
Abstract
Introduction
Hyponatremia is a common condition of varying etiology among hospitalized patients and is associated with adverse outcomes. Treatment to normalize serum sodium is advisable. Tolvaptan received European Union marketing authorization for hyponatremia secondary to the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). Post-marketing pharmacovigilance activities were required to characterize the safety profile of tolvaptan more fully in this population, which is often elderly with a high burden of comorbid illness.
Methods
This was a prospective, observational, multinational, post-authorization pharmacovigilance study (NCT01228682) in seven European countries. Hospitalized patients were enrolled who received tolvaptan for hyponatremia associated with SIADH and consented to data collection. Tolvaptan was initiated and assessments performed at physician discretion per local standards of care. To reflect actual clinical practice, no assessments or procedures were required outside the standard of care. Patients who continued to receive long-term tolvaptan following hospital discharge and provided consent received follow-up from their community physicians.
Results
A total of 252 patients (mean age 70.6 years) enrolled. Mean tolvaptan treatment duration was 139.4 days, median 18.5 (range 1–1130) days; most frequent dose was 15 mg/day (used in 75% of patients). Serum sodium increased from baseline (mean 123.2 mmol/l) during treatment week 1 and remained stable during follow-up, with little difference across doses of 7.5, 15, and 30 mg/day. Hyponatremia symptoms (e.g., confusion, unsteady gait, lethargy) were present in 122/252 (48.4%) patients at pre-treatment baseline, decreasing to 46/252 (18.3%) during treatment. Sixty-two patients (24.6%; mean baseline serum sodium 120 mmol/l) experienced rapid correction of hyponatremia within 72 h. No osmotic demyelination syndrome occurred.
Conclusion
In clinical practice, tolvaptan improved serum sodium and decreased hyponatremia symptoms in hyponatremia secondary to SIADH. Serum sodium should be monitored during treatment to minimize risk of rapid correction.
Trial Registration
Clinicaltrials.gov identifier NCT01228682.
Plain Language Summary
Hospitalized patients often experience abnormally low blood sodium levels (hyponatremia), which can cause significant symptoms and poses a serious health risk (Wald et al. in Arch Intern Med 170:294–302, 2010). Yet, increasing sodium levels too rapidly in these patients can unintentionally cause osmotic demyelination syndrome, resulting in long-term neurologic damage or death. Tolvaptan was approved in the European Union to treat one type of hyponatremia caused by a hormonal imbalance known as the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). Tolvaptan effectively increased patient sodium levels in clinical trials, but real-world data are needed to understand tolvaptan treatment more fully in everyday clinical practice. We evaluated patterns of use, efficacy, and safety of tolvaptan in patients treated in hospitals and after discharge for hyponatremia due to SIADH. Tolvaptan was correctly used to treat only hyponatremia caused by SIADH in nearly all of the 252 patients studied. Patient sodium levels increased in the first week of tolvaptan treatment and then stabilized. Hyponatremia symptoms, such as confusion, nausea, tiredness, and dizziness, were present in 48.4% of patients before treatment and in 18.3% after starting tolvaptan. Consistent with earlier studies, some patients (24.6%) experienced excessively rapid correction of hyponatremia. However, no subsequent neurologic problems or deaths were attributed to the rapid correction, which suggests that medical providers were carefully monitoring and managing sodium levels to prevent serious consequences. Our study indicates that tolvaptan is being used safely and effectively to treat hyponatremia due to SIADH in a patient population with complex medical needs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Hospitalized patients with hyponatremia secondary to the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) have a high burden of illness and require careful management to safely normalize serum sodium levels. |
This pharmacovigilance study was conducted to provide real-world safety and efficacy data on the use of tolvaptan in this population. |
What was learned from the study? |
In clinical practice, tolvaptan was used in the correct indication of hyponatremia secondary to SIADH, increasing patient serum sodium and reducing the frequency of hyponatremia symptoms. |
Cases of overly rapid correction of hyponatremia occurred, but no osmotic demyelination syndrome was reported. |
Data from real-world clinical practice support clinical trial findings that tolvaptan improves serum sodium in hyponatremia secondary to SIADH with appropriate monitoring to reduce the risks associated with overly rapid correction of hyponatremia. |
Introduction
Hyponatremia is a common sodium disturbance among hospitalized adults. Prevalence estimates vary according to the threshold serum sodium level selected [1], but in recent studies, hyponatremia was present in 20–38% of hospital patients [2,3,4]. The condition also frequently occurs in elderly residents of long-term care facilities, with prevalence estimates of 18–31% [5, 6]. The etiology is variable, with causes including congestive heart failure, liver cirrhosis, and endocrine disorders [7]. One of the most frequent causes of hyponatremia, accounting for 31–36% of cases, is the syndrome of inappropriate secretion of antidiuretic hormone (SIADH), in which release of antidiuretic hormone (vasopressin) despite hypotonicity of the serum leads to antidiuresis and hyponatremia [8,9,10]. SIADH itself may be caused by multiple conditions, the most common of which are malignancies, pulmonary disorders, central nervous system disorders (e.g., encephalitis, head trauma), and use of certain medications (e.g., thiazide diuretics, selective serotonin reuptake inhibitors), with a substantial number of idiopathic cases [11]. Essential diagnostic criteria for SIADH include decreased serum osmolality (< 275 mOsm/kg of water), clinical euvolemia, urinary osmolality > 100 mOsm/kg of water, urinary sodium > 40 mmol/l with normal dietary sodium intake, normal thyroid and adrenal function, normal kidney function, no recent use of diuretics, and no hypokalemia or acid-base disorders [7]. Treatment of hyponatremia of any etiology is advisable, as abnormally low serum sodium levels are associated with an increased risk of mortality and other adverse outcomes such as longer hospital stays and higher medical costs [3, 4, 12,13,14,15,16].
