would characterize a polymer’s gas diffusivity accurately, except where there are strong interactions between the polymer and the gas molecules (e.g., water vapor and hydrophilic polymers) [18]. Many polymers, especially polar polymers, show a “nonideal” behavior for the permeation of water vapor. Due to the fact that at polar polymers the water vapor permeability coefficient increases with higher RH due to swelling, their sorption isotherms deviated from Henry’s law, so the applied equations for the sorption experiments are just approximations and the WVP is valid only for a defined RH on both sides of a film [24, 25]. Therefore, studying of WVP of such polymers with an alternative method is of great interest to show their potential use in food packaging applications. In this study, simple water vapor sorption experiments (non-steady state) were applied to measure water vapor diffusion coefficients and sorption coefficients of polymers. It is also worth to mention that relevant polymer properties such as density, microscopic polymer structures, etc., which can influence the sorption behavior or WVP, and data about these properties were not available. Therefore, the aim of this study is to discuss how and which extend chemical composition, and RH affects water vapor barrier properties of synthesized films and compares the results of alternative method with standard gravimetric method.
Materials and Method
Materials
Castor oil (CO) (functionality of 2.67) with a hydroxyl number of 161.01 mg KOH g−1 and an acid number of 0.99 mg KOH g−1 was purchased from Aldrich (Aldrich, Milwaukee, USA). Polyethylene glycol (PEG) with a number-average molecular weight of 1500 g mol−1 and 1,6-hexamethylene diisocyanate (HDI) were obtained from Fluka (Fluka, Seelze, Germany). Technical grade 1,4-butanediol (BDO) was purchased from Sigma-Aldrich (Sigma, Seelze, Germany).
Synthesis of Polyurethane Films
Thermoplastic polyurethane (PU) films were prepared by one-step bulk polymerization technique according to Akkas et al. [1]. Prior to the synthesis, PEG was dried on a rotary evaporator (RV 10 IKA, Staufen, Germany) for 6 h at 90–95 °C, and CO was dried at 80 °C under vacuum for 24 h. BDO was dried overnight at 50 °C in a vacuum oven (Vacucell MMM, Graefelfing, Germany).
PEG and/or CO was added into a flask and mixed at a rotary evaporator with 80 rpm. Then, BDO was added into the flask and mixed until a homogeneous mixture was obtained. Neither a catalyst nor a solvent was used with the intention to get a material suitable for food contact. The mixture was poured into a three-necked reaction flask equipped with a mechanical stirrer, dropping funnel, and nitrogen inlet and outlet. The temperature was then increased to 50 °C. HDI was added to the mixture during stirring at 300 rpm, and the reaction was continued for 150 s. All PU films were prepared at an equimolar amount of diisocyanate relative to hydroxyl groups. Polyurethane films were prepared by a casting-evaporation technique. The reaction mixture was poured into glass petri dishes and placed in an oven at 80 C for 24 h.
Films were prepared with 1500 g mol−1 molecular weight of PEG, at two different weight ratios of CO/PEG (50/50 and 70/30) and at three different ratios of BDO/ (PEG+CO) (40/60, 50/50, and 60/40) as mole percent of the hydroxyl groups. They are named using the abbreviation PU 1500-b-c, where 1500 indicates molecular weight (g mol−1) of PEG, b indicates the weight ratio of CO to PEG (CO/PEG), and c indicates the weight ratio of BDO to PEG and CO [BDO/ (PEG+CO)]. Each film production was replicated two times.
Characterization of Polyurethane Films
The film thickness was measured by a precision thickness gauge (Model FT3 by Rhopoint Instruments, Bexhill-on-Sea, UK) at five different positions. The arithmetic average of the film thickness was used to determine WVP.
The density of the films was measured by applying the Archimedes’ principle using a density kit (Mettler-Toledo AG; Greifensee, Switzerland) with an analytical balance (AT261 DeltaRange®; Mettler-Toledo AG; Greifensee, Switzerland) according to ASTM standard test method D792-A. Density of samples ρ was measured according to Eq. (1) at 20 °C and calculated from the weight of the sample in air A and the weight of the sample in distilled water (0.9975 g/cm3) B. Air density was neglected because of its minor influence on the results.
where ρ0 is density of water at 20 oC (0.9982 g/cm3), A is the apparent weight of specimen in air, and B is the apparent weight of specimen completely immersed in water. Experiments were done as quintuplicates.
