
Abstract
Cancer diagnosis and therapeutics have been traditionally based on pathologic classification at the organ of origin. The availability of an unprecedented amount of clinical and biologic data provides a unique window of opportunity for the development of new drugs. What was once treated as a homogeneous disease with a one-size-fits-all approach was shown to be a rather heterogeneous condition, with multiple targetable mutations that can vary during the course of the disease. Clinical trial designs have had to adapt to the exponential growth of targetable mechanisms and new agents, with ensuing challenges that are closer to those experienced with rare diseases and orphan medicines. To face these problems, precision/enrichment and other novel trial designs have been developed, and the concept of histology-agnostic targeted therapeutic agents has emerged. Patients are selected for a specific agent based on specific genomic or molecular alterations, with the same compound used to potentially treat a multiplicity of cancers, granted that the actionable driver alteration is present. There are currently approved drugs for such indications, but this approach has raised issues on multiple levels. This review aims to address the challenges of this new concept and provide insights into possible solutions and frameworks on how to tackle them.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cancer is a heterogeneous condition, with potential targetable alterations. |
Precision/enrichment trials and histology-agnostic targeted therapies have emerged. |
Patients are selected for therapies based on specific genomic/molecular alterations. |
The same agent is used to treat several cancers if the alteration is present. |
This new approach carries several challenges that must be addressed. |
Introduction
Cancer diagnosis and therapeutics have been traditionally based on pathologic classification and organ of origin. However, the availability of an unprecedented amount of data regarding cancer molecular biology [1], combined with high throughput screening [2] and combinatorial chemistry [3] has provided a unique window of opportunity for the development of new drugs. It is now possible to identify several targets with potential impact on cancer cells. Advances in computational power and technology have also allowed refining drug development by designing molecules complementary to the molecular target to which they interact and bind, exploiting new cancer targets, with therapeutic benefit for patients [4]. Frequently, this relies on computational modeling and the use of approaches as structure- or ligand-based design. The former is based on knowledge of the three-dimensional structure of the biologic target, to which candidate drugs that predictably bind with high affinity and selectivity are then tested. The latter relies on knowledge of other molecules that bind to the target of interest, which may help build a model of the biologic target that can be used to design new molecular entities (NME) that interact with it. This has allowed the development of a generation of cancer treatment drugs, the so-called targeted therapies. Just like conventional chemotherapy, they are used to inhibit growth or increase cell death, but instead of relying on non-specific mechanisms that ultimately result in excess toxicity, they focus on specific molecular changes, sometimes unique to particular cancers.
Advances in the molecular understanding of cancer in recent years have supported the development of several new targeted breakthrough drugs for cancer treatment. What was once treated as a homogeneous disease with a one-size-fits-all approach was shown to be a rather heterogeneous condition with potential targetable mutations that can vary during the course of the disease. Clinicians are currently making major treatment decisions early in the continuum of care based on tumor molecular analysis.
This article is a review of previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Histology-Agnostic Treatments: Promises and Pitfalls
Both the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) have granted approval for biomarker-driven and histology-agnostic (HA) targeted therapeutics [4]. The concept of HA therapies is distinct from conventional therapies and represents a paradigm shift in cancer treatment. Patients are selected for a specific agent based on specific genomic or molecular alterations rather than on their tissue of origin. Therefore, the same compound can be used to treat a multiplicity of cancers, granted that the driver genomic or molecular alteration is present, with tumor genomic and molecular signatures superseding histology in treatment decisions (highlighting the importance of precision medicine) [5]. This fueled both basic and clinical research and became a key developmental area for an increasing number of pharmaceutical companies, expanding the opportunity to improve cancer treatment. However, this new approach has raised several issues.
When targeting a specific biomarker across tumor types, complex interactions between signaling pathways may differ among specific histologies, with potential implications in response heterogeneity. BRAF V600 mutations provide the most paradigmatic example of this complexity. These mutations usually lead to constitutive activation of the mitogen-activated protein kinase (MAPK) pathway, promoting tumor growth [6]. Targeted inhibition with agents as vemurafenib was expected to provide therapeutic benefit across a range of tumors. While this was observed in melanoma, where the use of single-agent vemurafenib in BRAF-mutated patients provided an objective response rate (ORR) of 48% [7], only one partial response (< 5% of patients) was observed among 21 patients with colorectal cancer (CRC) [8]. This was later explained at the molecular level by feedback activation of the epidermal growth factor receptor (EGFR) in CRC, which rendered these tumors primarily resistant to single BRAF blockade, as opposed to melanoma, where EGFR is barely expressed [9]. This finding led to the concomitant use of EGFR and BRAF inhibition in CRC patients to circumvent this molecular pitfall [10] and highlighted the relevance of the histologic context in some diseases.
