
Abstract
Introduction
Among the various types of progressive fibrosing interstitial lung diseases (PF-ILDs), substantial survival data exist for idiopathic pulmonary fibrosis (IPF) but not for other types. This hinders evidence-based decisions about treatment and management, as well as the economic modelling needed to justify research into new treatments and reimbursement approvals. Given the clinical similarities between IPF and other PF-ILDs, we reasoned that patient survival data from four major IPF trials could be used to estimate long-term survival in other PF-ILDs.
Methods
We used propensity score matching to match patients with IPF taking either nintedanib or placebo in the TOMORROW, INPULSIS-1, INPULSIS-2 and INPULSIS-ON trials to patients with PF-ILDs other than IPF in the INBUILD trial. Seven models were fitted to the survival data for the matched patients with IPF, and the three best-fitting models were used to generate informative priors in a Bayesian framework to extrapolate patient survival of the INBUILD population.
Results
After propensity score matching, the analysis included data from 1099 patients with IPF (640 nintedanib patients; 459 placebo patients) and 654 patients with other PF-ILDs (326 nintedanib patients; 328 placebo patients). Gamma, log-logistic and Weibull models best fit the survival of the matched patients with IPF. All three models led to consistent Bayesian estimates of survival for the matched patients with other PF-ILDs, with median rates of overall survival ranging from 6.34 to 6.50 years after starting nintedanib. The corresponding control group survival estimates were 3.42 to 3.76 years.
Conclusion
We provide the first estimates of long-term overall survival for patients with PF-ILDs other than IPF, and our analysis suggests that nintedanib may prolong their survival. Our Bayesian approach to estimating survival of one disease based on clinical trial data from a similar disease may help inform economic modelling of rare, orphan and newly defined disorders.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Why carry out this study? |
Uncertainty about survival in patients suffering with progressive fibrosing interstitial lung diseases (PF-ILDs) hinders clinical decision-making and economic modelling for developing new treatments and bringing them to market |
We reasoned that we could use trial data from patients with idiopathic pulmonary fibrosis (IPF) to estimate survival of patients with other PF-ILDs |
What was learned from this study? |
Various models led to consistent estimates of overall survival in patients with PF-ILDs other than IPF, suggesting the reliability of our approach |
Median overall survival since starting treatment with nintedanib was 6.34–6.50 years. The equivalent estimate was 3.42–3.76 years if PF-ILDs were left untreated |
These survival estimates may help clinicians and patients make evidence-based decisions about treating and managing PF-ILDs other than IPF, and they may accelerate development of new treatments |
Introduction
Progressive fibrosing interstitial lung diseases (PF-ILDs) are relatively rare conditions in which patients experience a decline in lung function that reduces their health-related quality of life and often leads to premature death [1,2,3]. PF-ILDs include idiopathic pulmonary fibrosis (IPF), idiopathic non-specific interstitial pneumonia, autoimmune interstitial lung diseases, sarcoidosis and exposure-related diseases such as asbestosis. These diseases show limited response to standard anti-inflammatory and immunosuppressive therapies [4, 5].
IPF is the best-studied PF-ILD [1, 2, 6]. Much less is known about the survival of patients with other PF-ILDs. The most extensive clinical data on other PF-ILDs has come from the phase 3 INBUILD trial involving 663 patients. That work reported that nintedanib significantly slows disease progression, based on analysis of forced vital capacity (FVC) during 1 year. Whether the drug increases survival remains unclear.
Uncertainty about the survival of patients with PF-ILDs other than IPF makes it difficult for clinicians and patients to make evidence-based decisions about their treatment and management. It also prevents reliable economic modelling of costs and benefits of potential treatments for PF-ILDs, which payers, healthcare insurers, and pharmaceutical companies use to justify long-term investment in treatment allocation and drug development. Indeed, the ability of a new treatment to improve survival may be regarded by regulatory agencies, insurers and other stakeholders as a stronger argument than its ability to slow disease progression. However, survival data that have been collected over a sufficiently long period from an adequate number of patients are lacking for PF-ILDs other than IPF. A similar problem exists for rare and orphan diseases more generally, as well as for diseases that have only recently been defined clinically.
The Bayesian framework relies on a synthesis of evidence, such as historical data or subjective beliefs, to generate an informative prior, which is a prior belief of what the trial evidence (likelihood) may show. Bayesian extrapolation has been proposed to predict drug efficacy for children based on efficacy reported for adults [7], and the US Food and Drug Administration and the European Medicines Agency have integrated this approach into their guidance [8, 9]. The Decision Support Unit of the National Institute for Health and Care Excellence in the UK acknowledges the importance of “flexible survival models”, based on extrapolation using Bayesian methods or other techniques, in health technology assessments [10].
