
Abstract
Introduction
Tazemetostat is an enhancer of zeste homolog 2 (EZH2) inhibitor recommended for patients with relapsed/refractory (R/R) follicular lymphoma (FL) after demonstrating single-agent, antitumor activity in patients with wild-type or mutant EZH2. The phosphoinositide 3-kinase (PI3K) inhibitors idelalisib, copanlisib, umbralisib and (formerly) duvelisib are indicated for third-line, fourth-line, and later (3L/4L+) treatment of R/R FL. The objective of this analysis was to provide an indirect treatment comparison of tazemetostat with each PI3K inhibitor for 3L/4L+ R/R FL treatment.
Methods
A systematic literature review was conducted to identify trials for idelalisib (DELTA), duvelisib (DYNAMO), copanlisib (CHRONOS-1 Part B), and umbralisib (UNITY-NHL) in 3L+ R/R FL. Matching-adjusted indirect comparisons were conducted by weighting tazemetostat individual patient data with available baseline characteristics from each comparator trial: age, Eastern Cooperative Oncology Group performance status, disease stage, histology, prior treatment lines, prior stem cell therapy, progression within 24 months, and refractory status to last therapy. Only the tazemetostat trial included patients with grade 3b or transformed FL, or recorded EZH2 mutation status. Primary safety outcomes included risk of grade ≥ 3 treatment-emergent adverse events (TEAEs); primary efficacy outcomes included objective response rate (ORR).
Results
Matched patients treated with tazemetostat had lower relative risk (RR) for all grouped safety outcomes, including any grade ≥ 3 TEAEs, compared with idelalisib (RR = 0.45), duvelisib (RR = 0.35), copanlisib (RR = 0.37), and umbralisib (RR = 0.65; all, p < 0.01), any serious TEAE, and any TEAE leading to dose reduction, drug discontinuation, or interruption. The ORR was not significantly different for tazemetostat versus other treatments (idelalisib 43% vs 56%, p = 0.16; duvelisib 48% vs 47%, p = 0.91; copanlisib 49% vs 61%, p = 0.11; and umbralisib 57% vs 47%, p = 0.10).
Conclusions
In this statistically adjusted comparison, tazemetostat was associated with lower RR for safety outcomes versus idelalisib, duvelisib, copanlisib, and umbralisib, while achieving similar efficacy outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tazemetostat, a first-in-class, oral enhancer of zeste homolog 2 (EZH2) inhibitor, was recently approved by the US Food and Drug Administration in patients with relapsed/refractory (R/R) follicular lymphoma (FL) after demonstrating single-agent, antitumor activity in patients with wild-type (WT) or mutant (MT) EZH2. |
The objective of this analysis was to provide a matching-adjusted indirect treatment comparison of tazemetostat against the phosphoinositide 3-kinase (PI3K) inhibitors idelalisib, duvelisib, copanlisib, and umbralisib for the third-line, fourth-line, and later treatment of R/R FL. |
Primary safety outcomes included risk of grade ≥ 3 treatment-emergent adverse events (TEAEs), while primary efficacy outcomes included objective response rate (ORR). |
Matched patients treated with tazemetostat had lower relative risk (RR) for all grouped safety outcomes, including any grade ≥ 3 TEAEs, any serious TEAE, and any TEAE leading to dose reduction, drug discontinuation, or interruption. The ORR was not significantly different for tazemetostat versus other treatments. |
Introduction
Follicular lymphoma (FL) is a common indolent lymphoma that accounts for approximately 20% of non-Hodgkin lymphoma (NHL) and up to 70% of indolent NHL cases [1]. The Surveillance, Epidemiology, and End Results (SEER) program registry estimates that the incidence of FL in the USA from 2013 to 2017 was 2.7 per 100,000, and the reported incidence has risen since 1975, with an increase of 1.7 incident cases per 100,000 [1].
