
Abstract
Follicular lymphoma (FL) is one of the most common subtypes of non-Hodgkin lymphoma worldwide. Improved survival outcomes with rituximab-based therapy in clinical trials led to the establishment of rituximab-based immunochemotherapy as standard of care for first-line (1L) treatment of FL. In the GALLIUM trial, obinutuzumab-based immunochemotherapy demonstrated improved progression-free survival (PFS), prolonged time-to-next antilymphoma treatment (TTNT) and comparable overall survival (OS) compared with rituximab-based immunochemotherapy as 1L treatment for FL. Using GALLIUM as an example, this article aims to explain how improved outcomes in 1L treatment of FL have changed the landscape for the design and interpretation of future trials. As approved therapies for 1L FL already achieve good responses, it is becoming more difficult to design trials that demonstrate further treatment benefits with the currently accepted primary endpoints. New endpoints are needed to reflect the long remission times, low relapse rates, and impact of subsequent therapies in FL. PFS is used as a primary efficacy endpoint in registrational clinical trials for indolent malignancies like FL, where improvement in OS is not always observed due to the large number of patients and long study duration required to demonstrate a clear survival benefit. However, there are limitations to using PFS as the primary endpoint. Other potential endpoints, including TTNT, progression of disease within 2 years, response rate, and minimal residual disease status are explored.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Rituximab chemotherapy and obinutuzumab chemotherapy are effective treatments for patients with previously untreated follicular lymphoma. |
There are challenges in designing modern clinical trials to demonstrate further benefit achieved with new therapies. |
Endpoints such as overall survival are often not feasible to achieve due to the need for large numbers of patients and long follow-up. |
Progression-free survival is often used as the primary measure of efficacy in registrational clinical trials, although it has some limitations. |
Other endpoints, e.g. complete response rate at 30 months and progression of disease within 24 months, may be considered for future trials, but they too have limitations and their clinical relevance should be explored further. |
Risk-adaptive trials to direct treatment should be explored. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.14381117.
Introduction
Follicular lymphoma (FL) is one of the most common types of non-Hodgkin lymphoma (NHL), accounting for ~ 20% of all NHL cases globally [1]. It is often stated that FL has no cure. However, patients can be disease-free after 20 years, even without treatment. FL can therefore be a slowly progressing disease with pseudo-progression and spontaneous regression. As such, the benefit–harm assessment of new treatments needs to incorporate changing patterns in the natural evolution of the disease.
Clinical trials of the type I anti-CD20 antibody rituximab in the early-2000s showed a significant improvement in patient outcomes that led to rituximab-based immunochemotherapy becoming the standard of care for 1L treatment of FL. Rituximab with chemotherapy was shown to prolong overall survival (OS) when given as induction therapy, and to delay the time to disease progression when rituximab was used as maintenance therapy [2, 3]. Obinutuzumab, a type II anti-CD20 antibody, in combination with chemotherapy was granted approval in 2017 by the US Food and Drug Administration (FDA), European Medicines Agency (EMA), and many other agencies for the 1L treatment of FL. Approval was based on the results of the multicentre, randomised, Phase III, GALLIUM trial, which demonstrated improved progression-free survival (PFS) with obinutuzumab-based immunochemotherapy compared with rituximab-based immunochemotherapy in this patient population [4].
This article explains how improved outcomes in 1L treatment of FL have changed the landscape for the design and interpretation of future trials. Relevant outcome measures used in contemporary clinical trials are discussed, using the GALLIUM trial as an example. Finally, the design, population and endpoints of future trials in 1L treatment of FL are considered. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Evolution of First-Line Follicular Lymphoma Treatment
Prior to 2000, the median survival among patients with advanced FL increased from 63 to 72 months (from 1983–1989 to 1990–1999) in the US, an increase thought to be due to the availability of new therapeutic options and better supportive care [5, 6].
Rituximab
Several randomised Phase III trials were conducted to evaluate rituximab in combination with a variety of chemotherapy regimens, including cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), cyclophosphamide, vincristine, and prednisone (CVP), cyclophosphamide, adriamycin, teniposide, and prednisone (CHVP), and mitoxantrone, chlorambucil, and prednisone (MCP) regimens (Table 1) [2, 7,8,9]. In 2007, a meta-analysis showed consistently superior OS with rituximab plus chemotherapy versus chemotherapy alone [10]. Based on the evidence provided by these studies, rituximab therapy plus chemotherapy has been adopted as the standard of care for the 1L treatment of patients with FL.
Two years of rituximab maintenance was subsequently found to improve PFS in patients responding to induction immunochemotherapy [3, 11]. The PRIMA trial compared rituximab maintenance with observation in patients who had responded to induction with immuno-chemotherapy [3, 12]. The final analysis of PRIMA, performed after a median of 9 years of follow-up, demonstrated the clinically relevant long-term benefit of rituximab maintenance on PFS in patients who had responded to induction with immunochemotherapy [12]. Rituximab maintenance led to a significant improvement in PFS compared with observation [10.5 vs. 4.1 years, hazard ratio (HR) 0.61; 95% confidence interval (CI) 0.52–0.73], but there was no evidence of a difference in OS (HR 1.04; 95% CI 0.77–1.40), and the 10-year OS rate was approximately 80% in both arms. A 2017 individual patient data meta-analysis showed that rituximab maintenance improved OS versus observation in patients with FL [13], although this analysis included heterogeneous trials in the relapsed setting, with and without rituximab induction therapy.
Obinutuzumab-Based Compared with Rituximab-Based Immunochemotherapy
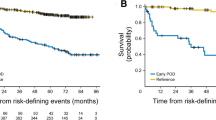
Nonclinical studies have shown that obinutuzumab mediates superior induction of direct cell death and effector cell-mediated, antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis, together with reduced CD20 internalisation, compared with the type I CD20 antibodies rituximab and ofatumumab [14, 15]. The GALLIUM trial was designed to show the superiority of obinutuzumab over rituximab in 1L FL. Overall, 1202 patients were randomised 1:1 to receive obinutuzumab-based or rituximab-based immunochemotherapy (with CHOP, CVP or bendamustine) as induction treatment, and those exhibiting a response continued with the same antibody as maintenance treatment for up to 2 years. Details of the trial have been published previously [16, 17]. Investigator-assessed PFS was the primary endpoint, with a target HR of 0.74, and, at the time of the primary analysis, the observed HR was 0.66 with 95% CI 0.51–0.85 (p = 0.001; Fig. 1) [17]. Similarly, in later analyses after a median 76.5 months of follow-up, the HR was 0.76, 95% CI 0.62–0.92 (p = 0.0043) [18]. This is the only trial to date that compares obinutuzumab-based versus rituximab-based immunochemotherapy in patients with FL.