FTIR spectra, obtained with a PerkinElmer (Waltham, MA, USA) Spectrum One instrument in the range from 650 to 4000 cm-1, were used to characterize the functional groups of the synthesized PU films and also to investigate hydrogen bonding in the hard segment.
For the scanning electron microscopic (SEM) imaging, specimens of PU1500-50-40, PU1500-70-40, and PU1500-50-60 coded materials have been prepared. Topographic images of film surface were taken using FEI model Nova NanoSEM 450 (Thermo Fisher, Eindhoven, Netherlands) by using the low vacuum (LVD) detector. The cross-sections of the films were generated by freezing them in liquid nitrogen and breaking them. The images of the cross-sections were taken using another scanning electron microscope (Vega-3, Tescan, AS, Brno, Czech Republic) with various settings. In order to reduce electrical charging of the nonconducting samples by the electron beam, all film specimens were coated with a gold layer prior to observation. Cross-section images have been obtained by using the secondary electron detector (SE mode).
Water Vapor Sorption of Thermoplastic Polyurethane Films (Non-steady State)
Water vapor sorption measurements were carried out at 50, 85, and 97% relative humidities (RHs) at 23 °C. Saturated solutions of calcium nitrate tetrahydrate (Ca(NO3)2.4H2O) and potassium sulfate (K2SO4) salts with residue (undissolved salt crystals) were placed in a desiccator to provide atmospheres of 50% and 97% RHs, respectively, at 23 °C [11]. A climatic chamber (Binder GmbH, Tuttlingen, Germany) was used to test moisture sorption at 85% RH and 23 °C. Temperature and relative humidity were recorded by a data logger to check and validate the targeted conditions. Prior to testing, the square film samples of 3 cm × 3 cm in size were dried in a vacuum oven (Vacucell MMM, Graefelfing, Germany) at 80 °C for 4 h to a constant weight. One side of the film was covered with a self-adhering aluminum foil (Reinaluminium-Klebeband, ISODEAL Steinmetz GmbH, Eichenau, Germany) with a window of area of 2.5 cm × 2.5 cm. By this way, water vapor absorption took place from only one side of the film, and side permeation was prevented (Fig. 1a). The weight of the thermoplastic polyurethane films was measured twice a day, for a period of 7 days with laboratory scales (Mettler Toledo, Gießen, Germany, Delta Range AT 261). Due to possible water absorption of the adhesive of the self-adhering aluminum foil itself, reference masks without sample were stored at the same conditions. Their weight increase was deducted from the weight increase of the samples in the masks. The mass fraction of absorbed water is presented in relation to the dry mass of the films.

Aluminum masking of polyurethane film for water vapor absorption determination, absorption from one side a, and test arrangement for measuring the water vapor transmission rate of the film by gravimetric method b
Theoretical Considerations
When studying the sorption of small molecules by a matrix, we must consider the assumptions underlying the applied theoretical models. Fundamental models describing water absorption in films are usually based on Fick’s second law of diffusion, which is in the specific case of mass transport through a plane sheet with constant boundary conditions. The absorption and desorption of water vapor by films from two sides reduce to the following Eq. (2) [8]:
where m∞ is the amount of water absorbed at equilibrium, mt is the amount of absorbed water at the time t, D is the diffusion coefficient, mt/m∞ is the relative mass fraction of water in the film, and l is the average thickness of the film. Edge effects being neglected, the early stages of diffusion-controlled uptake can be predicted in a simplified way as Eq. (3) [9]:
The mass fraction mt/m∞ as a function of the square root of time t is approximately linear until a value of mt/m∞ of 0.5–0.7 is reached [29]. Equations (2) and (3) are only valid for the case that the absorption by a sample can be described by the solution-diffusion model [8, 13]. Boundary conditions:
-
(1.)
Henry’s law can be applied (Eq. (4)), i.e., the concentration (cwater) of the absorbed water vapor in the polymer depends linearly on the partial pressure (pwater) of the water vapor. The proportionality constant is the sorption coefficient or solubility coefficient (S) [13].
-
(2.)
The adsorption of water vapor on the surface of the film is fast. The diffusion in the polymer is the time-limiting step for the absorption rate of the film.
-
(3.)
The polymer does not react with the absorbed water.
-
(4.)
The diffusion and sorption coefficients are constants.
-
(5.)
The film material is isotropic on a macroscopic scale.