Current treatments in FL (e.g., anti-CD20-based chemoimmunotherapy regimens with rituximab) may induce durable remissions and/or slow disease progression, but treatment is not expected to be curative [1]. Thus, patients with FL eventually require retreatment following relapse, and/or they become refractory to anti-CD20 therapy. However, response rates associated with anti-CD20-based therapy retreatment become lower with each successive attempt [2]. Another treatment class, phosphoinositide 3-kinase (PI3K) inhibitors, is indicated for patients with relapsed/refractory (R/R) FL who have received at least two prior systemic therapies, However, the use of these therapies has been associated with adverse reactions and serious toxicities, including hepatotoxicity, infections, severe diarrhea, colitis, pneumonitis, and intestinal perforation [3,4,5]. The PI3K/casein kinase 1 (CK1) inhibitor umbralisib, which is indicated for treatment of patients with relapsed or refractory (R/R) FL who have received at least three prior systemic therapies, appeared to show lower rates of adverse events in clinical trial, albeit with some of the same toxicity issues observed [6].
Approximately 17–29% of patients with FL are reported to have gain-of-function mutations in the EZH2 gene, an oncogenic driver of FL [7,8,9,10,11]. Enhancer of zeste homolog 2 (EZH2) signaling has been shown to sustain or promote tumor cell growth in FL and other forms of cancer [12]. In FL specifically, inhibition of EZH2 has been associated with the downregulation of oncogenic polycomb repressive complex 2 signaling activity and promotion of differentiation and apoptosis, even among patients whose tumors are not positive for gain-of-function mutations in EZH2 [9, 13, 14].
Tazemetostat (Tazverik; Epizyme, Inc.) was approved for the treatment of adult patients with R/R FL whose tumors are positive for an EZH2 mutation, as detected by a US Food and Drug Administration (FDA)-approved test, and who have received at least two prior systemic therapies; and for the treatment of adult patients with R/R FL who have no satisfactory alternative treatment options regardless of EZH2 status [15]. Tazemetostat is also listed in the National Comprehensive Cancer Network (NCCN) Guidelines for B-cell Lymphomas as a category 2A recommended treatment for patients with FL in this indication [16].
Data on the efficacy of tazemetostat compared with other treatments are needed to inform treatment decision-making by patients and health care providers. Given the absence of head-to-head trials among approved treatments in R/R FL, an indirect treatment comparison (ITC) was conducted to compare the safety and efficacy profile of tazemetostat versus agents approved for the treatment of R/R FL in the third or fourth line or later (3L/4L+), adjusting for potential differences in baseline characteristics. On the basis of the most recent NCCN guidelines, treatments specifically FDA approved for R/R FL in 3L are idelalisib and copanlisib, while duvelisib was formerly approved (all 3 are PI3K inhibitors), and umbralisib is approved in 4L; therefore, these four treatments were considered most relevant for indirect comparison with tazemetostat.
Methods
Evidence Base
A systematic literature review was conducted to identify published clinical trials to support the ITC. Overall, 514 publications were identified and screened on the basis of their titles and abstracts; of these, 20 full-text articles were reviewed, and six were selected for data extraction and inclusion in this analysis. On the basis of the literature review, four comparator clinical trials were identified for the ITC: DELTA (idelalisib) [17], DYNAMO (duvelisib) [18], CHRONOS-1 Part B (copanlisib) [19, 20], and UNITY-NHL (umbralisib) [6]. The E7438-G000-101 trial clinical study report (data on file; Epizyme, Inc.) and two published articles were reviewed for tazemetostat [21, 22]. In all of the evaluated trials, patients with R/R FL represented a subset of the enrolled NHL trial population. Where trial publication data for the R/R FL subpopulation were unavailable, supplemental targeted searches were conducted to fill in evidence gaps. For the DELTA trial, the National Institute for Health and Care Excellence (NICE) submission for idelalisib as a treatment for FL refractory to two treatments (TA604) was identified as the primary source for efficacy and safety data in place of the trial publication, as FL-specific data were available [23]. For the DYNAMO trial, the FDA submission for duvelisib was used to supplement baseline information not reported in the trial publication; however, separate FL population data were not found [24]. Follicular lymphoma-specific population baseline data were also not found for CHRONOS-1 Part B. Additional targeted searches were conducted to identify prognostic factors otherwise not available in the full trial publications, including progression of disease within 24 months of first treatment (POD24) [25]. POD24 data for idelalisib were obtained from an idelalisib-only study, where the patient population was comparable to the study population from the DELTA trial.