Preliminary results of the NCI-MATCH trial represented another case study. In this complex HA multi-arm trial, patients selected for phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA)-activating mutations regardless of histology were treated with the PI3K inhibitor taselisib. However, the PI3K blockade did not translate into objective response or benefit [11]. Also BRCA1 and 2 mutations have been identified as unreliable HA biomarkers for the use of poly ADP-ribose polymerase (PARP) inhibitors [12]. Overall, this raises the question of how to interpret next-generation sequencing (NGS) results and adequately select patients when molecular alterations are identified.
Challenges in Building Evidence for HA Treatments
When considering these new approaches, it should be considered that clinical trial evidence has classically been the cornerstone of benefit-risk assessment in the regulatory approval of new therapies and the health technology assessment. However, as the understanding of the disease and new potential drugs increases, challenges to the traditional drug evaluation and approval paradigm arise. Clinical trial designs have had to adapt to the exponential growth of targetable mechanisms and new agents [13]. Given the increasing number of potential treatment combinations, it has become harder to design trials for every single disease, target, biomarker, or agent. With the emerging concept of tumor heterogeneity and the idea that molecular phenotypes can be exploited through targeted therapy and potentially be response-predictive, biomarkers have gained increasing relevance to select patients for trials of new therapies [14]. Unfortunately, this has also narrowed target populations, with ensuing challenges close to those experienced in the setting of rare diseases and orphan medicines.
To overcome this hurdle, novel precision/enrichment trial designs have been developed (Table 1) [13, 15,16,17]. Umbrella and basket trial designs are the most common and seem to fit well to targeted agents, avoiding exposure of biomarker-negative patients to treatments from which they would most likely not benefit. Umbrella trials typically focus on a single tumor type and enroll parallel marker-driven cohorts, thus harboring multiple enrichment designs within the same protocol. This facilitates screening of a large number of patients for multiple biomarkers and recruitment of low-prevalence phenotypes. In contrast, basket trials focus on the molecular alteration, enrolling patients based on biomarker status irrespective of tumor type or histology. These mostly non-randomized trials allow flexibility through the inbuilt possibility of continuously opening or closing arms based on preliminary efficacy data of the different cohorts through master protocols and platform trials. Additionally, this trial design also enables the inclusion of very rare cancer types, otherwise difficult to include in trials with classic randomized designs.
While these precision trial designs enable rapid evolution of knowledge regarding investigational targets and drugs, their flexible and adaptive nature has the drawback of significantly increasing workload and operational burden, with ultimately only a few arms being successful [18, 19]. Moreover, disparities have been observed in response across different tumors, and the regulatory approval pathway for agents investigated through these approaches is yet to be determined [20]. This represents a challenge for healthcare systems and regulatory agencies.
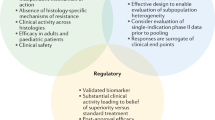
Several methodologic questions must be addressed when designing HA trials (Fig. 1), and this currently represents one of the major issues faced by regulators for HA drug approval. Molecularly targeted therapies may not achieve the expected efficacy levels across all tumor histologies harboring a specific molecular alteration. From a regulatory standpoint, the number of cancer types included in these studies and in those including common and rare tumors is of utmost importance for early HA drug approval. Regulatory agencies may have to consider different drug approval frameworks and standards to deal with the uncertainty that comes with using HA trial designs in clinical development [18, 21]. Uncertainties stem from the absence of traditional randomization, inclusion of small heterogeneous populations, and limited surrogate-outcome data regarding prognosis and standard of care. Approvals based on this level of evidence presume a provisional favorable risk-benefit assessment that may require additional data from expanded and more informative trials and real-world evidence to maintain the HA indication. How many tumor types should be included to enable HA approval and how the emergence of post-approval negative data should impact the HA indication remain unanswered.