However, in real life these conditions are often not fulfilled, because the polymer can interact with absorbed water vapor. Therefore, diffusion and sorption coefficients are often dependent on the amount of absorbed water vapor [20]. The water vapor sorption isotherm cannot be described by Henry’s law in such a case. In this case, Eqs. (2) and (3) are simplifications. In this work, in order to distinguish exact values from the coefficients of these simplifications, they are labelled as effective. Water vapor absorption experiments with thermoplastic polyurethane films were evaluated at a relative mass fraction of absorbed water of between 0 and 0.5. The effective diffusion coefficients (Deff) were calculated using the slope of the initial absorption-time curve (according to Eq. (3)) and the thickness of the film. For the absorption by the film from only one side, the double thickness has to be taken. The effective sorption coefficients (Seff) were calculated from the water vapor absorption at equilibrium (Eq. (4)). The WVP was calculated using Eq. (5).
Experiments were done as quintuplicates.
Water Vapor Transmission Rate (Steady State)
For measuring the water vapor transmission rate (WVTR) through films, the standard gravimetric method according to ASTM E 96 was used with slight modification [3]. The specimens were cut into circles with test area of 10 cm2 and were dried in a vacuum oven (Vacucell MMM, Graefelfing, Germany) at 80 °C for 4 h to a constant weight. For some cases, the films were conditioned in a climatic chamber maintained at 23 °C and 50% RH for at least 2 days. During this period, the samples reached equilibrium conditions. After that, film specimens were mounted on a screw cup filled with dry silica gel. The cup and the lid were treated with technical vacuum grease, to fix and seal the sample on the cup. The specimen was placed in the screw cup, which was closed with a ring-shaped, rubberized lid (Fig. 1b). Silica gel was used for adsorbing water. It created nearly 0% RH in the headspace of the cup. Each test arrangement was weighed and stored at 50, 85, and 97% RHs at 23 °C. Because of the moisture gradient, between the inside of the cup (nearly 0% RH) and the environment (50–85–97% RH), water vapor permeated through the specimen towards the inside of the cup. Water vapor uptake by silica gel causes an increase in weight of the test arrangement. Each test arrangement was weighted twice a day, for a period of 4–5 days. Tests were done as triplicates. The water vapor transmission rate was calculated according to Eq. (6):
where t is the period of time between two weight measurements in h, Δm represents the weight difference between two weight measurements in g, and A is the test area in cm2. The units were g·m-2∙d-1. Finally, by using Eq. (7), water vapor permeability (WVP) was obtained [5].
In the above equations, WVP is water vapor permeability, l is the average thickness of polyurethane samples, P is the vapor pressure of water at 23 °C, and R1 and R2 are the moisture gradients.
Statistical Analysis
Statistical analysis was performed using Minitab 16.0 (Minitab Inc., State College, PA, USA). The permeability data were subjected to analysis of variance and Tukey method to evaluate the differences between chemical formulations and RHs (p < 0.05).
Results and Discussion
Polyurethane Film Characterization
The polyurethane film thicknesses and densities applied for calculation of WVP properties were shown in Table 1. All casted film samples have around 1 mm thickness. Density of the polyurethane films tended to decrease with increasing CO content. There was no significant difference in densities at different BDO contents. PU1500-70-40 coded film had the lowest density. At this point, the differences of density and structure of the monomers are needed to be noted (ρCO = 0.96 g/cm3, ρPEG1500 = 1.23 g/cm3, and ρBDO = 1.015 g/cm3) [6]. Density is a function of chemical composition, being dependent on the weight of individual molecules and the way they pack together. The decrease in density can be attributed to a lower alignment of polymer chains as a result of weak formation of polymer crystals [21]. Previous study also showed that increasing CO content resulted in decreased crystallinity of polymer films [32]. Reduced density and crystallinity in polyurethane film composition might result in free volume increase which enhances the WVP.