Copyright © 2017, Massachusetts Medical Society
Kaplan–Meier estimates of investigator-assessed PFS in the GALLIUM trial [17]. Chemotherapy was stipulated at each site, with all patients at that site receiving the same regimen; options included CHOP, CVP, or bendamustine. CHOP cyclophosphamide, doxorubicin, vincristine, and prednisone; CI confidence interval; CVP cyclophosphamide, vincristine, and prednisone; HR hazard ratio; PFS progression-free survival.
Rationale for Using PFS as a Primary Outcome Measure in Phase III Clinical Trials of First-Line Follicular Lymphoma
In most cases, regulatory agency guidelines recognise that endpoints other than OS may represent a benefit to the patient and that these alternative endpoints may potentially be used for accelerated and full approval of new therapies [19, 20]. EMA guidelines state that “prolonged PFS is considered to be of benefit to the patient”, and that the choice of primary endpoint should be guided by the relative toxicity of the experimental therapy. Additional factors such as expected survival after progression, availability of next-line therapies, the prevalence of the condition and health-related quality of life, while the magnitude of treatment effect on all relevant measures must also be taken into account, and may help to identify what is the real clinical benefit for the patients [20]. FDA guidelines note that PFS has served as a primary endpoint for drug approval, and that it has the advantage of not being confounded by subsequent therapy [19]. These FDA and EMA guidelines provide methodological considerations for using PFS.
The finding of significantly improved PFS, but not OS, in the PRIMA trial after a median 9 years of follow-up illustrates the difficulty in demonstrating OS advantages in the setting of indolent FL, and why PFS is sometimes considered an appropriate primary measure of efficacy in 1L FL [12]. Some of the lack of effect on OS in the PRIMA trial may have been due to treatment crossover in the maintenance setting and/or use of subsequent therapies. Although data regarding the use of crossover maintenance treatment in the relapse setting were not available, it is likely that imbalances in using rituximab at relapse may have also influenced survival outcomes. In total, 81.5% of patients in the observation arm received rituximab-based therapy at relapse or progression. This probably affected the time from progression to death, and diluted the OS HR [12]. Crossover can be accounted for in statistical analyses, although this requires various assumptions to be made about the data, and may not be accepted by regulators and payers [21], especially when a substantial number of patients in the control arm cross over to the experimental arm. Additionally, OS data take much longer to mature than PFS in FL due to the indolent nature of the disease.
The justification for considering PFS as the primary endpoint rests on the limitations of OS in light of current excellent patient prognoses, trial treatment crossovers and multiple effective additional anti-cancer therapies, all of which together makes it difficult, if not impossible, to show a clear improvement in OS. A trial that could never show a clinically meaningful and statistically significant benefit for OS is unlikely to be funded, as having a long and expensive trial outweighs the likely small chance of showing an OS improvement. This may be an unsatisfactory view, but cancer therapy is a rapidly changing environment (e.g. changes in background patient care and the available therapeutic options), so a 12- to 15-year trial is unlikely to have sufficient appeal. Nevertheless, investigators are encouraged to consider higher-risk patient groups (poor prognosis), potentially using modern novel biomarkers for risk stratification, for which OS is appropriate.
There is extensive published literature discussing the strengths and limitations of PFS as a surrogate endpoint for OS in various cancers, as well as how to evaluate surrogacy [22,23,24,25,26]. Zhu et al. [27] examined trial-level data on various surrogate markers in FL, but were unable to evaluate the correlation between PFS and OS due to an insufficient number of trials.
PFS is likely not a surrogate for OS in FL. Statistical modelling indicates that the association between PFS and OS tends to weaken with increasing post-progression survival time, and this period is prolonged with indolent malignancies such as FL [28]. Historically, payers have been reluctant to accept PFS as a patient-relevant endpoint in FL. However, future trials of 1L treatments for FL are expected to demonstrate benefits on PFS but not on OS.
There are several limitations to using PFS as a primary endpoint in clinical trials of FL. In lymphoma, progression is generally defined using either Cheson 2007 [29] or Lugano 2014 criteria [30], both of which have a subjective element. Progression assessment may also be influenced by the size and location of the target lesions, and the timing of the assessment. Several other considerations apply to using PFS as a primary endpoint, particularly what its clinical value is to patients and clinicians when the trial cannot show a benefit on OS and only an improvement in PFS, and whether this is complemented by other benefits such as improvements in health-related quality of life and/or cost savings due to lower costs associated with the management of disease progression. For example, a trial might show that PFS is improved using combination immunochemotherapy compared to rituximab monotherapy, but if most patients survive for many years on both arms and 30% never need additional chemotherapy [31], the advantage of PFS might be less clear cut to patients and payers.
Work is currently ongoing in some cancer types to ascertain how patients interpret PFS, and there is also research on whether improvements in PFS are directly linked to improvements in quality of life or reduction in resource use [32]. If such associations can be demonstrated in FL, this could give more support to the value of PFS in this disease. Although using PFS might be uncertain in some cases, it does allow a comparison between two trial arms, and avoids issues that might occur when patients cross over between treatment arms or receive effective subsequent therapies.
Different Measures of Efficacy
Clinical trial efficacy results can be summarised in several different ways: HRs, differences in event rates (proportions) at a specific time point, or differences in medians. They each indicate different aspects of the effect of a treatment.
