
Abstract
Introduction
Lubiprostone capsules are approved for managing three different chronic constipation conditions. A “sprinkle” formulation may facilitate use in individuals with difficulty swallowing capsules. Our objective was to evaluate the bioequivalence, pharmacokinetics (PK), and bioavailability of lubiprostone sprinkles vs lubiprostone capsules, compared with placebo.
Methods
A 1-week randomized, placebo-controlled, double-blinded, bioequivalence study (study 302) and a single-dose PK and bioavailability crossover study (study 304) were conducted. In study 302, 522 subjects with chronic constipation were randomized to lubiprostone sprinkle 24 μg twice daily (BID), lubiprostone capsule 24 μg BID, or placebo. The primary efficacy endpoint was observed spontaneous bowel movement (SBM) counts (equivalence defined as showing the 90% confidence interval [CI] of the “between-group SBM ratio” to be contained within 0.8–1.25). Study 304 included two cohorts of healthy volunteers randomized to a single 48-μg lubiprostone dose, sprinkle, or capsule (n = 35) or to a single 48-μg sprinkle dose, in fed or fasted state (n = 14).
Results
Both lubiprostone formulations significantly improved SBM count (sprinkle, 4.82 ± 3.66, P = 0.002; capsule, 5.74 ± 3.79, P < 0.0001) vs placebo (3.68 ± 2.16), but equivalent efficacy was not demonstrated, with a 90% CI for the SBM count ratio of 0.69–0.95. Quantifiable PK data on lubiprostone were limited; however, overall exposure to the M3 metabolite was approximately 44% higher with sprinkles vs capsules under fasted conditions (geometric mean ratio 1.441 [90% CI, 1.166, 1.782]), and exposure with the sprinkle formulation was 11% lower in the fed state vs the fasted state (geometric mean ratio 0.888 [90% CI, 0.675, 1.168]). Both formulations were generally well tolerated.
Conclusion
Despite the significant improvement in SBM counts vs placebo, the sprinkle formulation did not demonstrate bioequivalence to the capsule formulation in either pharmacodynamic or PK key parameters.
Trial Registration
Study 302: ClinicalTrials.gov identifier, NCT03097861; https://www.clinicaltrials.gov/ct2/show/NCT03097861; Study 304: ClinicalTrials.gov identifier, NCT03010631; https://www.clinicaltrials.gov/ct2/show/NCT03010631.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Most prescription-based constipation medications, including lubiprostone, are available in oral formulations. Given that constipation affects a wide variety of patients, including patients at both ends of the age spectrum (pediatric and elderly) and patients with a range of comorbidities, optimizing the acceptability and ease of ingestion of oral medications may be important to optimizing adherence in certain populations. |
Two randomized studies were conducted to evaluate whether sprinkled lubiprostone formulation, designed as an alternative to swallowing capsules, showed biological equivalence to the currently available capsules. |
What was learned from the study? |
In the pharmacokinetic study, evaluation of concentrations of the drug metabolite showed greater systemic exposure with the sprinkle formulation than with the capsule formulation. |
Although the sprinkled lubiprostone formulation caused improvement in bowel function, it did not appear to be truly biologically equivalent. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.14199803.
Introduction
Constipation is a gastrointestinal (GI) disorder characterized by infrequent stools, difficult stool passage, abdominal pain, and abdominal discomfort [1]. Chronic constipation is a common and widespread condition, with an estimated global prevalence of 14% [2] and an estimated US prevalence of up to 27%; however, its prevalence can vary from region to region [3, 4]. Chronic constipation typically affects more female than male individuals and its prevalence increases with age [3]. In most patients, chronic constipation is idiopathic and caused or exacerbated by a multitude of factors, including genetic predisposition, psychosocial factors, diet (e.g., lack of adequate fluid or fiber intake), or lack of mobility; secondary constipation can stem from concomitant conditions or medications [1].
While many patients manage constipation satisfactorily with non-prescription interventions or lifestyle/dietary adjustments, others cannot reach adequate symptom control/resolution with these approaches and may report substantial distress and experience significant reductions in their quality of life [5, 6]. These patients may benefit from additional pharmacological therapy.
Lubiprostone, a synthetic analogue of naturally occurring prostone compounds, addresses chronic constipation by stimulating the type 2 chloride channel (ClC-2) in the apical membrane of intestinal epithelia, thereby enabling secretion of intestinal fluid into the lumen of the small bowel without affecting serum electrolyte concentrations. By increasing intestinal fluid secretion, lubiprostone reduces intestinal transit time, facilitating the passage of stool and alleviating symptoms associated with constipation [7].
The efficacy of lubiprostone has been demonstrated in several randomized clinical trials in adult patients with chronic idiopathic constipation, constipation associated with irritable bowel syndrome, and opioid-induced constipation [8,9,10,11]. Lubiprostone significantly increased the frequency of spontaneous bowel movements (SBMs) and improved stool consistency, as well as reduced straining, abdominal bloating, and abdominal discomfort [8,9,10,11].