We attempted to use Bayesian methods to exploit the long-term survival data available for IPF, reflecting follow-up of up to 5.90 years [11], in order to estimate the survival of patients with other PF-ILDs. IPF is similar to other PF-ILDs in terms of the rate of FVC decline [2, 3, 12, 13]. We hypothesised that the survival would also be similar. First, we used propensity score matching to match patients with IPF in the TOMORROW, INPULSIS-1, INPULSIS-2 and INPULSIS-ON trials to patients with other PF-ILDs in the INBUILD trial. Next, we modelled the survival of the matched patients with IPF, and we used the best models from those patients to generate informative priors in Bayesian estimation of the survival of matched patients with other PF-ILDs, based on the method outlined in Soikkeli et al. [14]. The resulting survival estimates provide a foundation to understand the progression of other PF-ILDs and to promote the development of new treatments. Our approach may also be helpful in other rare, orphan or newly defined diseases for which survival evidence is limited.
Methods
Data Sources and Outcome
We considered patients with IPF who received nintedanib (300 mg/day) or placebo in the 52-week phase 2 TOMORROW trial (NCT00514683) [15] and the 52-week phase 3 INPULSIS-1 and INPULSIS-2 trials (NCT01335464 and NCT01335477) [15]. Long-term IPF data for patients receiving nintedanib were obtained from the open-label INPULSIS-ON extension trial (NCT01619085) [11]. Across the trials, patients with IPF received nintedanib for a median of 2.1 years (range 0.1–5.9 years).
We considered patients with PF-ILDs other than IPF who received nintedanib (300 mg/day) in the 52-week phase 3 INBUILD trial (NCT02999178) [16].
Overall survival (OS) was defined as the time between a patient’s first and last study visits, which corresponded to the time from treatment initiation until death or the last visit. Patients who were alive at the time of their last study visit were censored at that visit. This corresponded to the time when patients started treatment with nintedanib or control.
Our analysis were based on previously conducted studies and did not involve the conduct of any new studies with human participants or animals.
Propensity Score Matching
Patients with IPF were matched to patients with other PF-ILDs on the basis of their propensity score to ensure similar baseline characteristics and disease severity between the two populations. The following baseline characteristics were used in the matching: age, sex, ethnicity (Asian vs other), time since diagnosis, haemoglobin-corrected percentage of predicted CO diffusion capacity (DLco), percentage predicted FVC, and smoking status (never smoked, used to smoke, currently smokes). Patients for whom data were unavailable for any of these characteristics were excluded from the analysis.
Scores were matched using a kernel approach, in which patients with IPF with a propensity score closer to that of a patient with another PF-ILD were weighted more than those with scores farther away; and using a radius approach, in which all patients with IPF within a certain radius of a patient with another PF-ILD were weighted the same [17]. In both approaches, we tested radii of 0.1 and 0.05. Matching was performed separately for patients who received nintedanib or placebo. Propensity score matching was conducted using Stata IC version 14.2 (StataCorp, College Station, TX, USA).
The ability of the matching to minimise differences between patients with IPF or other PF-ILDs was assessed in terms of bias; Rubin’s B, defined as the absolute standardised difference in mean linear propensity score index between the two groups; and Rubin’s R, defined as the ratio of propensity score index variances in the two groups. We considered the matching successful if bias was less than 5%, Rubin’s B was less than 25 and Rubin’s R was between 0.5 and 2.0 [18].
Bayesian Survival Analysis
We aimed to extend the approach of Soikkeli et al. [14] to the extrapolation of data from one disease (IPF) to clinically similar ones (other PF-ILDs). First, we tested the following seven standard frequentist survival models to determine the best fit model of the survival data for the matched patients with IPF: exponential, Weibull, log-normal, log-logistic, generalised gamma, gamma and Gompertz. All these models are routinely used in health technology assessments of new treatments [19, 20]. Modelling was performed using the “flexsurv” function in R 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria) [21]. Model fit was assessed by visual inspection and in terms of the Akaike and Bayesian information criteria, where lower values indicate better model fit. We identified the three best-fitting models and used them to generate informative priors for the “shape” parameter of the Bayesian model. Bayesian modelling was carried out using OpenBUGS 3.2.3 (revision 1012; MRC Biostatistics Unit, Cambridge, UK). The OpenBUGS code and further details of the analysis are provided in the online Supplementary Material. Number of iterations and burn-in are detailed in Table S1 in the online Supplementary Material. If autocorrelation was high, thinning factors were applied to determine whether they affected parameter estimates. If not, estimates without the thinning factor were used.