The Fourier transform infrared (FTIR) spectra of all samples with characteristic polyurethane peaks are presented in Fig. 2. Although a number of vibrational modes are sensitive to hydrogen bonding in these systems, there were two regions that the most information can be obtained from the amide (N-H) and carbonyl (C=O) stretching vibrations near 3200–3400 and 1600–1700 cm-1, respectively. Hydrogen-bonded urethane C=O and N-H vibration peaks appear around 1680 cm-1 and 3320 cm-1, respectively. In the N-H stretching region, peak at around 3320 cm-1 was proposed for H-bonding with urethane C=O [37]. In fact, if the mixed phase or non-H-bonded N-H stretching is available, a shoulder band on the high-frequency side around 3350 cm1 would appear as shown in Fig. 2a [6, 10]. In the C=O stretching region, a strongly hydrogen-bonded C=O peak at about 1682 cm-1, indicating the presence of H-bonded urethane C=O groups for the ester structure, was observed for all film samples, and poorly H-bonded carbonyl was observed at about 1715 cm-1 for PU1500-70-50 and PU1500-70-60 coded samples (Fig. 2b). The most noticeable was the systematic decrease in the intensity of the C=O···H-N hydrogen bonds between chains with increasing CO content. Moreover, increasing BDO content at 70% CO level resulted in lower and split peak. This is due to the long alkyl groups which break hydrogen bonding between the polymer chains [4]. It is also stated that interaction of water molecules is greater for polyurethanes with higher content of C=O and N-H groups as they are accepted as active absorption centers [10]. Thus, increasing CO and BDO content may reduce sorption of polyurethane films.

FTIR spectra of polyurethane film samples
Microscopic Images
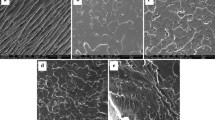
The surface and cross-section morphologies of polyurethane films obtained from SEM are shown in Fig. 3. PU1500-50-40 coded film sample showed clustered porous shapes on the surface (Fig. 3a), and the cross-sectional surface morphology of the film showed wrinkles to appear (Fig. 3b). These clustered structures and wrinkles can arise through micro-phase separation of the hard and soft segments [17]. It is also stated that micro-phase separation can improve waterproof capability of polyurethanes [35]. Thus, PU1500-50-40 may have lower VWP. The surface of PU1500-70-40 appeared to be rough (Fig. 3c), and the cross-sectional surface showed less wrinkles than the cross-sectional surface of PU1500-50-40 (Fig. 3d). Increasing CO content might be contributed to chain mobility within the polymer and inhibited cluster formation by distributing pores uniformly. PU1500-50-60 coded sample showed smoother and microporous surface (Fig. 3e). These porous structures may promote the water vapor permeability. Increasing BDO content caused increasing hard segment [32]. At higher hard segment content, a morphology with a dispersed soft segment phase was observed. PU1500-50-60 also showed a dense and nonporous cross-sectional surface (Fig. 3f). As we know that the mass transfer occurs in nonporous structure by sorption, diffusion, and desorption, therefore sorption data are important for characterization of WVP [19].

SEM images of polyurethane films coded as PU1500-50-40 surface a; cross-section b; PU1500-70-40 surface c; cross-section d; PU1500-50-60 surface e; and cross-section f
Water Vapor Sorption and Permeation
To determine the effective diffusion coefficients (Deff) according to Eq. (3), the relative mass fraction (mt/m∞) of sorbed water vapor is described as a function of the square root of time divided by the thickness for measurements at 50% RH, 85% RH, and 97% RH. Measurements at 50% RH were depicted in Fig. 4, representatively. At 50% RH, 85% RH, and 97% RH for the film samples coded as PU1500-70-60, PU1500-50-40, and PU1500-50-50, the mass fraction of water increased not immediately with a linear slope but with a time lag, due to the low reaction rate of structural changes caused by BDO and CO content. The water vapor sorption was approximately linear at a mass fraction between 0 and 0.6 and progressed regressively to the absorption equilibrium value of one. This behavior is typical for materials that follow Fick’s law. The slope of the curve decreased when the relative humidity reached targeted equilibrium, which can be explained by saturation [24, 25]. From the initial linear section slope, Deff was calculated. The changes in Deff values for the film samples against RH were shown in Fig. 5. As expected, the Deff increased with higher RH because the samples absorbed water vapor at a higher absorption rate except for PU1500-50-40. In this case, the explanation might be that at higher RHs the water in excess of the dynamic solubility is clustered and does not contribute substantially to the diffusion process [28]. Furthermore, the size of the cluster might be not the only factor which determines its ultimate mobility within the polymer, but the specific polymer-water vapor interactions may also determine ultimate mobility. For example, a stiff polymer may totally immobilize a water “dimer,” whereas a more flexible polymer may permit transport of dimerized water species [36]. PU1500-70-40 together with PU1500-70-50 showed higher Deff at all RHs, and the rate of increase in Deff was also high at higher RH. Water vapor absorption widens polymer chains. This increases the diffusivity of water vapor [16]. Reduced density or crystallinity of those PU samples and increased chain mobility due to the higher CO content in the structure may also help diffusion process. Moreover, increasing BDO content at 50% CO level helped to improve Deff. The obvious reason for the faster water vapor absorption by PU1500-50-60 coded film might be the creation of an open porous structure which is also visible in the microscopic pictures in Fig. 3.