Hazard Ratio
In the primary analysis of the GALLIUM trial, the HR for PFS was 0.66, which represents a 34% reduction in the risk (hazard) of progression, relapse or death at any time point among patients treated with obinutuzumab [17]. It is a measure of relative effect. Because the HR summarises information for all patients over the entire time they have each been followed, it is often considered to be a reliable measure of efficacy. It takes into account the number and timing of events, and therefore reflects whether few or many patients benefit from a new treatment. Relying on the assumption of proportional hazards over time, the HR can be estimated early on in a trial. This implies that it is particularly useful when the median PFS (or OS) has not been reached, which can occur with FL, especially in 1L studies.
The assumption underlying the use of proportional hazards is that the ratio of hazard functions between two treatment groups is the same at all time points (i.e. the hazards are proportional). However, it is likely that the treatment effect will not last forever, so the hazards will start to approach each other over time. Therefore, this assumption may not be valid during the later years of follow-up that are reported in a trial paper, when, for example, curves overlap each other. One needs to then carefully assess whether the HR still provides a meaningful quantifier of the effect and an estimate of the ‘risk’ (e.g. of progression) when comparing outcomes between treatments. Also, when long-term benefits are seen in plateauing survival curves, this effect may not be appropriately summarised using a single HR. However, quantification of such effects can be important for understanding the effects of 1L treatments for FL.
Event Rates (Proportions)
If we specify a particular milestone time point, the estimated event rates at that point can be compared. In the GALLIUM trial, the PFS rates at 3 years were 80.0% (95% CI 75.9–83.6) and 73.3% (95% CI 68.8–77.2) for the obinutuzumab and rituximab arms, respectively [17]. This represents an absolute risk difference of 6.7 percentage points (i.e. among 100 patients given obinutuzumab, an estimated extra 6.7 are alive and progression-free at 3 years compared with 100 patients given rituximab). A limitation of using event rates is that they only summarise the treatment effect at a single specified milestone. Furthermore, estimates at one time point typically come with higher uncertainty than a HR, meaning they can be influenced by chance ‘blips’ on the curve. A sufficient number of patients need to have been followed up to the chosen milestone. However, event rates at 3 or 5 years can describe the longer-term benefits of treatments for FL in an easily accessibly way, so they are useful when considered alongside HRs.
Medians
Median PFS (or OS or other time-to-event endpoints) is typically measured on the scale of the actual endpoint, i.e. months or years, so comparing two median PFS times is often easily understood by patients and clinicians. However, in order to give a meaningful and sufficiently precise estimate, enough trial participants need to have been followed up long enough to reach the median in both groups, and, as with event rates, the median represents only one point on a Kaplan–Meier curve. Taking an early snapshot of median PFS increases the degree of uncertainty with that estimate, and for a long-term endpoint, e.g. PFS in clinical trials such as GALLIUM, it may take a long time until a reasonably precise estimate of the median difference is available. In the GALLIUM trial (Fig. 1), no accurate assessment of the median PFS could be made, even at a median follow-up of 54 months. The situation is even more pronounced for OS, where we expect the median could take many years to be observed, and, even if the OS median was observed earlier, it remains a very imprecise estimate. Therefore, unlike advanced cancers, evaluating new 1L treatments for FL would rarely involve examination of the median PFS, and only HRs or event rates at early milestones are used.
Interpretation of Subgroup Analyses
Subgroup analyses are commonly reported for Phase III clinical trials, but they are sometimes misinterpreted. Investigators use subgroup analyses to provide some assurance that the size of a treatment effect (e.g. HR) seems consistent across groups with different patient or disease characteristics. This is a reasonable approach; however, great care is required when these types of analyses are used to make claims that a new treatment is beneficial (or harmful) for one group of patients but not another [33, 34].
There are two commonly used but fundamentally different statistical approaches for subgroup analyses [35]. The first involves observing whether any of the CIs for the subgroups exclude the overall treatment effect (investigators sometimes incorrectly compare the CIs to HR = 1). Examining the subgroup analyses for the GALLIUM trial (Fig. 2), the HR for male patients was 0.82, with a 95% CI 0.59–1.15, and for female patients was 0.49 with 95% CI 0.33–0.74 [17]. Both of these CIs clearly overlap the overall HR of 0.66, so there is insufficient evidence to suggest the size of the treatment effect of obinutuzumab among either males or females differs from the effect seen in all patients. Statistical evidence of a subgroup effect would occur if either of the 95% CIs excluded 0.66, but further confirmation is required.

Copyright © 2017, Massachusetts Medical Society
GALLIUM trial prespecified subgroup analyses [17]. ADL activities of daily living; Chemo chemotherapy; CHOP cyclophosphamide, doxorubicin, vincristine and prednisone; CI confidence interval; CVP cyclophosphamide, vincristine and prednisone; ECOG Eastern Cooperative Oncology Group; FL follicular lymphoma; FLIPI Follicular Lymphoma International Prognostic Index; G obinutuzumab; HR hazard ratio; IADL instrumental activities of daily living; IPI International Prognostic Index; KM Kaplan–Meier; NE not estimable; R rituximab.
The second approach is a test for interaction that compares the HRs between two subgroups [35]. For example, this tells us whether the HR of 0.82 (observed in males) is statistically significantly different from the HR of 0.49 (females) in GALLIUM. Here, the p value was 0.056 [17], so again there is insufficient evidence of a subgroup effect using this statistical test [36]. Small p values (≤ 0.05 and ideally ≤ 0.01) provide some statistical evidence of a subgroup difference, and in GALLIUM none of the tests of interaction for any factor had a p value ≤ 0.05.
Subgroup analyses have limitations. First, any subgroup is by definition smaller than the overall study sample, so the results in each subgroup are less reliable by construction. Second, the more subgroups that are investigated, the more likely it is that we will find a differential treatment effect just by chance alone. This is a multiplicity issue. The following criteria should be used to assess the validity of subgroup claims [34, 35]:
-
1.
Have both statistical tests been met (could chance alone explain the finding[s])?
-
2.
Is the subgroup effect consistent across independent studies?
-
3.
Was the subgroup hypothesis one of a small number of hypotheses developed a priori?
-
4.
Is there strong underlying biological support?