Patient and Public Involvement
Patients were not involved in the design or conduct of this analysis.
Results
Patients
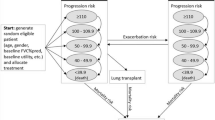
Of the 1239 patients with IPF in the TOMORROW, INPULSIS-1, INPULSIS-2 and INPULSIS-ON trials, 140 were removed during propensity score matching, leaving 1099 matched patients, of whom 640 received nintedanib and 459 received placebo (Fig. 1). Of the 663 patients with other PF-ILDs in the INBUILD trial, nine were removed during propensity score matching, leaving 654 matched patients, of whom 326 received nintedanib and 328 received placebo. Of all the radius matching algorithms tested for the nintedanib and placebo groups, the algorithm with a radius of 0.1 produced the best results: a Rubin’s B score of 13.3 for nintedanib and 12.8 for placebo, and respective Rubin’s R scores of 0.96 and 0.89. After matching, small bias was observed in the percentage of predicted FVC and the percentage of predicted DLco. Nevertheless, the residual differences between the matched patients with IPF or other PF-ILDs were not clinically significant (Table 1). There was sufficient overlap of propensity scores within each group of matched patients (Fig. S1 in Supplementary Material), and the matching process reduced the standardised percentage bias across covariates (Fig. S2 in Supplementary Material).

Summary of the study procedure. IPF idiopathic pulmonary fibrosis, PF-ILD progressive fibrosing interstitial lung disease
Bayesian Estimation of OS
On the basis of the Akaike and Bayesian information criteria, three survival models gave good fits to the survival data for matched patients with IPF who received nintedanib or placebo: gamma, log-logistic and Weibull (Table S3 in Supplementary Material). For matched patients with IPF who received nintedanib, the log-logistic model estimated the highest median OS (6.48 years), while estimates were lower with the gamma model (6.13 years) and Weibull model (6.06 years). For patients who received placebo, the gamma and log-logistic models gave similar median OS estimates (2.93 or 3.00 years), while the Weibull model gave a lower median OS (2.61 years).
Since the three models visually fit the matched IPF data (Fig. 2), all three were considered in the Bayesian analysis to estimate OS for patients with other PF-ILDs. Diagnostic plots for the three models suggested good convergence across model parameter estimates (Figs. S3–S8 in Supplementary Material).

Modelling of overall survival of matched patients with IPF using gamma, log-logistic or Weibull models. Model output is shown against the corresponding trial data. IPF idiopathic pulmonary fibrosis
The complete results for the different model parameters estimated in the Bayesian analysis are provided in Table S4 in the Supplementary Material. For patients who received nintedanib, all three models gave similar estimates of median OS (6.34–6.50 years) and 5-year OS rates (59–60%) (Table 2, Fig. 3). For patients who received placebo, the three models gave similar estimates of median OS (3.42–3.76 years), but the Weibull model estimated a substantially lower 5-year survival rate (21%) than the other models (32% or 34%).

Estimation of overall survival of patients with progressive fibrosing interstitial lung diseases (PF-ILDs) other than idiopathic pulmonary fibrosis (IPF), based on Bayesian extrapolation from trial data for patients with IPF. Extrapolation was performed according to a gamma, b log-logistic, or c Weibull models
Discussion
Drawing on available survival data for patients with IPF, we used a Bayesian methodology to provide the first long-term OS estimates for patients with other PF-ILDs whose disease is left untreated or is treated with nintedanib. These estimates may help clinicians and patients make informed decisions about disease treatment and management, and it may help drug manufacturers and healthcare agencies more reliably estimate the costs and benefits associated with proposed treatments.
We estimated that when untreated, patients with PF-ILDs other than IPF show median OS of 3.42–3.76 years and 5-year survival of 21–34%. When treated with nintedanib, their median OS increases to 6.34–6.5 years and 5-year survival to 59–60%. While the three best-performing models for Bayesian extrapolation gave consistent results for median OS, the Weibull model gave a substantially lower 5-year survival estimate for placebo (21%) than the gamma and log-logistic models gave (32–34%). Given that a systematic review has reported a ≥ 5-year survival rate of 31% for patients with IPF not receiving antifibrotic treatment [22], it is likely that the Weibull model underestimates survival compared with the other two options. Our estimates may be refined in the future, as the ongoing open-label INBUILD-ON extension trial (NCT03820726) continues to provide new data. Our analysis suggests that to observe at least 50% patient death in clinical trials or registries, follow-up would need to exceed 3 years in the placebo arm and 6 years in the nintedanib arm.