Challenges in designing, conducting, and analyzing HA trials
Summary of HA Drugs Currently Approved and in the Pipeline
As of early 2022, four molecules have been approved by the FDA with a HA indication: pembrolizumab, for tumors with high microsatellite instability (MSI-H)/mismatch repair deficiency (dMMR) and with high tumor mutational burden (TMB-H), dostarlimab, for tumors with MSI-H/dMMR, and entrectinib and larotrectinib, for tumors harboring neurotrophic tyrosine receptor kinase (NTRK) fusions. EMA only approved pembrolizumab for MSI-H tumors and entrectinib and larotrectinib for tumors with NTRK fusions (Table 2).
Pembrolizumab
Pembrolizumab is a monoclonal antibody targeting the programmed cell death protein 1 (PD‐1). Interaction of this receptor with its ligands (PD-L1/PD-L2) downregulates T-cell function and is crucial in keeping the balance between T-cell activation and immune-mediated tissue damage. Therefore, it was postulated that blocking this receptor would enhance T-cell activation and lead to immune-mediated tumor response [22]. This approach led to highly favorable results in several tumor types, with non-small cell lung cancer (NSCLC) and melanoma leading the way with practice-changing data [23, 24].
Tumor mutational burden (TMB) has been investigated as a biomarker of immune checkpoint inhibitor (ICI) response. The rational underlying this hypothesis was that missense mutations generate neo-antigens that, in turn, increase immune system recognition [25]. MSI-H and dMMR have been traditionally considered biomarkers of high neoantigen burden [26], leading to increased sensitivity to immune checkpoint inhibition. This was explored in several trials investigating the use of pembrolizumab in patients with MSI-H tumors. In a first pivotal trial, results were very promising. The study included 11 dMMR and 21 mismatch repair (MMR)-proficient CRC patients and an additional cohort of 9 dMMR non-CRC patients and served as a proof of concept. ORR was 40% in the dMMR CRC cohort, 0% in the MMR-proficient CRC cohort, and a staggering 71% in the dMMR non-CRC cohort [27]. FDA approval was ultimately based on the pooled analysis of five independent clinical trials of the KEYNOTE clinical development program (KEYNOTE-012, KEYNOTE 016, KEYNOTE-028, KEYNOTE-158, and KEYNOTE-164), which included 149 patients and 15 different histologies and achieved a pooled ORR of roughly 40% (7.4% of complete responses [CR]) and significant response durability (78% at 6 months) with pembrolizumab treatment, with no additional safety signs to the previously reported in pivotal trials. This led to the first FDA HA approval in May 2017 for pembrolizumab in the treatment of adult and pediatric patients with unresectable or metastatic MSI-H or dMMR solid tumors progressing after prior treatment and with no satisfactory alternative treatment options [28].
On June 16, 2020, the FDA additionally granted pembrolizumab accelerated approval for the treatment of adult and pediatric patients with unresectable or metastatic non-CRC and high TMB (TMB-H, defined as ≥ 10 mutations/megabase) as per the FDA-approved test FoundationOneCDX progressing after prior treatment and with no valid treatment alternatives. This approval was based on the retrospective analysis of 10 TMB-H cohorts in the KEYNOTE-158 trial. In a total of 102 patients with TMB-H tumors, ORR was 29% (4% CR) and responses were significantly sustained (57% at 12 months) [29].
EMA ultimately granted approval for the use of pembrolizumab to treat dMMR cancers in March 2022, although it should be noted that the TMB-high indication was only granted by the FDA. With positive review still lacking from EMA, the use of this drug in Europe for TMB-high tumors remains off-label.
Dostarlimab
Similarly to pembrolizumab, dostarlimab is an IgG4-k humanized monoclonal antibody that binds with high affinity to PD-1, resulting in inhibition of PD-L1 and PD-L2 binding.
Dostarlimab efficacy was assessed in the non-randomized, multicenter, open-label, multi-cohort GARNET trial in 209 patients with dMMR recurrent or advanced solid tumors who progressed following systemic therapy and had no satisfactory alternative treatment. The ORR in this cohort was 41.6% (95% CI: 34.9–48.6), with 9.1% CR. The median DoR reached 34.7 months, with an impressive 95.4% of patients having a sustained response for > 6 months.
Based on these data, the FDA granted accelerated approval to dostarlimab for adult patients with dMMR recurrent or advanced solid tumors who have progressed on or following prior treatment and lack satisfactory alternative treatment options. Notably, the FDA also approved the VENTANA MMR RxDX Panel as a companion diagnostic device to select patients with dMMR solid tumors for treatment with this agent [30].