None of the factors in Fig. 2 meet all these criteria. In the GALLIUM trial, patients with a low Follicular Lymphoma International Prognostic Index (FLIPI) score had a PFS HR of 1.17 [17], and it might seem biologically plausible that the benefit of obinutuzumab is smaller in patients with a good prognosis, compared with those who had a worse prognosis (high FLIPI score, HR 0.58). However, there is no statistical evidence to support this from the trial (both statistical tests for subgroup analyses mentioned above do not provide sufficient evidence of an effect of FLIPI).
Endpoints Other Than PFS
Several endpoints other than PFS may be useful for trials of 1L treatments for FL. Such endpoints are not accepted by regulatory authorities for the registration of new drugs, but they might be candidates for primary or co-primary endpoints in academic trials. Key considerations regarding the value of alternative endpoints include whether they are clinically relevant to patients, whether they can be standardised when measured, and whether they can serve as a surrogate for OS and/or PFS. Any conclusion about whether an endpoint is a valid surrogate depends on the type/class of drug, type of cancer and line of therapy. They all have limitations.
Time to Next Antilymphoma Treatment
Time to next antilymphoma treatment (TTNT) is defined as time from randomisation to the start of any subsequent antilymphoma therapy, for whatever reason (usually progression). In the primary analysis of the GALLIUM trial, the TTNT HR was 0.68 (95% CI 0.51–0.91; p = 0.009), meaning that patients in the obinutuzumab group were less likely to receive a subsequent anticancer treatment than patients in the rituximab group [17]. With longer follow-up, 84.2% of the obinutuzumab group versus 76.7% of the rituximab group had not started further therapy at 4 years, indicative of a worthwhile longer-term benefit [37]. However, the criteria for initiation of subsequent therapy can vary, introducing a degree of subjectivity to TTNT (and hence potential bias). TTNT is therefore likely to remain as a secondary (supportive) endpoint for regulatory purposes, especially in open-label studies such as GALLIUM, and needs to be precisely defined upfront in study protocols in order to act as a valuable endpoint. Nevertheless, TTNT might be considered a clinically relevant endpoint because it reflects a direct change in how a patient is managed and treated. Indeed, the NICE review of GALLIUM commented that TTNT may be more meaningful to patients than PFS, and that TTNT is expected to be longer in clinical practice than in research trials, because patients are assessed less frequently in the real world [38]. Extending TTNT using a new therapy might therefore be useful.
Progression of Disease Within 24 Months
Disease progression or death due to progressive disease within 24 months of randomisation (POD24) is a potential early endpoint. It was explored in FL in an analysis of pooled data from 13 randomised trials [39]. Studies conducted before or after the introduction of rituximab were included in the analysis, and POD24 was found to be strongly associated with poor OS (HR 5.24 95% CI 4.63–5.93, p < 0.01) [39]. More recently, the risk of POD24 was evaluated in an exploratory analysis of the GALLIUM trial, and obinutuzumab-based chemotherapy was associated with a lower risk of POD24 than rituximab-based chemotherapy (average risk reduction 46.0%, 95% CI 25.0–61.1) [40]. More patients with POD24 received a new antilymphoma treatment compared with patients with later disease progression (64.5% vs. 27.3%, respectively) [40]. In addition, the earlier the POD24 event, the higher the mortality observed [40].
While POD24 correlates with OS at the patient level, surrogacy for OS at the trial level has not been demonstrated. Notably, Bachy et al. [41] recently reported that POD24 was a correlate, but not a surrogate, for OS in the PRIMA study. Another limitation is the paucity of POD24 events with contemporary therapies, which will make it difficult to demonstrate a difference with a new therapy using this endpoint. In the GALLIUM trial, less than 10% of patients treated with obinutuzumab-based chemotherapy experienced an event within 2 years, but statistical significance for the POD24 HR was reached [40].
Complete Response at 30 Months
Achieving durable remission is an important goal in the 1L treatment of FL, and a complete response as assessed by computed tomography (CT) scan at 30 months (CR30) is proposed as a surrogate for PFS in trials in this setting. Shi et al. assessed this endpoint using data from 13 randomised trials [42]. The results (Fig. 3) show a high correlation between the treatment effects on CR30 and PFS (correlation coefficient 0.88; a good surrogate marker should have values ≥ 0.8). The authors concluded that CR30 could be a primary endpoint in trials of 1L treatment for FL.

Copyright © 2017, ASCO
Trial-level correlation between CR30 and PFS in a FLASH analysis of 13 randomised trials [42]. Gold rituximab trials, blue non-rituximab trials; triangles induction trials, circles maintenance trials; the size of the triangles and circles is proportional to the trial population size. The fitted weighted least squares regression line (solid line) is log(HRPFS) = – 0.093 – 0.636 × log(ORCR30); dashed lines 95% prediction limits, horizontal dashed line log(HRPFS) of 0 (i.e. HR of 1), vertical dashed line log(ORCR30) of 0 (i.e. OR of 1). CR30 complete response at 30 months, FLASH follicular lymphoma analysis of surrogacy hypothesis, HR hazard ratio, OR odds ratio, PFS progression-free survival.
However, in an analysis of the GALLIUM trial, the HR for PFS (0.66) was not accompanied by any material improvement in CR30 (obinutuzumab-based chemotherapy 44.3%, rituximab-based chemotherapy 42.1%; difference of 2.2% [Roche Data on file]). It appears, therefore, that CR30 may not be a good surrogate when evaluating obinutuzumab-based combinations, which appear to exert their anti-lymphoma activity through extension of relapse-free intervals. As such, while CR30 may be a good alternative to PFS for some compounds, it may not be wise to use it to replace PFS as a registration endpoint for all compounds.
Minimal Residual Disease
Minimal residual disease (MRD) status provides an indication of the depth of tumour response, and many studies have shown a correlation between MRD and PFS [43,44,45]. Chronic lymphocytic leukaemia (CLL) was the first mature B-cell malignancy for which MRD was used [46]. Over the last two decades, MRD has been investigated in haematological disease including acute leukaemias, CLL, chronic myeloid leukaemia and FL to enable a better understanding of response patterns in different clinical settings [45, 47, 48]. There is accumulating evidence that MRD is a strong independent prognostic factor in FL [49]. In GALLIUM, MRD status in peripheral blood or bone marrow was defined as negative (“MRD response”) if real-time quantitative polymerase chain reaction (RQ-PCR) and subsequent nested PCR were negative at the respective time point [50]. Overall, 696 of 1202 enrolled patients were evaluable for MRD status. Significantly higher MRD response rates were seen with obinutuzumab-based chemotherapy versus rituximab-based chemotherapy at mid-induction (92.0% vs. 84.9%; p = 0.0041, in peripheral blood and/or bone marrow) and end of induction (94.3% vs. 88.9%; p = 0.013) [50].