The antifibrotic drug nintedanib has been shown to slow FVC decline of IPF and other PF-ILDs [23], and it may also improve OS of patients with IPF, based on an exploratory extrapolation of trial data [24]. The present extrapolation suggests that the same may be true for other PF-ILDs: the difference in median OS between the nintedanib and placebo groups was 2.61–3.03 years across all three models that we used to generate IPF-based priors for Bayesian extrapolation. While our results require validation with real-world evidence, they justify further investment into nintedanib.
In fact, our results may help accelerate the development of new treatments for other PF-ILDs, since nintedanib is currently the only drug licensed for this condition. Our approach may also prove useful for rationalising and stimulating investment in new treatments for other rare, orphan or newly described diseases where the lack of patient data may create too much uncertainty for drug or device manufacturers to justify investment. The reliability of such extrapolations from one disease to another depends on clinical similarity between the two. Here we relied on the demonstrated similarity in FVC decline between IPF and other PF-ILDs. Indeed, Simpson et al. showed that survival rates were similar between patients with IPF or with other PF-ILDs during follow-up of approximately 2.5 years [13].
Our estimates should be treated with caution as no long-term IPF survival data were available to generate priors for the placebo arm. These patients either discontinued or crossed over to nintedanib in the open-label extension and were therefore censored in the present study. In addition, we did not take into account potential confounding due to the use of anti-inflammatory medications by patients with other PF-ILDs in the INBUILD trial. That trial allowed patients to take such medications in addition to the study drug.
Despite these limitations, our analysis demonstrates the feasibility of extrapolating from long-term survival data for patients with one disease to estimate survival of patients with a clinically related disease. The resulting estimates can improve treatment decisions as well as payer treatment allocation decisions.
Conclusions
We used a Bayesian approach to estimate survival of one disease based on clinical trial data from a similar disease, allowing us to provide the first estimates of long-term overall survival for patients with PF-ILDs other than IPF. Our analysis suggests that nintedanib may prolong their survival, justifying further investment in the drug. Our approach may prove useful for economic modelling of rare, orphan and newly defined disorders for which only limited survival data are available.
References
Cottin V, Wollin L, Fischer A, et al. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev. 2019;28(151):180100.
Kolb M, Vašáková M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. 2019;20(1):57.
Wollin L, Distler JHW, Redente EF, et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur Respir J. 2019;54(3):1900161.
Cottin V, Hirani NA, Hotchkin DL, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
Makino S. Progressive fibrosing interstitial lung diseases: a new concept and indication of nintedanib. Mod Rheumatol. 2021;31(1):13–9.
Olson AL, Gifford AH, Inase N, Fernández Pérez ER, Suda, T. The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. Eur Respir Rev. 2018;27(150):180077.
Gamalo-Siebers MA-O, Savic J, Basu C, et al. Statistical modeling for Bayesian extrapolation of adult clinical trial information in pediatric drug evaluation. Pham Stat. 2017;4(16):232–49.
Agency EM. Extrapolation of efficacy and safety in paediatric medicine development. 2018. https://www.ema.europa.eu/en/extrapolation-efficacy-safety-paediatric-medicine-development. Accessed 2021.
Center for Devices and Radiological Health. Leveraging existing clinical data for extrapolation to pediatric uses of medical devices. 2016. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/leveraging-existing-clinical-data-extrapolation-pediatric-uses-medical-devices. Accessed 2021.
Rutherford MJ, Lambert PC, Sweeting MJ, et al. NICE DSU Technical Support Document 21: Flexible methods for survival analysis. 2020. http://www.nicedsu.org.uk. Accessed 2021.
Crestani B, Huggins JT, Kaye M, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med. 2019;7(1):60–8.
Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020;55(6):2000085.
Simpson T, Barratt SL, Beirne P, et al. The burden of progressive fibrotic interstitial lung disease across the UK. Eur Respir J. 2021;58(1):2100221.
Soikkeli F, Hashim M, Ouwens M, Postma M, Heeg B, et al. Extrapolating survival data using historical trial-based a priori distributions. Value Health. 2019;22(9):1012–7.
Richeldi L, Kreuter M, Selman M, et al. Long-term treatment of patients with idiopathic pulmonary fibrosis with nintedanib: results from the TOMORROW trial and its open-label extension. Thorax. 2018;73(6):581–3.
Flaherty KR, Brown KK, Wells AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res. 2017;4(1):e000212.