Most prescription-based constipation medications, including lubiprostone, are available in oral formulations. Given that constipation affects a wide variety of patients, including patients at both ends of the age spectrum (pediatric and elderly) and patients with a range of comorbidities, optimizing the acceptability and ease of ingestion of oral medications may be important to optimizing adherence in certain populations [12, 13]. Populations that may particularly benefit from an easily ingestible oral medication include patients with physiological difficulties in swallowing, young patients with negative cognitive responses (e.g., recalcitrance) to ingesting medication, patients dependent on caregivers for administration, and patients using multiple medications.
Lubiprostone is currently available in a standard soft gel capsule formulation. In this article we describe the methodology, results, and implications of two phase 3 studies undertaken to evaluate the bioequivalence of the pharmacokinetic (PK) and the standard pharmacodynamic (PD) outcome measures (SBM counts) of a new “sprinkle” formulation designed to provide an alternative for patients who are unable to swallow or who prefer to avoid swallowing capsules.
Methods
Study Design and Population
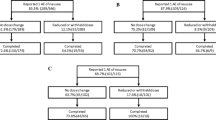
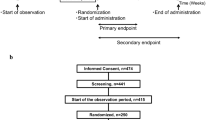
Two phase 3 studies (studies 302 and 304) were conducted to assess the PK and PD bioequivalence of an oral sprinkle and oral capsule formulation of lubiprostone and to evaluate the effects of food on the PK of the sprinkle formulation (Fig. 1). The oral capsule formulation used in studies 302 and 304 was the standard marketed formulation of lubiprostone. The sprinkle, or microbead, formulation evaluated in both studies was designed to very closely match the release profile of lubiprostone observed with the marketed capsule formulation.

Design and flow of the bioequivalence study, study 302 (a), and the pharmacokinetic study, study 304 (b). BID twice daily
Compliance with Ethics Guidelines
Studies reported here were conducted in accordance with Good Clinical Practice (GCP) standards and applicable country and/or local statutes and regulations regarding ethical committee review, informed consent, and the protection of human subjects participating in biomedical research. The study protocols were approved by the participating centers’ institutional review boards (IntegReview IRB, Austin, TX).
Study 302
The first study, study 302, was a randomized, placebo-controlled, double-blinded, double-dummy 1-week study comparing the bioequivalence of the capsule and sprinkle formulations of lubiprostone in subjects with chronic idiopathic constipation (CIC) (ClinicalTrials.gov identifier, NCT03097861; https://www.clinicaltrials.gov/ct2/show/NCT03097861). The trial was conducted in 66 centers in the USA between March and August of 2017. Subjects were eligible for the trial if they were at least 18 years of age and met the Rome III diagnostic criteria for CIC. According to the Rome III diagnostic criteria, chronic constipation is defined by the presence of at least two of the following signs or symptoms for more than 25% of defecations: straining, lumpy or hard stools, a sensation of incomplete evacuation, a sensation of anorectal obstruction or blockage, manual maneuvers to facilitate evacuation, and fewer than three SBMs per week. The Rome III criteria also specify that loose stools are rarely present in chronic constipation without the use of laxatives, and that the criteria for the diagnosis of irritable bowel syndrome are not met. The diagnosis was confirmed using the Rome III constipation module questionnaire during the screening period. Subjects were excluded from the trial if they had known or suspected disorders of the large or small bowel, if they were candidates for or had undergone surgery affecting GI motility, if they had reported weight loss, anemia, and/or rectal bleeding potentially related to another GI condition, or if they had previous use of lubiprostone or systemic antibiotic use 30 days prior to study entry. Detailed eligibility criteria for both studies are listed in Table S1 in the electronic supplementary material.
After eligibility was confirmed and baseline evaluations were completed, subjects were randomized to receive their first dose of study medication of either lubiprostone sprinkle (24 μg twice daily [BID]), lubiprostone capsule (24 μg BID), or placebo (BID) in a 1:1:1 ratio. Because of a lack of balance from the specification used for the randomization system, subjects were actually randomized in a 2:1:1 ratio. At each investigational site, subjects were randomized to treatment groups using a randomization code and stratification (SAS® software, SAS Institute, Cary, NC).
Eligible subjects received lubiprostone sprinkle 24 μg BID, lubiprostone capsule 24 μg BID, or placebo BID for 1 week in a double-dummy setting. The use of daily fiber supplements and antihypertensive medications was permitted if subjects were stable for the 30 days prior to screening. Subjects using medications known to affect GI motility were required to discontinue these medications at the screening visit.
Details regarding bowel movements (time of occurrence and associated subject ratings of consistency and straining), as well as details regarding rescue medication administration, were collected via an electronic diary completed by the subject during the screening period and throughout the trial. The use of rescue therapy was recorded in the electronic diary by the subject, and the site personnel subsequently recorded the information on an electronic case report form.