Neurotrophic Tyrosine Receptor Kinase Inhibitors
NTRK genes (NTRK1, NTRK2, and NTRK3) encode the tropomyosin-related kinase (TRK) glycoproteins TRKA, TRKB, and TRKC, which are membrane receptors with a decisive function in central and peripheral nervous system physiology. These receptors are activated by ligands of the neurotrophin family, as NGF, BDNF, NT-3, and NT-4. Binding of these ligands ultimately results in receptor dimerization and kinase domain phosphorylation, leading to cell proliferation and survival by activating downstream intracellular signaling pathways [31].
Despite the significant number of genomic alterations in NTRK genes, NTRK fusions are currently the only clinically targetable ones [32]. They were first identified as an oncogenic phenotype in 1986 and later shown to retain this feature independently of tissue of origin, with these tumors presenting very few secondary mutations compared to tumors without NTRK fusions [33, 34]. Regarding incidence, cancers can be grouped into two general categories according to the frequency at which these fusions are detected. In the first category, rare cancer types are highly enriched in NTRK fusions. For example, the ETV6-NTRK3 fusion is considered practically pathognomonic in secretory breast carcinoma, mammary analog secretory carcinoma (MASC), congenital mesoblastic nephroma (cellular or mixed subtypes), and infantile fibrosarcoma, with a prevalence > 90%. In the second category, NTRK fusions are found at lower frequencies in more common tumors. Cancers such as papillary thyroid cancer, Spitzoid neoplasms, and gastrointestinal stromal tumors (GIST) lacking the canonical KIT, PDGFRA, or RAS alterations have been shown to harbor NTRK fusions with a frequency of 5—25% of cases. In the remaining tumors, the incidence of these alterations is < 5% (predominantly < 1%) [31, 35].
By preserving an intact TRK domain that is key to oncogenic activity, NTRK fusions are amenable to selective targeting and drug development. Two agents are currently approved for NTRK targeting: larotrectinib and entrectinib.
Larotrectinib
Larotrectinib is an oral and highly selective pan-TRK inhibitor and was the first TRK-specific inhibitor. The first data published on this agent go back to 2015 [36]. Clinical development was accomplished through collaborative work between regulators and researchers to design a trial that would allow to correctly identify activity signals. The predetermined efficacy endpoint of ORR was reported for the first time in a pooled analysis of three clinical trials including 55 pediatric and adult patients [37]. The investigator-assessed ORR was 80% (16% CR), with a median time to response of 1.8 months and 71% of responses ongoing at 12 months [38]. Regarding tolerability and safety, only 5% of patients presented with grade 3 or higher treatment-related adverse events (TRAE).
Based on these data, in November 2018 the FDA granted accelerated approval to larotrectinib for the treatment of adult and pediatric patients with tumors harboring NTRK fusions that are metastatic or unamenable to surgical resection. However, this was contingent on post-marketing requirements of response assessment across other tumors. Accordingly, a supplementary dataset of an additional 67 patients (122 patients in total) was later presented at the European Society of Clinical Oncology (ESMO) Congress 2018 [39], confirming high ORR (81%) across studies, tissue types, and age ranges. These data were further expanded and presented at the ESMO Congress 2019 through evaluation of 98 patients (153 patients in total), showing an ORR of 79% (16% CR), a median progression-free survival (PFS) of 28 months, and a median duration of response (DoR) of 35 months [38]. Larotrectinib was subsequently approved by EMA in September 2019, with a similar indication.
Entrectinib
Entrectinib is also a potent TRKA, TRKB, and TRKC inhibitor, but has a broader action spectrum, with demonstrated activity against other kinases, such as c-ros oncogene 1 (ROS1) and anaplastic lymphoma kinase (ALK).
A pooled analysis of three clinical trials investigating entrectinib (ALKA-372-001, STARTRK-1, and STARTRK-2) included adult patients harboring NTRK fusions, in a total of 10 tumor types and 19 histologies [40]. Fifty-four subjects were evaluated, with a median follow-up of 15.5 months. An ORR of 57% (7% CR) was reported, with most responses occurring within the first two treatment cycles and a median DoR of 10 months (29) [40]. Data submitted for regulatory approval were based on an expanded analysis with a median follow-up of 14.2 months including 74 patients, which reported an ORR of 63.6%, median DoR of 12.0 months, and median PFS of 11.2 months [41]. Regarding safety, most adverse events (83%) were grade 1 or 2 and reversible with drug discontinuation or dose reductions [40, 42]. Data from 35 children and adolescent patients included in the STARTRK-NG study were updated at the American Society of Clinical Oncology (ASCO) Annual Meeting 2020, showing an ORR of 76% and a median DoR not yet reached [43].