Despite these promising findings, MRD is likely to remain a secondary or exploratory endpoint in new trials in FL until it can be properly validated as a surrogate. Furthermore, despite ongoing development of laboratory assays, evaluation of MRD status is currently largely confined to academic research centres and not used in routine practice. MRD status is not evaluable for a sizeable number of patients using currently available assays. These issues limit its potential for use as a regulatory endpoint for now. Changes in circulating tumour DNA levels have shown promise as a sensitive early marker of efficacy in a number of indications, including diffuse large B-cell lymphoma [51], and should be investigated in FL.
Complete Metabolic Response by Positron Emission Tomography
Positron emission tomography (PET) is recognised as a standard post-treatment evaluation tool in FL, due to its ability to distinguish between viable residual tumour and necrosis or fibrosis in residual mass(es) often present after treatment, thereby allowing more precise response evaluation than CT [52]. Several studies have reported significantly longer PFS in patients with negative PET status at end of treatment compared with those with positive PET status [53,54,55]. In GALLIUM, PET was compared with contrast-enhanced CT at the end of induction therapy, with independent central review of the images. Among patients in the PET-evaluable population (n = 595), the complete response rate for CT (using International Harmonisation Project 2007 criteria [29]) was 29.9% (27.5% with rituximab-based immunochemotherapy vs. 32.3% with obinutuzumab-based immunochemotherapy), but when using PET (Lugano 2014 criteria [30]), the complete metabolic response rate was 75.6% (72.5% with rituximab-based immunochemotherapy vs. 78.8% with obinutuzumab-based immunochemotherapy). When comparing PFS between complete responders and patients with less than a complete response, the HR was 0.50 (p = 0.001) for CT but 0.20 (p < 0.0001) for PET, indicating a greater reduction in risk of PFS [56]. Achieving a complete metabolic response by PET was also associated with better OS [HR 0.2 (95% CI 0.1–0.5) log-rank p < 0.0001]. The authors concluded that PET is a better imaging method than contrast-enhanced CT to measure response, although it should be noted that patients with a partial metabolic remission were considered with those who achieved a non-metabolic response. Also, OS rates for PET-positive patients were still good. A further analysis of the GALLIUM trial examined PET response together with MRD response and their association with PFS [57]. The risk of progression or death in patients who had either PET complete metabolic response or MRD negativity (but not both) was 2.5-fold higher than in patients who had both outcomes. This suggests that PET and MRD responses could provide complementary information for prognosis [57]. Overall, the available PET data are promising, and a formal assessment of whether PET response can be used as a surrogate for PFS would now be valuable.
Future Clinical Trials in First-Line Follicular Lymphoma
The positive results in GALLIUM raise the bar further in terms of efficacy in 1L FL, and the negative outcome of the Phase III RELEVANCE trial of rituximab plus lenalidomide highlights how challenging it is to demonstrate superiority over rituximab–CHOP in an FL patient population [17, 58]. The design of future 1L FL studies must account for the prolonged responses to existing treatment, such as the benchmark 3-year PFS rate of 80% reported in GALLIUM with obinutuzumab-based chemotherapy [17]. Even if the treatment benefit versus obinutuzumab were substantial, a large number of patients or a very long trial would be needed for a superiority study. It may be hypothesised that a new treatment can stop progression in 25% of the current 20% of patients who progress or die within 3 years. This would increase the 3-year PFS rate from 80% to 85%, and (based on an exponentiality assumption) the median PFS would increase from 9.3 to 11.9 years (HR 0.78). A trial with 80% power to detect this HR, with a 2-sided significance level of 0.05, would require approximately 517 events. To achieve this within a meaningful timeframe, the size of the trial would be similar to the requirements in early breast cancer (e.g. 2000 patients), but due to the rarity of the disease, recruitment time, and hence time to primary analysis, the trial duration would be longer (~ 4 and ~ 7 years, respectively). In this context, establishing valid surrogate endpoints that are measured earlier and that predict long-term treatment benefit is an important goal for future drug development. Furthermore, as patient prognoses improve, PFS may also become unfeasible as a primary endpoint and alternative endpoints are likely to be used.
Finding new therapies that are even more effective than current standards will be challenging. Therefore, non-inferiority studies are expected to be used where a new treatment has a better safety/tolerability profile than the existing standard of care (perhaps using treatment de-escalation) or is cheaper or easier to administer. Non-inferiority studies require a non-inferiority margin for the primary efficacy endpoint to be pre-defined, based on preservation of a certain proportion of the efficacy seen in the control arm. For comparison against obinutuzumab, a new study would need to show the preservation of a proportion of the benefit with obinutuzumab versus rituximab, and non-inferiority margins are often challenging to specify. In a previous 1L FL non-inferiority study, rituximab–bendamustine was compared with rituximab–CHOP, and a non-inferiority margin of 10% in PFS rate after 3 years (corresponding to HR of 1.32) was used for the primary endpoint [59]. However, few major non-inferiority studies have been performed in the 1L FL setting to date.