Samsca® (tolvaptan) was evaluated as a treatment for hyponatremia secondary to SIADH, given that it competitively blocks the binding of vasopressin to its V2 receptor, inhibiting antidiuretic activity [17]. This mechanism of action results in free water clearance without depletion of electrolytes and ultimately improvement of sodium balance in SIADH patients [18]. Two pivotal clinical trials demonstrated efficacy in correcting hyponatremia secondary to SIADH, resulting in EU marketing approval for this indication [17].
Given that drug regulatory approval is based on clinical trials, which enroll limited numbers of patients treated under carefully controlled conditions, pharmacovigilance activities conducted in the real-world clinical practice setting, after marketing authorization, are an integral component of comprehensive drug safety evaluation. In addition to detecting rare adverse events that may not have been observed in relatively small clinical trial populations, pharmacovigilance can identify patterns of inappropriate/erroneous medication use, drug interactions, and problems with lack of efficacy [19]. All of these issues are of particular relevance to patients with SIADH-related hyponatremia, who are often elderly and have a high burden of comorbid illness. Accordingly, the Committee for Medicinal Products for Human Use of the European Medicines Agency required a post-authorization safety study (PASS) of tolvaptan as a condition of marketing authorization for this indication. The objective of the PASS was to collect information on the safety of tolvaptan when used in the real-world setting for the treatment of hyponatremia secondary to SIADH. Results from this post-marketing study are presented here.
Methods
Design, Setting, and Participants
This was a prospective, observational, multinational, post-marketing, Phase 4, pharmacovigilance study (clinicaltrials.gov identifier NCT01228682) that enrolled a population of hospitalized adults treated with tolvaptan for hyponatremia secondary to SIADH. The inclusion criterion was treatment with tolvaptan, and the exclusion criterion was patients who had not signed the data consent form.
At each hospital site, when a prescription for tolvaptan was sent to the hospital pharmacy, the prescribing physician was asked about his or her interest in participating in the study. Physicians who agreed to participate could recruit their patients to enroll. It was expected that the size of the study population would be limited by the number of available SIADH patients. On the assumption that 50 hospital sites would allow a recruitment rate of about 100 SIADH patients per year, a total study population of at least 200 patients was anticipated for the planned 2-year accrual period. To obtain results in a timely manner, study sites were initially selected in EU countries with the earliest commercial availability of tolvaptan (UK, Germany, and Nordic countries). Sites from other EU countries were later added when tolvaptan became available in those areas.
Assessments
As an observational study, the study did not define a therapeutic strategy, and methods of assessment were not prespecified, because patient management was performed according to the standard of care of the participating hospitals. Use of tolvaptan was at the discretion of the treating physician in accordance with the drug Summary of Product Characteristics (SmPC), and no diagnostic, therapeutic, or monitoring procedures outside normal medical practice were required. The physician maintained the patient’s medical records to document observations that were part of the standard of normal care.
Data collection for each patient commenced after the patient had signed the data consent form and was to continue until the end of tolvaptan treatment. The target minimum duration of follow-up for patients who received long-term tolvaptan treatment was 12 months after treatment initiation. After hospital discharge, data collection could continue if the patient consented to the continuation of data collection by his/her community-based physician. Community-based physicians who agreed to participate would then assume responsibility for reporting and providing the data collected from consenting patients.