The identified trials were subsequently assessed for comparability in terms of study design and trial eligibility criteria. Trial designs were found to be sufficiently comparable, as detailed in Table 1. Each of the studies included in this analysis was conducted in accordance with the ethical standards of the local institutional review boards for each study site and with the Declaration of Helsinki and its later amendments or comparable ethical standards. Written informed consent was obtained from all participants before study participation.
Notable differences in trial design included the following:
-
The E7438-G000-101 trial (tazemetostat) was the only evaluated trial that identified patients’ EZH2 mutation status and reported results by mutant or wild-type EZH2 mutation status. Although EZH2 mutation was not found to be a treatment effect modifier in patients with R/R FL prior to the approval of tazemetostat (data on file; Epizyme, Inc.) [26], tazemetostat acts on EZH2 and does demonstrate differential results, depending on EZH2 status [22].
-
All trials except for the tazemetostat trial excluded grade 3b tumors. DELTA (idelalisib) and DYNAMO (duvelisib) also excluded transformed FL.
Baseline variables in the FL-specific population were available for the DELTA (idelalisib) comparison. For DYNAMO (duvelisib) and CHRONOS-1 Part B (copanlisib) baseline data were available only for the full-trial population (mixed histology) (64% and 73% of patients with FL, respectively). For UNITY-NHL (umbralisib), baseline data were available for the FL-specific population; however, safety outcomes were only reported for the full-trial population. Therefore, the full-trial population was used for the matching analysis (56% of patients with FL). Safety and efficacy outcomes evaluated corresponded to the populations with the matched baseline data.
Efficacy and Safety Parameters
The primary safety outcomes assessed in the ITC were the incidence of any grade ≥ 3 treatment-emergent adverse event (TEAE; grouped and individual); incidence of any adverse event of special interest (AESI); incidence of any treatment-emergent serious adverse event (TESAE); incidence of any TEAE leading to study drug interruption; incidence of any TEAE leading to dose reduction; and incidence of any TEAE leading to study drug discontinuation.
The primary efficacy outcome assessed was objective response rate (ORR; FL population for DELTA, and mixed histology population in line with baseline variables for DYNAMO, CHRONOS-1 Part B, and UNITY-NHL). The median duration of response (DOR) was also assessed.
A feasibility assessment for the analysis was conducted to determine if the studies were sufficiently comparable. Definitions of key safety and efficacy outcomes were found to be comparable. Among safety outcomes, all trials except DELTA explicitly specified that TEAEs were evaluated; adverse events (AEs) reported in DELTA were assumed to be equivalent to TEAEs. With respect to efficacy outcomes, response definitions were based on the 2007 International Working Group criteria for non-Hodgkin lymphoma (IWG-NHL) by Cheson et al. and were assessed by central radiographic review or an independent review committee in all trials except for UNITY-NHL, in which response definitions were assessed on the basis of the 2014 Lugano classification by Cheson et al., which was deemed comparable [27, 28]. The ORR was the primary endpoint in all trials. The median DOR was reported for the FL subgroup in the DELTA, DYNAMO, and UNITY-NHL trials only.
Follow-up duration differed across trials. Patients enrolled in the DELTA trial had a minimum follow-up time of 20 months (FL subpopulation); those who enrolled in the DYNAMO trial had a median follow-up time of 32.1 months, and patients enrolled in the CHRONOS-1 Part B trial had a median follow-up time of 6.7–31.5 months (follow-up differed by type of data: safety, 6.7 months; DOR, 16.1 months); and patients in UNITY-NHL had a median follow-up time of 21.4 months for safety outcomes, and 27.7 months for efficacy outcomes. In comparison, the median (interquartile range) follow-up time for the tazemetostat E7438-G000-101 trial was 22.2 months (12.0–26.7) and 35.9 months (32.2–39.0) for the mutant EZH2 and wild-type EZH2 cohorts, respectively [22].
The duration of exposure was roughly similar for all four comparator trials (DELTA 6.5 months [only patients with FL], DYNAMO 6.7 months, CHRONOS-1 Part B 26 weeks, UNITY-NHL 8.4 months), while the duration of exposure for the E7438-G000-101 trial was numerically higher (9.3 months).