Fractional moisture absorption at conditions 50% RH, 23 °C as function of the square root of time (t) divided by the sample thickness (d). Single results were plotted from fivefold determination.

Variation of effective diffusion coefficients for water vapor in polyurethane films related to relative humidity at 23 °C
The effective sorption coefficients (Seff) were calculated using the mass fraction of water in the films (Fig. 4), the density of the films (Table 1), and the water vapor saturation pressure of 28.08 mbar (2808 Pa) at 23 °C [26]. Figure 6 shows changes in Seff values for the film samples against RH. Due to the hydrophobic nature of CO, increasing CO content decreased Seff values of films at 70% CO level. However, all samples showed increasing Seff with increasing RH. PU films having higher content of H-bonding between C=O and N-H groups such as PU1500-50-40 showed higher interaction with water and depicted higher Seff values.

Variation of effective solubility coefficients for water vapor in polyurethane films related to relative humidity at 23 °C
In Fig. 7, the effective permeation coefficients (Peff) are shown that were calculated from Deff and Seff, using Eq. (5). The higher Peff for PU1500-50-60 can also be explained by channels, due to its porous structure. It can also be noted that Deff has more dominant role in permeability at higher RHs. At 50% RH, the water vapor uptake was lower; thus Seff was reduced. The Deff changed slightly; therefore Peff was reduced.

Variation of effective permeability coefficients for water vapor in polyurethane films related to relative humidity at 23 °C
To relate the results for the effective diffusion, sorption, and permeation coefficients, the Deff and Seff were depicted as pairs of values together with different polymers (Fig. 8). Pairs of values for Deff and Seff, were determined by sorption experiments; transverse lines represent Peff, determined by permeation measurements with the unit (mg.cm(cm2.s.Pa)-1) [2]. Nonpolar polymers have low Peff and Seff, namely, the opposite to polar polymers [27]. Moreover, all synthesized PU films have Peff and Seff values in between nonpolar and polar polymers. The Deff was in range of polymers with polar groups such as polyethylene terephthalate (PET) and polyamide (PA).

Comparison between water vapor sorption and diffusion coefficients of polyurethane films and different polymers at 23 oC and relative humidity 50%
Table 2 compares the result of WVP values obtained from sorption and permeation measurements of PU films. For steady-state permeation method, WVP values were calculated from WVTR divided by the relative humidity gradient and the water vapor saturation pressure of 2808 Pa at 23 °C. For polar polymers, it is known that the water vapor permeability and transmission rate depend on the RH [27]. In this study, WVP results from both sorption and permeation method at RHs 0➔50%, 50➔85%, and 50➔97% showed the same magnitude of order. At RH 0➔97% also showed similar results except for PU1500-50-40. A variability of the permeability by a factor of two was common for polymers of same specification or type [26]. In high RH cases especially at range of RH 0➔85%, the non-steady-state (sorption) method deviated by max up to factor 8 from the WVP values of steady-state (permeation) method especially if the PU films were not conditioned at 50% RH prior to test. Eighty-five percent RH might be a critical value for aggregation since the aggregates are being less permeable and slow down the diffusivity and the moisture storage. Moreover, the factor leading to increased water sorption at higher RH is entropy in nature; clustering of water might have occurred within the polyurethane matrix and, thereby, opening or altering its structure. Thus, it should be noted that preconditioning at 50% RH for PU films has great success to reach comparable results. Sorption experiments can be suitable for materials, where WVP of films is not measurable by standard permeation method due to the cracks or defects in the films, which can occur during drying and curing processes. Another application might be polymers with low amounts through small-scale laboratory production, e.g., biopolymers derived from natural sources.