At baseline, the data collected included patient eligibility per inclusion/exclusion criteria, patient consent, hospital setting, clinical diagnosis (hyponatremia and underlying disease), volume status, patient demographics, medical history and physical examination, laboratory values as available, hyponatremia symptoms, tolvaptan dosing, concomitant hyponatremia therapy, and adverse events. As per local standard of care, the treating hospital physician recorded data from follow-up assessments conducted up to the time of hospital discharge: serum sodium laboratory values, hyponatremia symptoms, tolvaptan dosing, concomitant medications, and adverse events (including abnormal laboratory parameters). The physician who monitored the patient after discharge recorded the same information as was collected during the hospital stay. Participating physicians documented all adverse events (AEs) on an electronic case report form regardless of the suspected causal relationship with tolvaptan.
Study Analyses
Data were summarized by descriptive statistics on an observed-case basis, with no imputation for missing data. All data analysis was performed using SAS® version 9.1.3. A descriptive analysis was selected for the reason that, although criteria for the diagnosis of SIADH are available [7, 20], diagnostic workup in patients with hyponatremia is often inadequate [10, 21], and misclassification of SIADH patients under other medical conditions is likely. Due to a lack of epidemiologic data on the true incidence of SIADH and patient characteristics, use of a historical control cohort for statistical comparison was not feasible.
An analysis of the effects of concomitant medication use on risk of treatment-emergent AEs (TEAEs) was conducted that accounted not only for use/non-use of concomitant medications, but also the timing of the medication use. Medications of interest were cytochrome P450 3A4 (CYP P450 3A4) inducers/inhibitors, serum potassium concentration increasing substances, and warfarin/antiplatelet agents. TEAEs were categorized as follows: (1) TEAEs that occurred in patients who did not take a concomitant medication of the selected category (non-users); (2) TEAEs that occurred in patients who took a medication concomitantly with tolvaptan or within 3 days after the concomitant medication was stopped or 2 days in the case of rapid correction of hyponatremia (during concomitant use); and (3) TEAEs that occurred in patients who took a medication concomitantly with tolvaptan but in whom the TEAEs did not occur during the time of co-administration or within 3 days after (outside of concomitant use).
Changes to the Study
In response to a query from the European Medicines Agency on the clinical study report, the definition of rapid correction of hyponatremia as used originally in the study was changed to align with the criteria used in the tolvaptan SmPC [18]. The occurrence of this AE was re-analyzed using the revised criteria, which are provided in the Supplementary Material. Furthermore, the definition of rapid correction of hyponatremia was extended from 48 to 72 h to include patients who were tested for serum sodium after 48 h. To account more accurately for cases where tests were not performed at one of the predefined intervals (i.e., at 6, 12, 24, and 48 h), linear extrapolations were performed.
Compliance with Ethics Guidelines
The study was conducted in accordance with Good Pharmacoepidemiology Practices, local national laws and regulations applicable to non-interventional studies, Volume 9A (in European Economic Area countries), and ethical principles that have their origins in the Declaration of Helsinki. Where required by local regulations for observational studies, the study was conducted under the auspices of an Independent Ethics Committee. The participating Ethics Committees were: Clinical Research Ethics Committee of Hospital Universitario “12 de Octubre,” Madrid, Spain; Comitato Etico Interaziendale A.S.O. Santa Croce e Carle, Cuneo, Italy; Research Ethics Committee Northwest 10 REC—GM North, Northwest Centre of Research Ethics Committees, Manchester, UK; Regionala etikprövningsnämnden, Göteborg, Göteborg, Sweden; Regional Committee for Medical and Health Research Ethics South East A (REK South East A), Oslo, Norway; Ethik-Kommission bei der Medizinischen Fakultät Universität Marburg, Marburg, Germany.
Patients provided informed consent separately for data collection within the hospital and for data collection by a community-based physician outside the hospital. Thus, patients could restrict their consent to the hospital phase of the study if desired. Patients had the right to withdraw data consent at any time; in such cases, data from withdrawn patients until the time of withdrawal from the study were used for analysis.
Results
Disposition
A total of 65 hospital sites participated in the study, including 48 active sites that enrolled 252 patients from 2 November 2010 to 15 November 2012. The last patient visit occurred on 15 November 2013. Sites were located in Denmark (4 patients), Germany (112 patients), Italy (8 patients), Norway (4 patients), Spain (49 patients), Sweden (11 patients), and the UK (64 patients). The 252 enrolled patients had a total of 271 enrollment periods in the study; 18 patients were enrolled more than once (including 17 enrolled twice and one enrolled 3 times), as multiple enrollment periods for a patient were allowed. At the time of data cutoff, 41 (15.1%) of the 271 enrollment periods were completed, and 176 (64.9%) had been discontinued. The most common reasons for discontinuation of enrollment periods were “other” (n = 64; 23.6%), death (n = 47; 17.3%), patient lost to follow-up (n = 31; 11.4%), and investigator discretion (n = 21; 7.7%) (Supplementary Table S2).