Similarly to larotrectinib, entrectinib was granted accelerated approval by the FDA in August 2019, with the same HA indication but a restriction regarding patient age (over 12 years only), followed by EMA approval in August 2020 [44]. After pembrolizumab having shown that a drug could be simultaneously approved for HA and non-HA indications, entrectinib corroborated this fact.
Besides inhibiting TRKA, TRKB, and TRKC, it is also a potent ROS1 inhibitor and was approved for ROS1-positive NSCLC. This was based on the results of a pooled analysis of a subgroup of 161 patients with ROS1-positive metastatic NSCLC enrolled in three multicenter single-arm, open-label clinical trials (ALKA, STARTRK-1, and STARTRK-2) [45]. In this ROS1-positive NSCLC population, efficacy-evaluable patients with ≥ 12 months of follow-up (n = 94) displayed an ORR of 73.4%, a median DoR of 16.5 months, and a median PFS of 16.8 months [45].
Pipeline of HA Drugs
The first successful HA approvals marked a paradigm shift in the development of cancer drugs, highlighting the importance of novel trial designs such as basket trials. Designed to include patients based on molecular or genomic features irrespective of tumor type, these trials can include rare cancers, usually underrepresented in clinical trials. This approach is extremely useful in cases where different molecular phenotypes have been identified, narrowing the target population per basket in a parallel setting to that of rare diseases. Basket trials typically have small sample size and can provide information on early signs of drug activity, facilitating go/no-go decisions regarding specific cohorts by focusing on endpoints as response rate [18]. With the groundbreaking success of the previously described molecules, many others are currently in investigation in trials with similar design.
RAS mutations are frequent alterations in cancer, with patients typically displaying high levels of the MAPK pathway due to constitutive activation of RAS protein kinases. Although traditionally deemed untargetable, small molecule inhibitors specifically for the KRAS G12C mutant have been developed (AMG510 and MRTX849). In ASCO Annual Meeting 2019, results from the CodeBreaK100 phase I trial of AMG510 have proven favorable in NSCLC (ORR 50%; disease control rate [DCR] 100%) [46] and modest in gastrointestinal cancers (ORR 7.1%; DCR 76.2%) [47]. Although promising, evidence is still lacking regarding tissue-independent activity.
For patients with RET alterations, the orally available selective RET inhibitor selpercatinib [48] was studied in the LIBRETTO-001 phase II basket trial. A total of 55 patients were enrolled, with an ORR of 69% and mostly grade 1–2 adverse events reported for patients with RET fusion-positive tumors [49]. Based on these results, in September 2018 the FDA granted selpercatinib breakthrough therapy designation for patients with RET fusion-positive NSCLC, papillary thyroid carcinoma, and RET-mutated medullary thyroid carcinoma (MTC), the tumor types of most patients enrolled in the LIBRETTO-001 trial (only one patient had pancreatic cancer). On December 2020, the Committee for Medicinal Products for Human Use (CHMP) also granted marketing authorization to selpercatinib for the treatment of cancers displaying RET alterations: RET-fusion positive NSCLC, RET-fusion positive thyroid cancer, and RET-mutant MTC.
Neuregulin 1 (NRG1) is a growth factor that binds to human epidermal growth factor 3 (HER3), inducing heterodimerization of HER3-HER2 and subsequent MAPK signaling. Despite being a very rare alteration, targeting NRG1 fusions seems to be a potentially useful approach, although still in a very preliminary phase [50]. The FDA has recently granted fast track designation to zenocutuzumab for the treatment of patients with metastatic solid tumors harboring NRG1 gene fusions who progressed following standard-of-care therapy. This compound is a bispecific antibody that binds to HER2 receptors, blocking the interaction of HER3 with NRG1, thus having the potential to be effective in NRG1 fusion-positive cancer setting. Data from a proof-of-concept clinical study presented at the 2019 AACR-NCI-EORTC Conference showed that three patients with this fusion experienced significant tumor shrinkage and symptom improvement with zenocutuzumab [51].