Statistical Methods for Matching-Adjusted Indirect Comparison
Given that all comparator trials were single-arm, and individual patient data (IPD) were available for the tazemetostat E7438-G000-101 trial [22], the matching-adjusted indirect comparison (MAIC) methodology was chosen for the ITC, as described by Signorovitch et al. [29]. The MAIC approach adjusts for baseline differences in potential effect modifiers between trials by reweighting the available IPD to match the average baseline characteristics reported in any trial with aggregate data. Outcomes for each treatment are then compared between balanced trial populations. In this analysis, the MAIC approach was used to reweight IPD from the tazemetostat E7438-G000-101 trial for each of the four pairwise comparisons, so that the reweighted tazemetostat E7438-G000-101 trial population was matched to the average baseline characteristics reported for each comparator trial in turn.
Baseline characteristics were identified on the basis of availability of data, and prognostic factors associated with efficacy and safety were identified for matching and confirmed by clinical input. Within demographic factors, only age was selected as a matching variable for population adjustment (as data were available and age is expected to be prognostic). Sex was assumed not to be prognostic for the ORR in FL [30]. Geographic region was not included, as it is not typically independently associated with disease or patient characteristics, although prior treatment use may vary by region. Disease severity was measured through objective disease-related characteristics such as those described below. Race could not be included owing to a lack of data availability in the E7438-G000-101 tazemetostat trial.
The following disease characteristics were available and were selected for inclusion because they were expected to be prognostic and/or treatment effect modifiers (identified by expert clinical input): Eastern Cooperative Oncology Group performance status (ECOG PS), disease stage at diagnosis (Ann Arbor), histology (tumor grade), number of prior lines of treatment (median), prior stem cell transplantation, POD24 (previously shown to be linked to survival [31]), and previous response to treatment. The presence of myelosuppression, EZH2 mutation status, Follicular Lymphoma International Prognostic Index (FLIPI) risk group, number of nodal sites, and presence of bulky disease were excluded because of a lack of published data in the comparators. For previous response to treatment, data were available on the number of patients who were double refractory as well as for patients who were refractory to the last line of therapy; however, as these are partially overlapping and, in order not to excessively reduce the sample size, only refractory status to last therapy was included in the analysis.
As EZH2 status could not be included as a matching variable because of a lack of comparator data, but may be a treatment effect modifier, a scenario analysis was carried out with an EZH2 weighting of 28.9% (using a published figure based on genetic sequencing of 159 patients recruited for the PRIMA clinical trial [9] of rituximab in previously untreated FL).
Comparison of Efficacy and Safety Outcomes Before and After Matching
For each head-to-head comparison, comparative analyses were conducted both before and after weighting. Before matching, binary outcomes, such as ORR, and safety outcomes were summarized in proportions and compared using the chi-square test. Risk differences comparing tazemetostat versus each of the comparator treatments were also reported. Using the weights generated in the MAIC, ORR and selected safety outcomes were compared between balanced trial populations. Risk differences comparing tazemetostat versus comparator treatments were reported for the ORR and selected safety outcomes. The standard error, 95% confidence interval, and p value for the indirect comparison were based on a robust estimate of the variance using a sandwich estimator that accounts for the variability in the propensity score weights [29, 32].
Results
Baseline Characteristics
Prior to matching, ECOG PS (0, > 0, and 1), tumor histology (grade 3b tumor and transformed FL), and refractory status to last therapy were the main characteristics that were significantly different when the E7438-G000-101 FL population was compared with that of each comparator arm.
After matching, all adjusted population baseline variables were successfully balanced between the reweighted tazemetostat data and the baseline population for each comparison trial. Baseline characteristics across trial populations, both before and after matching, are presented in Tables 2, 3, 4, and 5.
Safety Outcomes
Tazemetostat safety outcomes after population matching were compared with the outcomes from the idelalisib, duvelisib, copanlisib, and umbralisib trials, including any grade ≥ 3 TEAEs, any TESAEs, any AESIs, and any TEAE leading to dose reduction, drug discontinuation, or drug interruption. Unmatched safety comparisons are presented in Table 6.