Exposure
The mean duration of tolvaptan exposure was 139.4 (SD 239.68) days, with a median treatment length of 18.5 (range of 1–1130) days. The patient with 1130 days of tolvaptan use had this length of exposure during the 3-year study period because tolvaptan was started for idiopathic SIADH 2 months before the start of the study and the patient was enrolled after the study had commenced. A total of 79/252 patients (31.3%) had their dose increased, 82/252 (32.5%) had a dose reduction, and 78/252 (31.0%) had a temporary treatment interruption. The maximum daily dose ranged from 3.75 to 60.0 mg/day. The number of patients who received each dose level at least once during the study is shown in Table 1. A daily dose of 15 mg was the dose administered to the greatest number of patients (189/252; 75.0%).
Patient Characteristics
Patient baseline demographic and clinical characteristics are shown in Table 2. The mean age of patients was 70.6 (SD 13.37) years, with a nearly equal gender distribution. In total, 247/252 (98.0%) patients had a clinical diagnosis of hyponatremia, with a mean time from diagnosis to baseline study visit of 119.8 (SD 439.56; range 1–3771) days. The most frequent SIADH etiology was tumor (107/252; 42.5%), followed by idiopathic disease (66/252; 26.2%) and drug-induced (22/252; 8.7%). As the study eligibility criteria included all patients prescribed tolvaptan and who consented to data collection, nine non-SIADH patients to whom tolvaptan was prescribed off-label were captured, consisting of three patients diagnosed with hyponatremia due to liver cirrhosis, one with hyponatremia due to drug (spironolactone) and cirrhosis, two with congestive heart failure, two with disorders of the central nervous system, and one with post-surgery adrenal insufficiency. There was one additional case of differential diagnosis between polydipsia and SIADH.
At baseline, 245/252 patients (97.2%) had a concurrent diagnosis or abnormality. The most frequent current medical conditions by system organ class were vascular disorders (128 patients, 50.8%), neoplasms benign, malignant, and unspecified (including cysts and polyps) (120 patients, 47.6%), metabolism and nutrition disorders (101 patients, 40.1%), and gastrointestinal disorders (86 patients, 34.1%). The most frequent concurrent medical conditions by preferred term were hypertension (118 patients, 46.8%), chronic obstructive pulmonary disease (32 patients, 12.7%), constipation (27 patients, 10.7%), hyperlipidemia (27 patients, 10.7%), and hypothyroidism (27 patients, 10.7%). One patient had a prior medical history of chronic renal failure.
The hospital setting at time of patient enrollment (269 hospitalizations in total) was most often a hospital ward (216/269; 80.3%). Others included an intensive care unit (11; 4.1%), an emergency department (5; 1.9%), and intermediate care (2; 0.7%). Sixteen subjects (5.9%) were in outpatient care, most likely initiating tolvaptan during a hospital visit but without being admitted. During the study, 113/252 (44.8%) patients received a saline infusion and 117/252 (46.4%) had fluid restriction.
Serum Sodium
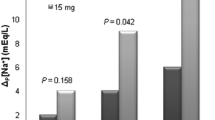
The mean serum sodium value at baseline for the study population was 123.2 mmol/l (SD 6.59). Mean change from baseline in serum sodium over time is shown in Fig. 1 for patients grouped according to the dose they received during their stable treatment phase (7.5, 15, or 30 mg/day). Increases from baseline in serum sodium were comparable across stable tolvaptan dose categories; no dose relationship was discernible. Following an increase during the first week of treatment (mean increase of 7.3 [SD 5.18] mmol/l for 7.5 mg/day, 11.8 [SD 7.76] mmol/l for 15 mg/day, and 7.3 [SD 7.17] mmol/l for 30 mg/day), serum sodium levels were stable in each dose group during the rest of the treatment period. Mean serum sodium values were more variable over time in patients receiving tolvaptan at stable doses of 45 mg/day (n = 1) or 60 mg/day (n = 1), but these data are not shown because of the small number of patients in these groups. Absolute serum sodium over time is shown in Supplementary Fig. S1.