Many 1L FL trials have included all patients, but, given the excellent prognoses in the majority of them, there is merit in trying to focus on subsets. Methods that identify patients who are at risk of shortened PFS and OS could be used to select more suitable patients for clinical trials (i.e. those with poorer prognosis and higher unmet need). If there is a patient group in whom OS is relatively short, it may then be feasible to design trials with OS as the primary endpoint [60]. In patients with newly diagnosed FL, FLIPI is the most widely used prognostic index. Although this index was originally based on data from the pre-rituximab era, an updated version (FLIPI2) includes refinements made since the introduction of rituximab, with low-, intermediate-, and high-risk groups according to FLIPI2 having estimated 3-year PFS proportions of 91%, 69%, and 51%, respectively [61]. A novel model based on clinical variables, the Follicular Lymphoma Evaluation Index, has demonstrated prognostic value for identifying patients receiving 1L immunochemotherapy at risk of poor PFS and early progression (POD24) [62]. A simple model based on two factors, bone marrow involvement and β2-microglobulin levels, the PRIMA-Prognostic Index, also stratifies patients according to low-, medium-, and high-risk of progression [63]. In future, gene-expression profiling at diagnosis may be more widely used to identify patients with high-risk disease. A prognostic model developed by Huet et al. [64] identified 23 genes expressed in high-tumour-burden FL capable of identifying patients at high risk of progression when treated with 1L rituximab-based immunochemotherapy. Similarly, evolution of the FLIPI risk score to include the mutation of seven genes associated with varying survival outcomes (m7-FLIPI) improved prognostication of FL compared with clinical or genetic predictors alone [65]. A limitation of models incorporating genetic markers is their apparent chemotherapy dependence. In exploratory analyses conducted in the GALLIUM trial, high-risk m7-FLIPI and a high-risk 23-gene signature were associated with an increased risk for disease progression or death only in patients receiving CHOP or CVP [66, 67]. The recruitment of patients with medium to high risk may make superiority trials feasible, although this could lead to consideration for approval only in the patient populations included in the superiority trial(s), with an assumption that current treatments would be adequate for low-risk patients.
Another approach to increasing the likelihood of demonstrating treatment improvements could be to use endpoints other than PFS and OS as surrogates. As discussed above, several endpoints such as CR30, MRD and PET status may have the potential to be used as primary endpoints in a superiority trial. However, it is not yet certain whether any of these endpoints will successfully increase our ability to detect a treatment benefit, while also satisfying the requirements of regulatory and health technology agencies for robust clinical evidence.
Conclusions
Prolonged PFS times are now achievable in 1L FL. While this is beneficial for patients, the challenges for designing trials to show that new treatments provide further benefits have increased. Available options already achieve very good results in FL, making it hard to differentiate new agents in development, which can delay access to new and effective drugs. Endpoints that were traditionally used to assess drugs in this setting (e.g. OS and PFS) may not be feasible in a realistic timeframe, and new endpoints might be needed to reflect the long remission times, low relapse rates, and the impact of subsequent anticancer therapies. Inclusion of quality of life endpoints is important, given the impact of lymphoma symptoms or therapies on a patient’s well-being. Subgroup analyses are standard practice, but there is a need for cautious interpretation, considering the small size of many subgroups and the likelihood of chance findings without a biological rationale. Finally, the potential for using risk-adapted trials to direct treatment, perhaps based on MRD or PET imaging, or restricting studies to patients with high-risk disease should be explored.
References
The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89:3909–18.
Hiddemann W, Kneba M, Dreyling M, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2005;106:3725–32.
Salles G, Seymour JF, Offner F, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377:42–51.
Highlights of Prescribing Information. GAZYVA® (obinutuzumab) injection, for intravenous use. Initial US approval: 2013. 2020, South San Francisco: Genentech, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125486s017s018lbl.pdf. Accessed Jan 2021.
Swenson WT, Wooldridge JE, Lynch CF, et al. Improved survival of follicular lymphoma patients in the United States. J Clin Oncol. 2005;23:5019–26.
Fisher RI, LeBlanc M, Press OW, et al. New treatment options have changed the survival of patients with follicular lymphoma. J Clin Oncol. 2005;23:8447–52.
Herold M, Haas A, Srock S, et al. Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol. 2007;25:1986–92.
Marcus R, Imrie K, Solal-Celigny P, et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol. 2008;26:4579–86.
Bachy E, Houot R, Morschhauser F, et al. Long-term follow up of the FL2000 study comparing CHVP-interferon to CHVP-interferon plus rituximab in follicular lymphoma. Haematologica. 2013;98:1107–14.
Schulz H, Bohlius JF, Trelle S, et al. Immunochemotherapy with rituximab and overall survival in patients with indolent or mantle cell lymphoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2007;99:706–14.
Hochster H, Weller E, Gascoyne RD, et al. Maintenance rituximab after cyclophosphamide, vincristine, and prednisone prolongs progression-free survival in advanced indolent lymphoma: results of the randomized phase III ECOG1496 Study. J Clin Oncol. 2009;27:1607–14.
Bachy E, Seymour JF, Feugier P, et al. Sustained progression-free survival benefit of rituximab maintenance in patients with follicular lymphoma: long-term results of the PRIMA study. J Clin Oncol. 2019;37:2815–24.
Vidal L, Gafter-Gvili A, Salles G, et al. Rituximab maintenance improves overall survival of patients with follicular lymphoma-Individual patient data meta-analysis. Eur J Cancer. 2017;76:216–25.
Tobinai K, Klein C, Oya N, Fingerle-Rowson G. A review of obinutuzumab (GA101), a novel type II anti-CD20 monoclonal antibody, for the treatment of patients with B-cell malignancies. Adv Ther. 2017;34:324–56.
Golay J, Da Roit F, Bologna L, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. 2013;122:3482–91.
Hiddemann W, Barbui AM, Canales MA, et al. Immunochemotherapy with obinutuzumab or rituximab for previously untreated follicular lymphoma in the GALLIUM study: influence of chemotherapy on efficacy and safety. J Clin Oncol. 2018;36:2395–404.
Marcus R, Davies A, Ando K, et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N Engl J Med. 2017;377:1331–44.
Townsend W, Buske C, Cartron G, et al. Sustained clinical benefit of obinutuzumab plus chemotherapy versus rituximab plus chemotherapy in patients with previously untreated follicular lymphoma: updated results from the GALLIUM study. EHA Libr. 2020;EP1170. Available at: https://library.ehaweb.org/eha/2020/eha25th/293659/william.townsend.sustained.clinical.benefit.of.obinutuzumab.plus.chemotherapy.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D1%2Asearch%3Dep1170. Accessed May 2021.
FDA. Clinical trial endpoints for the approval of cancer drugs and biologics. Guidance for Industry. 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics. Accessed May 2021.