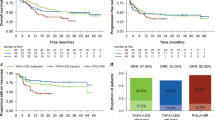
Overall, tazemetostat showed a lower incidence and lower relative risk for all grouped safety outcomes when data were available (Fig. 1; Table 7). In particular, for any grade ≥ 3 TEAE, tazemetostat had a relative risk of 0.45 (95% CI 0.30, 0.67) versus idelalisib, 0.35 (95% CI 0.22, 0.57) versus duvelisib, 0.37 (95% CI 0.28, 0.50) versus copanlisib, and 0.65 (95% CI 0.49, 0.86) versus umbralisib. For TEAEs leading to dose reduction, tazemetostat had a relative risk of 0.35 (95% CI 0.19, 0.65) versus idelalisib, 0.36 (95% CI 0.16, 0.82) versus duvelisib, 0.45 (95% CI 0.23, 0.85) versus copanlisib, and 0.67 (95% CI 0.29, 1.54) versus umbralisib. For TEAEs leading to drug discontinuation, tazemetostat had a relative risk of 0.23 (95% CI 0.08, 0.64) versus idelalisib, 0.28 (95% CI 0.07, 1.02) versus duvelisib, 0.42 (95% CI 0.19, 0.95) versus copanlisib, and 0.47 (95% CI 0.20, 1.11) versus umbralisib.

Summary of matching-adjusted key safety outcomes. Gray dots denote RR before adjustment and blue dots denote RR after adjustment. The horizontal line represents the 95% CI for each adjusted outcome. CI confidence interval, ESS effective sample size, TEAE treatment-emergent adverse event, TESAE treatment-emergent serious adverse event, RR relative risk
Several individual grade ≥ 3 TEAEs occurred at a significantly lower incidence with tazemetostat compared with matched patients treated with idelalisib, duvelisib, copanlisib, or umbralisib (Table 8). For example, the incidence of neutropenia was lower for tazemetostat versus all comparators (idelalisib 3% vs 22%; duvelisib 3% vs 25%; copanlisib 4% vs 24%; and umbralisib 2% vs 12%; all p < 0.05). Other individual grade ≥ 3 TEAEs that had a significantly lower incidence included hyperglycemia and hypertension (only copanlisib). There were no individual AEs with significantly higher incidence for tazemetostat in any comparison.
Efficacy Outcomes
After matching and adjustment for baseline characteristics, ORR for tazemetostat compared to each of idelalisib, duvelisib, copanlisib, and umbralisib showed no statistically significant difference between therapies (Fig. 2).

Summary of matching-adjusted key efficacy outcomes. Gray dots denote ORR before adjustment and blue dots denote ORR after adjustment. The horizontal line represents the 95% CI for each adjusted outcome. The ORR reported for duvelisib, copanlisib, and umbralisib reflect the full trial population, as per the availability of baseline variable data. FL-specific ORR is 42% for duvelisib, 59% for copanlisib, and 45% for umbralisib. CI confidence interval, ESS effective sample size, FL follicular lymphoma, ORR objective response rate
After adjustment to match the aggregate baseline characteristics of the idelalisib and duvelisib trials, the median DOR for tazemetostat decreased compared with the pre-adjustment value. The median DOR for tazemetostat in the idelalisib, duvelisib, copanlisib, and umbralisib comparisons was 7.5 (95% CI 3.8, 19.3), 7.5 (95% CI 3.4, 19.3), 11.3 (95% CI 7.2, not reached), and 13.1 (95% CI 7.2, not reached) months, respectively. Statistical tests for the difference in median DOR against the four comparators were not conducted, given the low effective sample size (defined as the remaining sample size after matching adjustment) and the lack of individual patient data for comparators.
In the scenario analysis where tazemetostat data were adjusted to an EZH2 mutation weight of 28.9% for all comparisons, ORR was slightly lower than in the base case. Results were as follows: tazemetostat versus idelalisib 37% (95% CI 24, 49) versus 56% (95% CI 44, 67), p < 0.05; tazemetostat versus duvelisib 39% (95% CI 20, 59) versus 47% (95% CI 39, 56), p = 0.49; tazemetostat versus copanlisib 44% (95% CI 33, 55) versus 61% (95% CI 53, 69), p < 0.05; and tazemetostat versus umbralisib 54% (95% CI 43, 64) versus 47% (95% CI 40, 54), p = 0.29. Safety outcomes were comparable to the base case analysis.