Mean (SD) change from baseline in serum sodium (mmol/l) in patients grouped by stable daily tolvaptan dose received. Patients who did not enter a stable treatment phase were grouped according to their most used dose. Data are not shown for patients who received tolvaptan 3.75 mg less than daily, 3.75 mg/day, 7.5 mg less than daily, 15 mg less than daily, 45 mg/day, or 60 mg/day as a stable daily dose, given the small numbers of patients receiving a specific dose within each category. BSL baseline, D day, M month, SD standard deviation, W week
Hyponatremia Symptoms
At baseline, 122/252 (48.4%) patients presented with ≥ 1 hyponatremia symptom (Fig. 2). The most common individual symptoms were fatigue and nausea (each 23 patients, 9.1%), confusion (22 patients, 8.7%), and disorientation and unsteady gait (each 21 patients, 8.3%). Symptom frequency decreased during treatment: 46/252 (18.3%) patients presented with a symptom at any time point post-baseline. At every individual post-baseline assessment time point, no individual symptom exceeded 1.2% in frequency.

Hyponatremia symptoms occurring in ≥1% of patients at either baseline or at any time during tolvaptan treatment. Baseline includes any symptoms starting in the 31 days up to tolvaptan initiation. Percentages are shown for the total number of patients (N = 252)
Adverse Events
A summary of adverse events is presented in Table 3. In results grouped by dose level (whether administered daily or less frequently than daily), there was no evident association of dose with frequency of TEAEs. In the 3.75 mg daily or less than daily dose group, 0 patients (0.0%) experienced TEAEs (there were only four patients in this group); in the 7.5 mg less than daily and 7.5 mg daily groups, 27/45 patients (60.0%) and 57/106 patients (53.8%), respectively, experienced TEAEs; in the 15 mg less than daily and 15 mg daily groups, 19/31 patients (61.3%) and 105/189 patients (55.6%), respectively, experienced TEAEs; in the 30 mg less than daily group no patient experienced any TEAEs (there were only three patients in this group); and in the 30 mg daily group 27/43 patients (62.8%) experienced TEAEs.
Regarding adverse event severity, there was no clear indication that higher daily tolvaptan doses were associated with increased reporting of serious TEAEs. In the 3.75 mg daily or less than daily group, 0/4 patients (0.0%) experienced serious TEAEs; in the 7.5 mg less than daily and 7.5 mg daily groups, 19/45 patients (42.2%) and 32/106 patients (30.2%) experienced serious TEAEs, respectively; in the 15 mg less than daily and 15 mg daily groups, 13/31 patients (41.9%) and 73/189 patients (38.6%) experienced serious TEAEs, respectively; in the 30 mg less than daily group, 0/3 patients (0.0%) experienced any serious TEAEs; and in the 30 mg daily group, 21/43 patients (48.8%) experienced serious TEAEs. There was no indication of any dose relationship for individual events.
TEAEs with a fatal outcome were experienced by 57 patients (22.6%), and a total of 105 events were reported. The number of TEAEs with a fatal outcome per 100 patient-years was 121.3. Regarding a potential causal relationship with tolvaptan, the physician considered the events with fatal outcome as not related to tolvaptan for 51 patients and to have an unknown relationship to tolvaptan for the other 6 patients. The preferred terms most commonly associated with a fatal outcome were: death not otherwise specified (eight patients, 3.2%), lung neoplasm malignant and malignant neoplasm progression (seven patients each, 2.8%), sepsis and respiratory failure (five patients each, 2.0%), and urinary tract infection, small-cell lung cancer, and small-cell lung cancer extensive stage (four patients each, 1.6%).
Twenty-one aquaresis-related TEAEs were experienced by 18/252 patients (7.1%), including 9 (3.6%) patients with thirst, 5 (2.0%) with dry mouth, 3 (1.2%) with orthostatic hypotension, and 2 (0.8%) with dehydration. One patient experienced a TEAE of increased blood creatinine.
A total of 62/252 patients (24.6%) experienced ≥ 1 episode of rapid correction of hyponatremia during the study. Mean baseline serum sodium for this group was 120 (range 105, 131) mmol/l. For the 190 patients who did not experience rapid correction of hyponatremia, mean baseline serum sodium was 125 (range 106, 141) mmol/l. No event of osmotic demyelination syndrome was reported during the study. Two patients experienced epilepsy/seizure related events, but in these two cases the events occurred very late after the episode of rapid correction of hyponatremia (11 months and 8 months, respectively) and were assessed as not related to tolvaptan by the physicians.