Conclusions
The water vapor permeability (WVP) of casted polyurethane films was determined successfully by two methods, sorption and permeation experiments. The WVP of polyurethane films showed significant dependence on relative humidity (RH). The Deff and Seff of the films were increased when RH increases. The significant differences among the values of WVP for polyurethane films were observed when chemical composition of the films was altered. Reduced density and crystallinity in polyurethane film composition due to high CO content enhanced the WVP. FTIR showed that PU films with higher H-bonding content of C=O···H-N groups showed higher solubility coefficient. High level of CO content decreased water sorption. According to SEM, porous structure of PU1500-50-60 can be the reason to promote the WVP. Moreover, micro-phase separation occurring in PU1500-50-40 can improve waterproof capability of film with better water sorption properties.
The WVP determined by sorption and permeation experiments yields results in the same magnitude of order. In most cases the non-steady-state method yielded higher WVP values compared to the steady-state method. In few cases, the WVP measured by the non-steady-state method was higher than the WVP values measured by the steady-state method up to factor 8. The observed differences between these methods are caused by the nonideal behavior of the analyzed films; their interaction with water vapor, time, and the humidity dependent on change of their effective diffusion; and their effective sorption coefficients. The difference of the results for the sorption method compared to the permeation method allows an indicative measurement for new materials and preconditioning at 50% RH, for PU films have great success to reach approximate results. In conclusion, the sorption experiment can be suggested as a suitable method for homogeneous, flat, monolayer samples where small sample areas are available which are too small for permeation measurements and to estimate the magnitude of order of their respective WVP.
References
Akkas T, Citak C, Sirkecioglu A, Guner FS (2013) Which is more effective for protein adsorption: surface roughness, surface wettability or swelling? Case study of polyurethane films prepared from castor oil and poly(ethylene glycol). Polym Int 62:1202–1209
Anonymous (2013) Fraunhofer IVV internal results
Astm International (2016) ASTM E96/E96M-16-Standard test methods for water vapor transmission of materials. In:ASTM International West Conshohocken, PA
Aydin A, Demirci F, Orhan M, Kocer HB (2019) Preparation of breathable polyurethane membranes with quaternary ammonium salt diols providing durable antibacterial property. J Appl Polym Sci 136
Babaee M, Jonoobi M, Hamzeh Y, Ashori A (2015) Biodegradability and mechanical properties of reinforced starch nanocomposites using cellulose nanofibers. Carbohyd Polym 132:1–8
Cakmak EG, Dalgakiran D, Guner FS (2018) Castor oil and PEG-based shape memory polyurethane films: effect of chain extender amount on some polymer properties and performance. Turk J Chem 42:1161–1173
Cameron AC, Talasila PC, Joles DW (1995) Predicting film permeability needs for modified-atmosphere packaging of lightly processed fruits and vegetables. Hortscience 30:25–34
Crank J (1979) The mathematics of diffusion. Clarendon Press, Oxford
Deb S, Braden M, Bonfield W (1995) Water absorption characteristics of modified hydroxyapatite bone cements. Biomaterials 16:1095–1100
Dilandro L, Pegoraro M, Bordogna L (1991) Interactions of polyether polyurethanes with water-vapor and water methane separation selectivity. Journal of Membrane Science 64:229–236
Greenspan L (1977) Humidity fixed-points of binary saturated aqueous-solutions. J Res Nbs Phys Ch 81:89–96
Krol P, Krol B (2012) Surface free energy of polyurethane coatings with improved hydrophobicity. Colloid and Polymer Science 290:879–893
Langowski HC (2008) Permeation of gases and condensable substances through monolayer and multilayer structures. Plastic packaging: interactions with food and pharmaceuticals, pp 297–347
Li W, Ryan AJ, Meier IK (2002) Effect of chain extenders on the morphology development in flexible polyurethane foam. Macromolecules 35:6306–6312
Li H, Freeman BD, Ekiner OM (2011) Gas permeation properties of poly (urethane-urea) s containing different polyethers. Journal of Membrane Science 369:49–58
Lim LT, Britt IJ, Tung MA (1999) Sorption and transport of water vapor in nylon 6,6 film. Journal of Applied Polymer Science 71:197–206
Lin CY, Liao KH, Su CF, Kuo CH, Hsieh KH (2007) Smart temperature-controlled water vapor permeable polyurethane film. Journal of Membrane Science 299:91–96