Discussion
Results from the ITC indicate that, after adjustment for baseline population differences, tazemetostat has better overall safety and tolerability when comparing grade ≥ 3 TEAEs and TEAEs leading to dose reduction or drug discontinuation than idelalisib, duvelisib, copanlisib, and umbralisib, while achieving similar response rates and duration of response. In particular, tazemetostat showed significantly lower relative risk in 13 out of 17 grouped AE categories (e.g., AEs that led to discontinuation, interruption, or dose reduction, TESAEs, and AESIs), and there was no category where point estimates were higher for tazemetostat.
This ITC provides a comprehensive evaluation of cross-trial heterogeneity and addresses potential sources of bias. Late-line cancer treatments are often approved on single-arm trials, and populations may often need adjustment in order to give a more accurate comparison between treatments and to provide clinicians with a more robust basis to compare treatments. This paper provides an illustration of such an adjustment for population differences across a group of comparators. Prior to adjustment, the E7438-G000-101 trial patients had lower ECOG PS, a higher proportion of grade 3b tumors and transformed FL, and fewer patients with a history of refractory status to therapy. These differences in the baseline population make the head-to-head comparison challenging. Using E7438-G000-101 trial IPD to adjust for observed cross-trial differences in patient characteristics allowed for estimates of the indirect treatment effect of tazemetostat, with sources of bias addressed.
There were several limitations associated with the ITC analysis. As with all such analyses, differences in some trial characteristics, such as operational design (e.g., different routes of treatment administration), cannot be addressed analytically. Similarly, another limitation concerns the suitability of using an ITC analysis to compare safety data that may not typically be of a dichotomous nature, but with a gradation in severity. The E7438-G000-101 trial had a longer duration of exposure, which may have led to an overestimation of tazemetostat safety events relative to those of the PI3K inhibitors (including the PI3K/CK1 inhibitor umbralisib). Additionally, adjustment variables for matching are limited to those for which published summary statistics from the comparator trials are available; thus, differences in unmeasured or unadjusted factors are not addressed and remain a potential source of bias. The evidence presented should thus be interpreted with caution until evidence from a direct randomized head-to-head trial between tazemetostat and each of the comparators is available.
There is evidence that EZH2 status is not prognostic for health outcomes in patients with FL treated beyond first line [7]; however, given the mechanism of action of tazemetostat and differential efficacy reported by EZH2 status, EZH2 status should be considered a potential effect modifier for treatment with tazemetostat. EZH2 mutation status was identified as a treatment effect modifier for tazemetostat, but the EZH2 mutation frequency for comparator trials was not available. The scenario analysis with a lower weighting of EZH2 patients shows that this has an influence on ORR results. Nonetheless, this variable is not likely to have had a major influence on safety outcomes, particularly since AEs are typically defined as treatment emergent.
The DOR point estimates decreased in two of the comparisons; however, the median DOR appears to remain adequate, validating the meaningfulness of the ORR. Effective sample size was reduced further for the DOR analysis, as only responders were included, which resulted in confidence intervals with wide ranges; the DOR results should therefore be interpreted with caution. We also note that the DOR was based on follow-up times as of the cutoff in published data, and the absolute median DOR values are likely to rise with additional follow-up data due to censoring involved.
Three of the comparator trials were mixed histology populations, for which separate FL data on baseline characteristics and safety were not published. An assumption can be made that comparator safety outcomes were driven primarily by study drug, and that there were no major differences in safety prognoses between different histologies included in the trial. This assumption regarding safety is reinforced first by the fact that most safety events were defined as treatment emergent across comparator trials, and second, because the three mixed histology trials still showed a clear majority of the population who had FL. Different histologies are more likely to have different prognoses for efficacy outcomes, suggesting that efficacy results for the comparisons should be interpreted with caution. Published ORR results in the FL-only population were slightly lower for duvelisib (FL only 42%, full-trial population 47%), copanlisib (FL only 59%, full-trial population 61%), and umbralisib (FL-only population 45%, full-trial FL/MZL/SLL population 47%). Although these differences are not large, and it remains possible that the patients with FL had baseline characteristics indicating a worse prognosis, the assumption that the full mixed histology results are comparable to tazemetostat’s FL-only results appears reasonable and is more likely to reflect unfavorably on tazemetostat.