Leuven E, Sianesi B. PSMATCH2: Stata module to perform full Mahalanobis and propensity score matching, common support graphing, and covariate imbalance testing. 2021;s432001. Available from: https://ideas.repec.org/c/boc/bocode/s432001.html. Accessed 2003.
Rubin DB. Using propensity scores to help design observational studies: application to the tobacco litigation. Health Serv Outcomes Res Method. 2001;2(3):169–88.
Canadian Agency for Drugs and Technologies in Health (CADTH). Procedures for the CADTH Common Drug Review and Interim Plasma Protein Product Review. 2020. https://www.cadth.ca/sites/default/files/cdr/process/Procedure_and_Guidelines_for_CADTH_CDR.pdf. Accessed 2021.
Latimer N. NICE DSU Technical Support Document14: Survival analysis for economic evaluations alongside clinical trials—extrapolation with patient-level data. 2011. http://nicedsu.org.uk/wp-content/uploads/2016/03/NICE-DSU-TSD-Survival-analysis.updated-March-2013.v2.pdf. Accessed 2021.
Jackson CH. Flexsurv: a platform for parametric survival modeling in R. J Stat Softw. 2016. https://doi.org/10.18637/jss.v070.i08.
Khor YA-O, Ng Y, Barnes HA-O, et al. Prognosis of idiopathic pulmonary fibrosis without anti-fibrotic therapy: a systematic review. Eur Respir Rev. 2020. https://doi.org/10.1183/16000617.0158-2019.
Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27.
Lancaster L, Crestani B, Hernandez P, et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res. 2019;6(1):e000397.
Acknowledgements
Funding
Boehringer Ingelheim funded this analysis and its publication and was involved at all stages from study design to manuscript preparation. The company was given the opportunity to review the manuscript for medical and scientific accuracy, as well as intellectual property considerations.
Medical Writing and Editorial Support
Julia Korell (Translational Medicine & Clinical Pharmacology, Boehringer Ingelheim Pharmaceuticals, Ridgefield, CT, USA) and Andreas Karpa (Translational Medicine & Clinical Pharmacology, Boehringer Ingelheim Pharma, Ingelheim, Germany) assisted with data management. Boehringer Ingelheim funded statistical analysis provided by Corinne LeReun (independent biostatistician, Guadeloupe, France) and writing support provided by Dr Joanna Todd (Stellar Medical Communications, Ely, UK) and Dr A. Chapin Rodríguez (Creaducate Consulting, Munich, Germany). Toby M Maher is supported by a Research Clinician Scientist Fellowship from the US National Institutes of Health (CS-2013-13-017) and a British Lung Foundation Chair in Respiratory Research (C17-3).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
BL performed statistical analysis and drafted the manuscript with the help of all co-authors. KBR, MB and AD provided methodological guidance, while TMM and YI provided clinical guidance. All authors revised the manuscript and agreed to its submission and publication.
Prior Presentation
Aspects of this research were previously presented at the European Respiratory Society (ERS) International Congress 2021; see Maher, T. Langford, B. Diamantopoulos, A. Rohr, K. Baldwin, M. Inoue, Y. 2021. Estimating the long-term survival in progressive fibrosing interstitial lung disease (PF-ILD) other than IPF using matched IPF data. Presented at: European Respiratory Society (ERS) International Congress 2021, 5–8 September 2021, virtual.
Disclosures
Bryony Langford and Alex Diamantopoulos are employees of Symmetron Limited. Toby M Maher has received, via his institution, industry-academic funding from GlaxoSmithKline and Union Chimique Belge, and has received consultancy or speaker fees from Apellis, AstraZeneca, Bayer, Blade Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Galapagos, GlaxoSmithKline, Indalo, Novartis, Pliant, ProMetic, Respivant, Roche, Samumed, and Union Chimique Belge for projects outside the work presented in this manuscript. Yoshikazu Inoue has worked as an advisor, steering committee member and speaker for Asahi Kasei, Boehringer Ingelheim, Galapagos, Nitto, Promedior, Roche, Shionogi Savara, and Taiho. Klaus B Rohr and Michael Baldwin are employees of Boehringer Ingelheim GmbH.
Compliance with Ethics Guidelines
This analysis was based on previously conducted studies and did not involve the conduct of any new studies with human participants or animals.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Langford, B., Diamantopoulos, A., Maher, T.M. et al. Using Data on Survival with Idiopathic Pulmonary Fibrosis to Estimate Survival with Other Types of Progressive Fibrosis Interstitial Lung Disease: A Bayesian Framework. Adv Ther 39, 1045–1054 (2022). https://doi.org/10.1007/s12325-021-02014-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-02014-z