Several other molecules are being studied in basket trials in HA setting, including Debio147 for fibroblast growth factor receptor (FGFR) alterations (NCT01948297 NCT03834220), entrectinib and repotrectinib for ALK or ROS-1 fusions (NCT02568267, NCT02650401, NCT03375437, NCT03093116, NCT04094610, NCT02568267, NCT02650401, NCT03375437, NCT03093116, NCT04094610), PLX8394 for BRAF mutations (NCT02428712), and atezolizumab plus rucaparib for DNA repair-deficient tumors (NCT04276376]), showing that this is an emerging approach that will surely be a paradigm changer in cancer treatment.
Selection of Patients for Targeted Therapies
Patient selection for HA therapies using biomarkers is a crucial aspect of the daily clinical practice, with key implications from the regulatory standpoint, namely in trial eligibility and indication requirements. Except for pembrolizumab in TMB-high tumors and dostarlimab in dMMR tumors, no HA drug approval has been granted based on the need for a specific companion test. The cost of genomic testing has significantly decreased over the past years and incorporating NGS into clinical care is not only useful and practical, but also potentially cost sparing in specific settings. When making such decisions, it is important to confront the amount and value of data provided by NGS with the cost, efficiency, and amount of tissue necessary for performing the test. With this in mind, NGS panels can be cost-effective, depending on the number of decision-driver molecular alterations in a specific disease and the number of market-approved molecules and available trials.
Based on this paradigm switch, international treatment guidelines and tools focusing on patient selection for targeted therapies have emerged and are slowly altering the clinical practice globally. One such example came from a collaborative project initiated by the ESMO Translational Research and Precision Medicine Working Group, which provided a systematic framework to rank molecular targets based on available evidence supporting their value as clinical targets, i.e., their actionability. The ranking scale, designated ESMO Scale for Clinical Actionability of molecular Targets (ESCAT), was published in 2018 in Annals of Oncology and classifies genomic alterations according to their relevance as markers for selecting patients for specific drugs based on the underlying strength of clinical evidence (Tier I-V, Table 3) [52]. As more and more patients are being offered multigene sequencing, these initiatives will facilitate the discussion of results among clinicians and ultimately with patients themselves.
According to a study presented by Patricia Romano at the ESMO Virtual Congress 2020, stratifying genomic alterations according to the ESCAT scale is useful to base treatment decisions in the clinical practice [53]. The Molecular Tumor Board (MTB) at Institute Gustave Roussy has evaluated genomic alterations in multigene panels since 2018. In the work by Romano and colleagues, NGS, whole-exome sequencing, and RNA-sequencing data from 27 different tumor types of 387 patients participating in the MOSCATO and MATCHR studies were prospectively assessed by the MTB according to ESCAT tiers. The authors reported clinically informative results for 32% of patients, leading to treatment selection in 20%, and concluded that ESCAT classification of genomic alterations through MTB assessment is feasible in clinical practice and helps adjust treatment in both standard-of-care and investigational settings [53].
Despite these data, both insurance payers and healthcare regulators are often unwilling to cover such tests, significantly restricting the number of patients benefiting from genomic testing. This will subsequently impair patient recruitment for basket trials and patient selection for market-approved molecules, as most of these treatments do not cover screening tests. Another crucial point is that test costs must be considered when reimbursing HA approvals.
What is Next?
The emerging concept of HA therapies marks the beginning of a new era in which patients are selected based on specific genomic or molecular alterations. However, as highlighted in this manuscript, this innovative approach comes with singular challenges and obstacles that must be addressed to be able to make optimal decisions at both a clinical and regulatory level.
In the real-world setting, the availability of several diagnostic assays poses challenges for clinical decision-makers. These assays are typically not standardized or validated, requiring expertise in their selection and result interpretation, for both clinical trial inclusion and real-world decisions. Enabling access to experienced molecular tumor boards and, most importantly, to initiatives such as ESMO-ESCAT might provide guidance to less experienced institutions and is key for the future development of this area. Advisory from scientific organizations will also be key in identifying which tests should be reimbursed by health technology evaluation committees of different countries, as most are neither reimbursed nor easily accessible as of today.
In the clinical development setting, precision/enrichment designs are potentially useful but narrow down the target populations to the level of rare diseases, potentially missing existing heterogeneity in treatment effect caused by factors as complex histology-dependent interactions in signaling pathways (e.g., BRAF-mutated colorectal cancer), prior treatment exposure, demographics, or tumor microenvironment (stromal and immune). Additionally, precision/enrichment designs often include ORR as primary endpoint, but the effect observed with surrogate markers of typically meaningfully endpoints in classical trials, as OS and PFS, should be interpreted with caution. Moreover, null hypotheses in these trials may require adaptation to provide the right perspective of results of a NME compared to the expected standard of care in studied indications. These limitations should be acknowledged when developing new agents, accepting that not all molecules may be suitable for HA development.