The tazemetostat trial was the only one without an exclusion criterion for prior allogeneic stem cell transplant, and this has not been adjusted for; the authors are not aware if this could have affected results in either direction. Separately, only the tazemetostat trial included patients with grade 3b or transformed NHL. Our methodology adjusts for this difference by excluding nine patients with these characteristics from the tazemetostat data. In light of this, the results should be taken as reflecting outcomes in FL without these types of patients. Although patients with these disease characteristics may be expected to have a poor prognosis, the tazemetostat individual patient data reveal that five out of nine of these patients showed a response on tazemetostat; therefore, not including these patients may result in potentially excluding positive outcomes for tazemetostat in patients with a worse prognosis.
Conclusions
More tolerable treatment options are needed for R/R FL because patients in this setting are often elderly and have exhausted multiple prior lines of treatment. Results from this ITC indicate that, after adjustment for baseline population differences, tazemetostat is associated with substantially lower relative risk for safety outcomes versus idelalisib, duvelisib, copanlisib, or umbralisib while achieving similar efficacy outcomes. This adjusted comparison, while an improvement over naive single-arm comparisons, is nonetheless based on statistical adjustment methods rather than direct evidence, and should ideally be confirmed by randomized controlled trials between tazemetostat and comparators in future.
References
Surveillance Epidemiology and End Results Program. SEER Cancer Stat Facts: NHL - Follicular Lymphoma. 2020. https://seer.cancer.gov/statfacts/html/follicular.html. Accessed Mar 27 2020.
Alperovich A, Batlevi C, Smith K, et al. Benchmark of progression-free survival for multiple lines of therapy in follicular lymphoma treated in the rituximab era [abstract]. Presented at: 58th American Society of Hematology Annual Meeting and Exposition; December 3–6, 2016; San Diego, CA.
Zydelig [package insert]. Foster City, CA: Gilead Sciences Inc., 2018.
Copiktra [package insert]. Needham, MA: Verastem, Inc., 2019.
Aliqopa [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, 2020.
Fowler NH, Samaniego F, Jurczak W, et al. Umbralisib, a dual PI3Kδ/CK1ε inhibitor in patients with relapsed or refractory indolent lymphoma. J Clin Oncol. 2021;39:1609–18.
Bodor C, Batlevi C, Nagy N, et al. EZH2 gain-of-function mutations are not associated with more favorable prognosis in relapsed/refractory follicular lymphoma: a preliminary analysis on 908 patients. 24th European Hematology Association Congress Amsterdam, Netherlands, 2019.
Bodor C, Grossmann V, Popov N, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122:3165–8.
Huet S, Xerri L, Tesson B, et al. EZH2 alterations in follicular lymphoma: biological and clinical correlations. Blood Cancer J. 2017;7: e555.
Guo S, Chan JK, Iqbal J, et al. EZH2 mutations in follicular lymphoma from different ethnic groups and associated gene expression alterations. Clin Cancer Res. 2014;20:3078–86.
Bodor C, O’Riain C, Wrench D, et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia. 2011;25:726–9.
Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–9.
Beguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9.
Epizyme Inc. TAZVERIK™ [Prescribing Information]. Epizyme, 2020.
National Comprehensive Cancer Network (NCCN). B-Cell Lymphomas (Version 4.2020). 2020.
Gopal AK, Kahl BS, Vos S, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–18.
Flinn IW, Miller CB, Ardeshna KM, et al. DYNAMO: a phase II study of duvelisib (IPI-145) in patients with refractory indolent non-Hodgkin lymphoma. J Clin Oncol. 2019;37:912–22.
Dreyling M, Santoro A, Mollica L, et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Amer J Hematol. 2019;95:362–71.
Dreyling M, Santoro A, Mollica L, et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clinl Oncol. 2017;35:3898–905.
Morschhauser F, Tilly H, Chaidos A, et al. Phase 2 multicenter study of tazemetostat, an EZH2 inhibitor, in patients with relapsed or refractory follicular lymphoma. Blood. 2019;134:123.