EMA. Guideline on the evaluation of anticancer medicinal products in man. 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-anticancer-medicinal-products-man-revision-5_en.pdf. Accessed May 2021.
EMA. Question and answer on adjustment for cross-over in estimating effects in oncology trials. 2019 [cited 2020 July 29]. https://www.ema.europa.eu/en/adjustment-cross-over-estimating-effects-oncology-trials. Accessed May 2021.
Jiménez-Ubieto A, Grande C, Caballero D, et al. Progression-free survival at 2 years post-autologous transplant: a surrogate end point for overall survival in follicular lymphoma. Cancer Med. 2017;6:2766–74.
Shi Q, Schmitz N, Ou FS, et al. Progression-free survival as a surrogate end point for overall survival in first-line diffuse large B-cell lymphoma: an individual patient-level analysis of multiple randomized trials (SEAL). J Clin Oncol. 2018;36:2593–602.
Lux MP, Böhme S, Hücherig S, et al. Surrogate threshold effect based on a meta-analysis for the predictive value of progression-free survival for overall survival in hormone receptor-positive, HER2-negative metastatic breast cancer. Breast Cancer Res Treat. 2019;176:495–506.
Liu L, Yu H, Huang L, et al. Progression-free survival as a surrogate endpoint for overall survival in patients with third-line or later-line chemotherapy for advanced gastric cancer. Onco Targets Ther. 2015;8:921–8.
Svensson S, Menkes DB, Lexchin J. Surrogate outcomes in clinical trials: a cautionary tale. JAMA Intern Med. 2013;173:611–2.
Zhu R, Lu D, Chu YW, et al. Assessment of correlation between early and late efficacy endpoints to identify potential surrogacy relationships in non-Hodgkin lymphoma: a literature-based meta-analysis of 108 phase II and phase III studies. AAPS J. 2017;19:669–81.
Broglio KR, Berry DA. Detecting an overall survival benefit that is derived from progression-free survival. J Natl Cancer Inst. 2009;101:1642–9.
Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86.
Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059–68.
Moccia AA, Taverna C, Schär S, et al. Prolonged rituximab maintenance in follicular lymphoma patients: long-term results of the SAKK 35/03 randomized trial. Blood Adv. 2020;4:5951–7.
Fuji S. Answer to simulation analysis of cost-effectiveness in 2012 has finally come in 2019. J Clin Oncol. 2020;38:522.
Dmitrienko A, Muysers C, Fritsch A, Lipkovich I. General guidance on exploratory and confirmatory subgroup analysis in late-stage clinical trials. J Biopharm Stat. 2016;26:71–98.
Sun X, Ioannidis JP, Agoritsas T, Alba AC, Guyatt G. How to use a subgroup analysis: users’ guide to the medical literature. JAMA. 2014;311:405–11.
Dehbi HM, Hackshaw A. Investigating subgroup effects in randomized clinical trials. J Clin Oncol. 2017;35:253–4.
Pocock SJ, McMurray JJ, Collier TJ. Making sense of statistics in clinical trial reports: part 1 of a 4-part series on statistics for clinical trials. J Am Coll Cardiol. 2015;66:2536–49.
Townsend W, Buske C, Cartron G, et al. Obinutuzumab-based immunochemotherapy prolongs progression-free survival and time to next anti-lymphoma treatment in patients with previously untreated follicular lymphoma: four-year results from the phase III GALLIUM study. Blood. 2018;132:1597.
NICE. Obinutuzumab for untreated follicular lymphoma. Technology appraisal guidance [TA513]. 2018. https://www.nice.org.uk/guidance/ta513. Accessed May 2021.
Casulo C, Le-Rademacher J, Dixon J, et al. Validation of POD24 as a robust early clinical endpoint of poor survival in follicular lymphoma: results from the Follicular Lymphoma Analysis of Surrogacy Hypothesis (FLASH) investigation using individual data from 5,453 patients on 13 clinical trials. Blood. 2017;130(Suppl. 1):412.
Seymour JF, Marcus R, Davies A, et al. Association of early disease progression and very poor survival in the GALLIUM study in follicular lymphoma: benefit of obinutuzumab in reducing the rate of early progression. Haematologica. 2019;104:1202–8.
Bachy E, Cerhan JR, Salles G. Early progression of disease in follicular lymphoma is a robust correlate but not a surrogate for overall survival. Blood Adv. 2021;5:1729–32.
Shi Q, Flowers CR, Hiddemann W, et al. Thirty-month complete response as a surrogate end point in first-line follicular lymphoma therapy: an individual patient-level analysis of multiple randomized trials. J Clin Oncol. 2017;35:552–60.
Landgren O, Devlin S, Boulad M, Mailankody S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transpl. 2016;51:1565–8.
Thompson PA, Wierda WG. Eliminating minimal residual disease as a therapeutic end point: working toward cure for patients with CLL. Blood. 2016;127:279–86.
Pott C, Sehn LH, Belada D, et al. MRD response in relapsed/refractory FL after obinutuzumab plus bendamustine or bendamustine alone in the GADOLIN trial. Leukemia. 2020;34:522–32.
Cheson BD, Mato AR. The evolving use of minimal residual disease (MRD) assessment in chronic lymphocytic leukemia. Clin Adv Hematol Oncol. 2020;18(Suppl.10):1–20.
Selim AG, Moore AS. Molecular minimal residual disease monitoring in acute myeloid leukemia: challenges and future directions. J Mol Diagn. 2018;20:389–97.
Azad NA, Shah ZA, Pandith AA, Rasool R, Jeelani S. Real-time quantitative PCR: a reliable molecular diagnostic and follow-up tool for ‘minimal residual disease’ assessment in chronic myeloid leukemia. Biosci Rep 2018;38:BSR20180974.
Lobetti-Bodoni C, Mantoan B, Monitillo L, et al. Clinical implications and prognostic role of minimal residual disease detection in follicular lymphoma. Ther Adv Hematol. 2013;4:189–98.
Pott C, Hoster E, Kehden B, et al. Minimal residual disease in patients with follicular lymphoma treated with obinutuzumab or rituximab as first-line induction immunochemotherapy and maintenance in the phase 3 GALLIUM study. Blood. 2016;128:613.