Regulatory agencies also face several challenges when evaluating HA drugs as the amount of uncertainty surrounding the approval process is substantial. Traditional randomized controlled trials (RCTs) are absent, small heterogeneous populations are typically included in trials, and there are limited surrogate-outcome data regarding prognosis and standard of care. With this in mind, approvals not based on RCT data should be conditional and rely on more informative clinical trials and, importantly, well-structured real-world data collection programs. This will be key to making a more precise risk-benefit assessment and sparing patients from potential exposure to ineffective treatments.
Customized approach to individual patient management—the so-called “precision medicine”—is currently a reality, and HA therapies represent an emerging concept gaining momentum. Collaboration between clinicians, pharmaceutical industry, patients, and regulatory agencies is crucial to address as early as possible the challenges and questions associated with this new concept and ensure that patients’ access to these drugs is done in an expedite and safe way.
References
Meldrum C, Doyle MA, Tothill RW. Next-generation sequencing for cancer diagnostics: a practical perspective. Clin Biochem Rev. 2011;32(4):177–95.
Mayr LM, Fuerst P. The future of high-throughput screening. J Biomol Screen. 2008;13(6):443–8.
Mario Geysen H, Schoenen F, Wagner D, Wagner R. Combinatorial compound libraries for drug discovery: an ongoing challenge. Nat Rev Drug Discov. 2003;2(3):222–30.
Cui W, Aouidate A, Wang S, et al. Discovering Anti-cancer drugs via computational methods. Front Pharmacol. 2020. https://doi.org/10.3389/fphar.2020.00733.
Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 2020;86: 102019.
Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–67.
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–8.
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3.
Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–43.
Krop IE, Jegede O, Grilley-Olson JE, et al. Results from molecular analysis for therapy choice (MATCH) arm I: taselisib for PIK3CA-mutated tumors. J Clin Oncol. 2018;36(15_suppl):101.
Curtin NJ, Drew Y, Sharma-Saha S. Why BRCA mutations are not tumour-agnostic biomarkers for PARP inhibitor therapy. Nat Rev Clin Oncol. 2019;16(12):725–6.
Hirakawa A, Asano J, Sato H, Teramukai S. Master protocol trials in oncology: review and new trial designs. Contemp Clin Trials Commun. 2018;12:1–8.
Horgan D, Ciliberto G, Conte P, et al. Bringing onco-innovation to Europe’s healthcare systems: the potential of biomarker testing, real world evidence, tumour agnostic therapies to empower personalised medicine. Cancers (Basel). 2021;13(3):583.
Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377(1):62–70.
Cunanan KM, Gonen M, Shen R, et al. Basket trials in oncology: a trade-off between complexity and efficiency. J Clin Oncol. 2017;35(3):271–3.
Tao JJ, Schram AM, Hyman DM. Basket studies: redefining clinical trials in the era of genome-driven oncology. Annu Rev Med. 2018;69(1):319–31.
Dittrich C. Basket trials: from tumour gnostic to tumour agnostic drug development. Cancer Treat Rev. 2020;90: 102082.
Lacombe D, Burock S, Bogaerts J, et al. The dream and reality of histology agnostic cancer clinical trials. Mol Oncol. 2014;8(6):1057–63.
Rodes Sanchez M, Henderson N, Steuten L. Bridging the gap: pathways for regulatory and health technology assessment of histology independent therapies. Consult. Reports 002290, Off. Heal. Econ. 2020.
Hierro C, Matos I, Martin-Liberal J, et al. Agnostic-histology approval of new drugs in oncology: are we already there? Clin Cancer Res. 2019;25(11):3210–9.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33.
Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908–18.
Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68(3):889–92.
Eshleman JR, Lang EZ, Bowerfind GK, et al. Increased mutation rate at the hprt locus accompanies microsatellite instability in colon cancer. Oncogene. 1995;10(1):33–7.
Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019;25(13):3753–8.
Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–65.
U.S. Food and Drug Administration. FDA grants accelerated approval to dostarlimab-gxly for dMMR advanced solid tumors. 2022. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors. Accessed 02 Oct 2022.