Morschhauser F, Tilly H, Chaidos A, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21:1433–42.
Food and Drug Administration. Application Number: 211155Orig1s000/211155Orig2s000. Silver Spring: Center for Drug Evaluation and Research; 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211155Orig1Orig2s000AdminCorres.pdf. Accessed 5 Apr 2020.
Food and Drug Administration. Application Number: 211155Orig1s000/211155Orig2s000, Product Quality Reviews. Silver Spring: Center for Drug Evaluation and Research, Food and Drug Administration, 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211155Orig1Orig2s000MultidisciplineR.pdf. Accessed July 6, 2021.
Gopal AK, Kahl BS, Flowers CR, et al. Idelalisib is effective in patients with high-risk follicular lymphoma and early relapse after initial chemoimmunotherapy. Blood. 2017;129:3037–9.
Okosun J, Bödör C, Batlevi C, et al. EZH2 gain-of-function mutations are not associated with more favorable prognosis in relapsed/refractory follicular lymphoma (FL): a preliminary analysis on 590 patients. Hematol Oncol. 2019;37:192–3.
Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86.
Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059–68.
Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15:940–7.
Nabhan C, Zhou X, Day BM, et al. Disease, treatment, and outcome differences between men and women with follicular lymphoma in the United States. Am J Hematol. 2016;91:770–5.
Casulo C, Byrtek M, Dawson KL, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the national lymphocare study. J Clin Oncol. 2015;33:2516–22.
Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28:935–45.
Acknowledgements
Funding
This study, including the journal’s Rapid Service and Open Access Fees, was sponsored by Epizyme, Inc.
Medical Writing, Editorial, and Other Assistance
The authors acknowledge Alex Wong, BS, and Jingru Wang, BA, of Analysis Group, Inc., for assistance with the literature review, statistical analysis, and medical writing in this study, and Angela Dai, BS, of Analysis Group, Inc., for assistance with the literature searches and review. Editorial support was provided by Peloton Advantage, an OPEN Health company, and funded by Epizyme, Inc.
Prior Presentation
The results from this study were previously presented at the virtual 62nd American Society of Hematology (ASH) Annual Meeting and Exposition 2020, and as an encore at the Academy of Managed Care Pharmacy (AMCP) Nexus 2021 meeting.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published
Author Contributions
David Proudman, Dave Nellesen and Khalid Mamlouk conceptualized the study. David Proudman and Deepshekhar Gupta conducted the data analysis. Deyaa Adib, Jay Yang and Khalid Mamlouk provided critical feedback on the study design and findings. David Proudman, Dave Nellesen and Deepshekhar Gupta developed the manuscript with input from all authors. All authors reviewed and approved the final manuscript
Disclosures
David Proudman, Dave Nellesen, and Deepshekhar Gupta are employees of Analysis Group, Inc., which was retained for consulting services to conduct the study. Deyaa Adib, Jay Yang, and Khalid Mamlouk are current or former employees of Epizyme, Inc and own stock.
Compliance with Ethics Guidelines
The results presented in this manuscript are based on previously published studies. All procedures performed in those studies were in accordance with the ethical standards of the local institutional review boards for each site and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the E7438-G000-101 (ClinicalTrials.gov identifier NCT01897571), DELTA (ClinicalTrials.gov identifier NCT01282424), DYNAMO (ClinicalTrials.gov identifier NCT01882803), CHRONOS-1 (ClinicalTrials.gov identifier NCT01660451), and UNITY-NHL (ClinicalTrials.gov identifier NCT02793583) trials.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available due to the E7438-G000-101 trial IPD being commercial-in-confidence.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Proudman, D., Nellesen, D., Gupta, D. et al. A Matching-Adjusted Indirect Comparison of Single-Arm Trials in Patients with Relapsed or Refractory Follicular Lymphoma Who Received at Least Two Prior Systemic Treatments: Tazemetostat was Associated with a Lower Risk for Safety Outcomes Versus the PI3-Kinase Inhibitors Idelalisib, Duvelisib, Copanlisib, and Umbralisib. Adv Ther 39, 1678–1696 (2022). https://doi.org/10.1007/s12325-022-02054-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-022-02054-z