Mangaraj S, Goswami TK, Mahajan PV (2009) Applications of plastic films for modified atmosphere packaging of fruits and vegetables: A Review. Food Eng Rev 1:133–158
Mondal S, Hu JL (2006) Structural characterization and mass transfer properties of nonporous segmented polyurethane membrane: influence of hydrophilic and carboxylic group. Journal of Membrane Science 274:219–226
Peterlin A (1985) The concentration dependence of the diffusion and permeability in a homogeneous membrane. Colloid and Polymer Science 263:35–41
Robertson GL (2012) Food packaging: principles and practice. CRC Press, Boca Raton
Sadeghi M, Semsarzadeh MA, Barikani M, Ghalei B (2010) The effect of urethane and urea content on the gas permeation properties of poly (urethane-urea) membranes. Journal of Membrane Science 354:40–47
Sandhya (2010) Modified atmosphere packaging of fresh produce: current status and future needs. LWT-Food Science and Technology 43:381–392
Sangerlaub S, Schmid M, Muller K (2018a) Comparison of water vapour transmission rates of monolayer films determined by water vapour sorption and permeation experiments. Food Packaging Shelf 17:80–84
Sangerlaub S, Seibel K, Miesbauer O, Pant A, Kiese S, Rodler N, Schmid M, Muller K (2018b) Functional properties of foamed and/or stretched polypropylene-films containing sodium chloride particles for humidity regulation. Polym Test 65:339–351
Sangerlaub S, Kucukpinar E, Muller K (2019) Desiccant films made of low-density polyethylene with dispersed silica gel water vapor absorption, permeability (H2O, N-2, O-2, CO2), and mechanical properties. Materials 12
Schmid M, Sangerlaub S, Wege L, Stabler A (2014) Properties of transglutaminase crosslinked whey protein isolate coatings and cast films. Packag Technol Sci 27:799–817
Schneider NS, Dusablon LV, Spano LA, Hopfenbe H, Votta F (1968) Sorption and diffusion of water in a rubbery polyurethane. Journal of Applied Polymer Science 12:527
Seo J, Han H (2001) Water diffusion studies in polyimide thin films. Journal of applied polymer science 82:731–737
Tano K, Oule MK, Doyon G, Lencki RW, Arul J (2007) Comparative evaluation of the effect of storage temperature fluctuation on modified atmosphere packages of selected fruit and vegetables. Postharvest Biol Tec 46:212–221
Togkalidou T, Karoglou M, Bakolas A, Giakoumaki A, Moropoulou A (2013) Correlation of water vapor permeability with microstructure characteristics of building materials using robust chemometrics. Transport Porous Med 99:273–295
Turan D, Gunes G, Guner FS (2016) Synthesis, characterization and O-2 permeability of shape memory polyurethane films for fresh produce packaging. Packag Technol Sci 29:415–427
Turan D, Sangerlaub S, Stramm C, Gunes G (2017) Gas permeabilities of polyurethane films for fresh produce packaging: response of O-2 permeability to temperature and relative humidity. Polym Test 59:237–244
Wang Y, Gupta M, Schiraldi DA (2012) Oxygen permeability in thermoplastic polyurethanes. Journal of Polymer Science Part B: Polymer Physics 50:681–693
Wang HY, Zhou YM, He M, Dai ZY (2015) Effects of soft segments on the waterproof of anionic waterborne polyurethane. Colloid and Polymer Science 293:875–881
Williams JL, Hopfenbe H, Stannett V (1969) Water transport and clustering in poly(vinyl chloride), poly(oxymethylene), and other polymers. J Macromol Sci Phys B 3:711–&
Yilgor E, Burgaz E, Yurtsever E, Yilgor I (2000) Comparison of hydrogen bonding in polydimethylsiloxane and polyether based urethane and urea copolymers. Polymer 41:849–857
Zhou H, Shi HH, Fan HJ, Zhou J, Yuan JX (2009) Thermo-sensitive polyurethane membrane with controllable water vapor permeation for food packaging. Macromol Res 17:528–532
Acknowledgments
The author gratefully acknowledges the Fraunhofer Institute for Process Engineering and Packaging and the German Academic Exchange Service (DAAD) for funds and support. Special thanks are due to Sven Saengerlaub and Marius Jesdinszki for their outstanding support. Furthermore, I thank Oliver Miesbauer for fruitful discussions and F. Seniha Güner for providing access to her laboratory equipment in order to synthesize the polymers. The author would also like to thank Muhammed Bekin and ITU NANO and TEMAG laboratories for their contribution by performing SEM analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Turan, D. Water Vapor Transport Properties of Polyurethane Films for Packaging of Respiring Foods. Food Eng Rev 13, 54–65 (2021). https://doi.org/10.1007/s12393-019-09205-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12393-019-09205-z