Klein R, Jing S, Nanduri V, et al. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65(1):189–97.
Kheder ES, Hong DS. Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res. 2018;24(23):5807–14.
Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319(6056):743–8.
Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1(2): e000023.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–47.
Doebele RC, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015;5(10):1049–57.
Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–9.
Hyman DM, van Tilburg CM, Albert CM, et al. Durability of response with larotrectinib in adult and pediatric patients with TRK fusion cancer. Ann Oncol. 2019;30:v162–3.
Lassen UN, Albert CM, Kummar S, et al. Larotrectinib efficacy and safety in TRK fusion cancer: an expanded clinical dataset showing consistency in an age and tumor agnostic approach. Ann Oncol. 2018;29:vii133.
Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):271–82.
Rolfo CD, De Braud FG, Doebele RC, et al. Efficacy and safety of entrectinib in patients (pts) with NTRK -fusion positive (NTRK-fp) solid tumors: an updated integrated analysis. J Clin Oncol. 2020;38(15_suppl):3605–3605.
Drilon A, Siena S, Ou S-HI, et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–9.
Desai AV, Robinson GW, Basu EM, et al. Updated entrectinib data in children and adolescents with recurrent or refractory solid tumors, including primary CNS tumors. J Clin Oncol. 2020;38(15_suppl):107–107.
Delgado J, Pean E, Melchiorri D, et al. The European Medicines Agency review of entrectinib for the treatment of adult or paediatric patients with solid tumours who have a neurotrophic tyrosine receptor kinase gene fusions and adult patients with non-small-cell lung cancer harbouring ROS1 rearr. ESMO Open. 2021;6(2): 100087.
Dziadziuszko R, Krebs MG, De Braud F, et al. Updated integrated analysis of the efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion-positive non-small-cell lung cancer. J Clin Oncol. 2021;39(11):1253–63.
Fakih M, O’Neil B, Price TJ, et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J Clin Oncol. 2019;37(15_suppl):3003–3003.
Strickler JH, Fakih M, Price TJ, et al. 83MO AMG 510, a novel small molecule inhibitor of KRAS(G12C), for patients (pts) with advanced gastrointestinal (GI) cancers: results from the CodeBreaK100 phase I trial. Ann Oncol. 2020;31:S1274–5.
Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29(8):1869–76.
Drilon AE, Subbiah V, Oxnard GR, et al. A phase 1 study of LOXO-292, a potent and highly selective RET inhibitor, in patients with RET-altered cancers. J Clin Oncol. 2018;36(15_suppl):102–102.
Jonna S, Feldman RA, Swensen J, et al. Detection of NRG1 gene fusions in solid tumors. Clin Cancer Res. 2019;25(16):4966–72.
MCLA-128 Fights NRG1 Fusion–Positive Cancers. Cancer Discov. 2019; 9(12):1636.2–1636.
Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol. 2018;29(9):1895–902.
Martin Romano P, Mezquita L, Lacroix L, et al. 1930O Genomic alterations in solid tumours according to ESMO scale for clinical actionability of molecular targets (ESCAT). Ann Oncol. 2020;31:S1092–3.
Acknowledgements
Funding
This work, including the journal’s Rapid Service and Open Access fees, was supported by Roche Farmacêutica Química Lda.
Medical Writing and Editorial Assistance
Editorial assistance in the preparation of this article was provided by Dr. Joana Cavaco-Silva (jo.cvsilva@gmail.com).
Author Contributions
Conceptualization: André Mansinho, Ricardo Miguel Fernandes, António Vaz Carneiro. Investigation: André Mansinho, Ricardo Miguel Fernandes, António Vaz Carneiro. Methodology: André Mansinho, Ricardo Miguel Fernandes, António Vaz Carneiro. Supervision: André Mansinho, Ricardo Miguel Fernandes, António Vaz Carneiro. Roles/writing—original draft: André Mansinho, Ricardo Miguel Fernandes, António Vaz Carneiro. Writing—review and editing: André Mansinho, Ricardo Miguel Fernandes, António Vaz Carneiro.
Disclosures
All named authors confirm that they have no conflicts of interest to declare.
Compliance with Ethics Guidelines
This article is a review of previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mansinho, A., Fernandes, R.M. & Carneiro, A.V. Histology-Agnostic Drugs: A Paradigm Shift—A Narrative Review. Adv Ther 40, 1379–1392 (2023). https://doi.org/10.1007/s12325-022-02362-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-022-02362-4