A search for predefined preferred terms related to hepatic disorders identified nine cases. Two cases, cholestasis secondary to hepatic metastases and jaundice by tumor progression, respectively, were clearly due to malignancy. The remaining seven cases (five serious and two non-serious) consisted of congestive liver (accompanying acute bilateral heart failure and pleural effusion), mild increases in alanine aminotransferase and gamma-glutamyl transferase versus baseline (in a patient with small cell lung cancer and liver metastasis), progression of pre-existing liver cirrhosis with encephalopathy and ascites, liver failure and septicemia (in a patient with liver cirrhosis), increased international normalized ratio/prolonged prothrombin time value (in a patient with a history of heart disease as atrial fibrillation, transient ischemic attacks, aortic stenosis, and cardioversion), liver values increased (in a patient with concomitant epilepsy, subcortical arteriosclerotic encephalopathy, and hypertension), and elevated liver values (transaminases > 450 U/l) most likely due to cardiac decompensation. Tolvaptan was temporarily interrupted following congestive liver with acute bilateral heart failure/pleural effusion, while no action regarding tolvaptan was taken for the other six events.
TEAEs were assessed in patients by concomitant medication use (CYP P450 3A4 inducers/inhibitors, serum potassium concentration increasing substances, and warfarin/antiplatelet agents). The frequency of TEAEs by system organ class is shown in Supplementary Tables S3, S4, S5, and S6 for each category of concomitant medication by non-users and users. For users of concomitant medications, TEAEs are further categorized by whether they occurred during or outside of the period of concomitant medication use. Differences in TEAE frequency occurred between users and non-users of concomitant medication; however, frequencies were generally similar between the subgroups of users who experienced the TEAEs during or outside the period of concomitant use. Accordingly, the TEAEs were most likely associated with the underlying condition rather than the medication used.
Discussion
The management of SIADH-related hyponatremia in clinical practice is often inadequate because of lack of appropriate diagnostic workup and underuse of available therapies [22]. Clinical trials of tolvaptan in SIADH-related hyponatremia conducted in Europe and North America, and, more recently, Japan, have demonstrated efficacy, leading to regulatory approval for this indication in these regions [17, 23]. Consistent with the clinical trial findings, tolvaptan in this post-marketing study was associated with increases in serum sodium in a population with hyponatremia secondary to SIADH and a high burden of comorbidity. After an initial increase during the first week of treatment, mean concentrations remained largely steady during follow-up. The increases from baseline (7–12 mmol/l) were similar across tolvaptan dose categories, with no dose relationship observed, a finding consistent with a previous pharmacodynamic study in patients with SIADH [24]. The magnitude of the increase from baseline in serum sodium at week 1 was comparable to improvements (10–14 mmol/l) seen in previous tolvaptan studies conducted in real-word settings among populations with varying baseline sodium levels and durations of follow-up [25,26,27]. The percentage of patients in the present study with ≥ 1 hyponatremia symptom decreased from baseline (48.4%) to the treatment period (18.3%), suggesting that improvement in sodium level was associated with improvement in hyponatremic symptoms.
The most frequent TEAEs were rapid correction of hyponatremia, nausea, and urinary tract infection. Aquaresis-related TEAEs were less frequent than observed in hyponatremia clinical trials, i.e., thirst 3.6% versus 14.3% and dry mouth 2.0% versus 12.6%, whereas there was a higher frequency of rapid correction of hyponatremia in this post-marketing study (62 of 252 patients within 72 h) compared to controlled clinical trials (4 of 223 patients during the first 24 h) [17]. The higher frequency of rapid correction of hyponatremia in the present study may be in part due to the low mean baseline serum sodium level (123 mmol/l) relative to the pivotal clinical trials (~ 129 mmol/l) and the even lower level in the patients who experienced a rapid correction of hyponatremia (120 mmol/l). Studies of tolvaptan conducted in real-world settings found rates of rapid correction ranging from 3 to 25% of patients, with lower baseline serum sodium associated with greater risk [25,26,27,28]. An additional possible reason for rapid correction in our study may include the composition of the population, which included a high proportion of patients with tumors (42.5%) and frequent concomitant use of saline solution (44.8%). Additionally, as noted in the SmPC, use of tolvaptan in conjunction with other treatments to correct hyponatremia may increase the risk of rapid correction [18]. Rapid correction did not lead to any neurologic sequelae in this study, and no cases of osmotic demyelination syndrome were reported in several previous real-world studies of tolvaptan, highlighting the importance of monitoring serum sodium [25,26,27,28].
Regarding patterns of use in this European study, tolvaptan was prescribed for the correct indication per the SmPC in nearly all patients; only 9/252 patients did not have SIADH. In addition, there was one case of differential diagnosis between polydipsia and SIADH. The use of doses lower than those specified in the labeling (minimum dose of 7.5 mg/day) was common (49/252 patients), which may have been due to the high burden of underlying disease in this population, necessitating careful dose titration by physicians.