Kurtz DM, Scherer F, Jin MC, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018;36:2845–53.
Gallamini A, Borra A. FDG-PET scan: a new paradigm for follicular lymphoma management. Mediterr J Hematol Infect Dis. 2017;9:e2017029.
Dupuis J, Berriolo-Riedinger A, Julian A, et al. Impact of [(18)F]fluorodeoxyglucose positron emission tomography response evaluation in patients with high-tumor burden follicular lymphoma treated with immunochemotherapy: a prospective study from the Groupe d’Etudes des Lymphomes de l’Adulte and GOELAMS. J Clin Oncol. 2012;30:4317–22.
Luminari S, Biasoli I, Versari A, et al. The prognostic role of post-induction FDG-PET in patients with follicular lymphoma: a subset analysis from the FOLL05 trial of the Fondazione Italiana Linfomi (FIL). Ann Oncol. 2014;25:442–7.
Trotman J, Fournier M, Lamy T, et al. Positron emission tomography-computed tomography (PET-CT) after induction therapy is highly predictive of patient outcome in follicular lymphoma: analysis of PET-CT in a subset of PRIMA trial participants. J Clin Oncol. 2011;29:3194–200.
Trotman J, Barrington SF, Belada D, et al. Prognostic value of end-of-induction PET response after first-line immunochemotherapy for follicular lymphoma (GALLIUM): secondary analysis of a randomised, phase 3 trial. Lancet Oncol. 2018;19:1530–42.
Pott C, Davies A, Hiddemann W, et al. Metabolic (PET) and MRD response confer reduced risk of progression or deathin patients treated within the phase III GALLIUM study. EHA Libr. 2018;PF436. Available at: https://library.ehaweb.org/eha/2018/stockholm/214899/christiane.pott.metabolic.%28pet%29.and.mrd.response.confer.reduced.risk.of.html. Accessed May 2021.
Morschhauser F, Fowler NH, Feugier P, et al. Rituximab plus lenalidomide in advanced untreated follicular lymphoma. N Engl J Med. 2018;379:934–47.
Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381:1203–10.
Friedberg JW. Progress in advanced-stage follicular lymphoma. J Clin Oncol. 2018;36:2363–5.
Federico M, Bellei M, Marcheselli L, et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol. 2009;27:4555–62.
Mir F, Mattiello F, Grigg A, et al. Follicular Lymphoma Evaluation Index (FLEX): a new clinical prognostic model that is superior to existing risk scores for predicting progression-free survival and early treatment failure after frontline immunochemotherapy. Am J Hematol. 2020;95:1503–10.
Bachy E, Maurer MJ, Habermann TM, et al. A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood. 2018;132:49–58.
Huet S, Tesson B, Jais JP, et al. A gene-expression profiling score for prediction of outcome in patients with follicular lymphoma: a retrospective training and validation analysis in three international cohorts. Lancet Oncol. 2018;19:549–61.
Pastore A, Jurinovic V, Kridel R, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22.
Jurinovic V, Passerini V, Oestergaard MZ, et al. Evaluation of the m7-FLIPI in patients with follicular lymphoma treated within the gallium trial: EZH2 mutation status may be a predictive marker for differential efficacy of chemotherapy. Blood. 2019;134(suppl 1):122.
Bolen C, Hiddemann W, Marcus R, et al. Treatment-dependence of high-risk gene expression signatures in de novo follicular lymphoma. 2019 [cited 2020 July 29]. https://library.ehaweb.org/eha/2019/24th/267301/christopher.bolen.treatment-dependence.of.high-risk.gene.expression.signatures.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D1550%2Aot_id%3D20953%2Amarker%3D530%2Afeatured%3D16435. Accessed May 2021.
Salles G, Mounier N, de Guibert S, et al. Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: results of the GELA-GOELAMS FL2000 study. Blood. 2008;112:4824–31.
Federico M, Luminari S, Dondi A, et al. R-CVP versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage follicular lymphoma: results of the FOLL05 trial conducted by the Fondazione Italiana Linfomi. J Clin Oncol. 2013;31:1506–13.
Rummel MJ, Maschmeyer G, Ganser A, et al. Bendamustine plus rituximab (B-R) versus CHOP plus rituximab (CHOP-R) as first-line treatment in patients with indolent lymphomas: Nine-year updated results from the StiL NHL1 study. ASCO. 2017;35. https://doi.org/10.1200/JCO.2017.35.15_suppl.7501. Accessed March 2021.
Acknowledgements
Funding
Funding for the journal’s Rapid Service and Open Access Fees was provided by F. Hoffmann-La Roche Ltd.
Medical Writing Assistance
Third-party medical writing assistance, under the direction of Emmanuel Bachy and Allan Hackshaw, was provided by David Wilson of DJW Stats Consulting and Scott Malkin of Ashfield MedComms, an Ashfield Health company, and was funded by F. Hoffmann-La Roche Ltd.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Emmanuel Bachy declares honoraria from Gilead Sciences, F. Hoffmann-La Roche Ltd, Amgen, and Janssen-Cilag; a consulting and/or advisory role for Roche; and receipt of travel, accommodation and expense support from Janssen-Cilag. Joana Parreira and Aino Launonen declare employment by F. Hoffmann-La Roche Ltd. Tina Nielsen and Kaspar Rufibach declare employment by F. Hoffmann-La Roche Ltd and stock ownership in F. Hoffmann-La Roche Ltd. Allan Hackshaw declares personal fees from Abbvie, Boehringer Ingelheim, Takeda, AstraZeneca, Daiichi Sankyo, Merck Serono, UCB, and F. Hoffmann-La Roche Ltd for delivering education/training in clinical trials; Merck Sharpe & Dohme as consultant; and F. Hoffmann-La Roche Ltd for member of Data Monitoring Boards.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bachy, E., Rufibach, K., Parreira, J. et al. Phase III Clinical Trials in First-Line Follicular Lymphoma: A Review of Their Design and Interpretation. Adv Ther 38, 3489–3505 (2021). https://doi.org/10.1007/s12325-021-01738-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01738-2