Limitations of this real-world study include data collection, which was dependent on local standards of practice rather than prespecified visit and assessment schedules, and the lack of a control group or randomized design. No standardized data-reporting protocols were used; rather, the investigators simply recorded the choices made for patient care. Post-marketing pharmacovigilance conducted in the context of routine clinical practice is inherently limited in data collection and patient follow-up relative to controlled clinical trials. Another limitation is that the study had a high proportion of patients (n = 112) enrolled at sites within Germany and a consequent large proportion of data collected from a geographically limited set of study sites and treating physicians. Furthermore, SIADH may be caused by different underlying conditions, resulting in a heterogenous study population.
Conclusion
This observational study found that tolvaptan is selected for use in patients with hyponatremia secondary to SIADH in a manner consistent with the drug SmPC and that tolvaptan is associated with improvement in serum sodium levels and hyponatremia symptoms. Use of tolvaptan to treat hyponatremia secondary to SIADH in the clinical setting requires careful monitoring of serum sodium levels to reduce the risk of rapid correction of hyponatremia, given that the target patient population is at risk because of the underlying disease and baseline sodium level.
References
Hauptman PJ, Greenberg A, Verbalis JG, et al. Design of a prospective, multinational registry to evaluate patients hospitalized with hyponatremia: the HN Registry. Open Access J Clin Trials. 2013;5:93–100.
Hoorn EJ, Lindemans J, Zietse R. Development of severe hyponatraemia in hospitalized patients: treatment-related risk factors and inadequate management. Nephrol Dial Transplant. 2006;21:70–6.
Waikar SS, Mount DB, Curhan GC. Mortality after hospitalization with mild, moderate, and severe hyponatremia. Am J Med. 2009;122:857–65.
Wald R, Jaber BL, Price LL, Upadhyay A, Madias NE. Impact of hospital-associated hyponatremia on selected outcomes. Arch Intern Med. 2010;170:294–302.
Miller M, Morley JE, Rubenstein LZ. Hyponatremia in a nursing home population. J Am Geriatr Soc. 1995;43:1410–3.
Chen LK, Lin MH, Hwang SJ, Chen TW. Hyponatremia among the institutionalized elderly in 2 long-term care facilities in Taipei. J Chin Med Assoc. 2006;69:115–9.
Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18:760–71.
Anderson RJ, Chung HM, Kluge R, Schrier RW. Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin. Ann Intern Med. 1985;102:164–8.
Padhi R, Panda BN, Jagati S, Patra SC. Hyponatremia in critically ill patients. Indian J Crit Care Med. 2014;18:83–7.
Tzoulis P, Bouloux PM. Inpatient hyponatraemia: adequacy of investigation and prevalence of endocrine causes. Clin Med. 2015;15:20–4.
Shepshelovich D, Leibovitch C, Klein A, et al. The syndrome of inappropriate antidiuretic hormone secretion: distribution and characterization according to etiologies. Eur J Intern Med. 2015;26:819–24.
Stelfox HT, Ahmed SB, Khandwala F, Zygun D, Shahpori R, Laupland K. The epidemiology of intensive care unit-acquired hyponatraemia and hypernatraemia in medical-surgical intensive care units. Crit Care. 2008;12:R162.
Gheorghiade M, Abraham WT, Albert NM, OPTIMIZE-HF Investigators and Coordinators, et al. Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE-HF registry. Eur Heart J. 2007;28:980–8.
Ruf AE, Kremers WK, Chavez LL, Descalzi VI, Podesta LG, Villamil FG. Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone. Liver Transplant. 2005;11:336–43.
Kim WR, Biggins SW, Kremers WK, et al. Hyponatremia and mortality among patients on the liver-transplant waiting list. N Engl J Med. 2008;359:1018–26.
Zilberberg MD, Exuzides A, Spalding J, et al. Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients. Curr Med Res Opin. 2008;24:1601–8.
Schrier RW, Gross P, Gheorghiade M, SALT Investigators, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–112.
Samsca [Summary of Product Characteristics]. January 28, 2020. https://www.ema.europa.eu/en/documents/product-information/samsca-epar-product-information_en.pdf. Accessed 12 July 2020.
World Health Organization. WHO pharmacovigilance indicators: a practical manual for the assessment of pharmacovigilance systems. Geneva: Switzerland; 2015. https://www.who.int/medicines/areas/quality_safety/safety_efficacy/EMP_PV_Indicators_web_ready_v2.pdf. Accessed 28 May 2021.
Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–72.
Laville M, Burst V, Peri A, Verbalis JG. Hyponatremia secondary to the syndrome of inappropriate secretion of antidiuretic hormone (SIADH): therapeutic decision-making in real-life cases. Clin Kidney J. 2013;6(Suppl 1):i1–20.
Verbalis JG, Greenberg A, Burst V, et al. Diagnosing and treating the syndrome of inappropriate antidiuretic hormone secretion. Am J Med. 2016;129:537.e9-537.e23.
Arima H, Goto K, Motozawa T, et al. Open-label, multicenter, dose-titration study to determine the efficacy and safety of tolvaptan in Japanese patients with hyponatremia secondary to syndrome of inappropriate secretion of antidiuretic hormone. Endocr J. 2021;68:17–29.
Shoaf SE, Bricmont P, Dandurand A. Low-dose tolvaptan PK/PD: comparison of patients with hyponatremia due to syndrome of inappropriate antidiuretic hormone secretion to healthy adults. Eur J Clin Pharmacol. 2017;73:1399–408.
Tzoulis P, Waung JA, Bagkeris E, et al. Real-life experience of tolvaptan use in the treatment of severe hyponatraemia due to syndrome of inappropriate antidiuretic hormone secretion. Clin Endocrinol. 2016;84:620–6.
Han SW, Yi JH, Kang KP, et al. Safety and efficacy of tolvaptan in Korean patients with hyponatremia caused by the syndrome of inappropriate antidiuretic hormone. J Korean Med Sci. 2018;33:e112.
Pose-Reino A, Runkle de la Vega I, de Jong-Laird A, et al. Real-world, non-interventional, retrospective study (SAMPLE) of tolvaptan in patients with hyponatraemia secondary to the syndrome of inappropriate antidiuretic hormone secretion. Adv Ther. 2021;38:1055–67.
Morris JH, Bohm NM, Nemecek BD, et al. Rapidity of correction of hyponatremia due to syndrome of inappropriate secretion of antidiuretic hormone following tolvaptan. Am J Kidney Dis. 2018;71:772–82.
Acknowledgements
We thank the participants of the study.
Funding
This study was funded by Otsuka Pharmaceutical Development & Commercialization, Inc. (Rockville, MD, USA) and Otsuka Pharmaceutical Co., Ltd. (Tokyo, Japan). Participating physicians, pharmacists, and, as appropriate, hospital administrative representatives received compensation for their study participation from Otsuka. The journal’s Rapid Service and Open Access Fees were provided by Otsuka.
Medical Writing and Editorial Assistance
Writing and editorial services in preparation of the manuscript, including drafting the first version, were provided by Andrew J. Horgan, PhD, of BioScience, Inc (New York, NY), activities that were funded by Otsuka.
Authorship
All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
All authors contributed to the study conception and design, material preparation, data collection and analysis. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Disclosures
Alvin Estilo, Linda McCormick, and Mirza Rahman are employees of Otsuka.
Compliance with Ethics Guidelines
The study was conducted in accordance with Good Pharmacoepidemiology Practices, local national laws and regulations applicable to non-interventional studies, Volume 9A (in European Economic Area countries), and ethical principles that have their origins in the Declaration of Helsinki. Where required by local regulations for observational studies, the study was conducted under the auspices of an Independent Ethics Committee. The participating Ethics Committees were: Clinical Research Ethics Committee of Hospital Universitario “12 de Octubre”, Madrid, Spain; Comitato Etico Interaziendale A.S.O. Santa Croce e Carle, Cuneo, Italy; Research Ethics Committee Northwest 10 REC – GM North, Northwest Centre of Research Ethics Committees, Manchester, United Kingdom; Regionala etikprövningsnämnden, Göteborg, Göteborg, Sweden; Regional Committee for Medical & Health Research Ethics South East A (REK South East A), Oslo, Norway; Ethik-Kommission bei der Medizinischen Fakultät Universität Marburg, Marburg, Germany. Patients provided informed consent separately for data collection within the hospital and for data collection by a community-based physician outside the hospital. Thus, patients could restrict their consent to the hospital phase of the study if desired. Patients had the right to withdraw data consent at any time; in such cases, data from withdrawn patients until the time of withdrawal from the study were used for analysis.
Data Availability
To submit inquiries related to Otsuka clinical research, or to request access to individual participant data (IPD) associated with any Otsuka clinical trial, please visit https://clinical-trials.otsuka.com/. For all approved IPD access requests, Otsuka will share anonymized IPD on a remotely accessible data sharing platform.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Estilo, A., McCormick, L. & Rahman, M. Using Tolvaptan to Treat Hyponatremia: Results from a Post-authorization Pharmacovigilance Study. Adv Ther 38, 5721–5736 (2021). https://doi.org/10.1007/s12325-021-01947-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01947-9