Study 304
The second study, study 304, was a randomized, open-label, two-way crossover study evaluating the PK effects of the two lubiprostone formulations, including the effects of administering the sprinkle formulation with and without food, in two cohorts of healthy volunteers (ClinicalTrials.gov identifier, NCT03010631; https://www.clinicaltrials.gov/ct2/show/NCT03010631). The study was conducted at a single inpatient center in the USA in November and December of 2016.
Subjects included healthy nonsmoking men and women (who are non-pregnant, non-lactating, and either postmenopausal for at least 1 year or surgically sterile for at least 3 months), 18–55 years of age, inclusive, with a body mass index (BMI) within the range of 18.0–30.0 kg/m2.
In cohort 1, subjects received either a 48-μg single dose of lubiprostone sprinkle or lubiprostone capsules in a fasted state, while in cohort 2, subjects received lubiprostone sprinkle under fasted or fed conditions. In this crossover study, subjects in cohort 1 who were randomized to receive lubiprostone sprinkle then crossed over to receive lubiprostone capsules and vice versa; in cohort 2, subjects randomized to receive lubiprostone sprinkle in a fasted state crossed over to receive lubiprostone sprinkle in a fed state. A randomization schedule was created by the study vendor to assign subjects in each treatment cohort to one of two treatment sequences in a 1:1 ratio.
In both cohorts, lubiprostone sprinkle or capsule was administered at a dose of 48 μg on days 1 and 8. This supratherapeutic dose is equivalent to the total daily dose of lubiprostone used in the treatment of CIC in adults, and was used to allow for a meaningful evaluation of the PK of these formulations.
Study Outcomes
Study 302
The primary endpoint in study 302, the bioequivalence study, was the demonstration of bioequivalence in efficacy as assessed by the number of SBMs at week 1 for lubiprostone sprinkle vs lubiprostone capsule. SBM was defined as any bowel movement other than those occurring within 24 h after the use of rescue medication. The SBM count was determined using the subjects’ electronic diaries. Bioequivalence was defined as achieving a ratio of mean week 1 SBM counts within a 90% confidence interval (CI) of 0.8–1.25 between the two active lubiprostone formulations.
Secondary endpoints included the demonstration of superiority of the observed SBM count at week 1 for lubiprostone sprinkle and lubiprostone capsule compared with matching placebo, the change from baseline in observed SBM count at week 1, overall mean scores and the change from baseline in stool consistency at week 1 as measured by the 7-point Bristol stool scale [14], overall mean scores and changes from baseline in straining at week 1 as assessed using a 5-point scale (ranging from 0, no straining at all, to 4, very severe straining), SBM frequency rate at week 1, and the mean change from baseline in SBM frequency at week 1. All assessments were conducted at baseline and at day 8. Subjects were also assessed 1 week following treatment.
Safety information was collected throughout the study, and any safety signal reported and not previously recorded as part of the subject’s medical history was defined as an adverse event (AE). AEs occurring from treatment initiation to 7 days after the last dose were defined as treatment-emergent AEs (TEAEs). AEs were described in detail, including their potential relationship to study drug, and were classified as “serious” or “non-serious.” Information on clinical laboratory parameters was also collected.
Study 304
For the PK evaluation in study 304, assessments of maximum observed concentrations (Cmax), time to maximum concentration (Tmax), and area under the concentration–time curve (AUC) from hour 0 to the last measurable concentration (AUC0–t), from hour 0 to 12 h (AUC0–12), and extrapolated to infinity (AUC0–∞) for lubiprostone and its M3 metabolite were conducted following lubiprostone administration. Plasma samples for lubiprostone (if measurable) and metabolite M3 concentration determination were collected predose and at approximately 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 h (day 2 for period 1; day 9 for period 2) post-dose for each period. PK samples consisted of 12 mL of venous blood. Plasma lubiprostone and the metabolite M3 concentrations were measured using a validated liquid chromatography tandem mass spectrometry (LC–MS/MS) method (HPLC system: Shimadzu 10A, Shimadzu Corporation; mass spectrometer (MS/MS system): API 4000™, AB Sciex; data management system software: Analyst ver. 1.6.1, AB Sciex; Laboratory: Shin Nippon Biomedical Laboratories, Ltd., Pharmacokinetics and Bioanalysis Center, Kainan, Wakayama, Japan).
Statistical Analyses
Study 302
The proposed sample size for study 302 was calculated on the basis of historical data for lubiprostone capsules generated for SBM counts at week 1, and the goal to contain the 90% CI for the active treatment arm ratio in SBM counts between 0.8 and 1.25 was in accordance with US Food and Drug Administration (FDA) guidance [15]. Prior clinical studies of the lubiprostone capsule formulation during the CIC development program had shown an average SBM count at week 1 for lubiprostone 24 μg BID of 5.62, with a standard deviation of 4.24. Under that assumption, demonstrating (with 90% power at a 10% significance level) that the ratio of mean week 1 SBM counts between groups fell within a 90% CI of 0.8–1.25 would require a sample size of 156 per group. Assuming 10% of subjects did not meet criteria for inclusion in the per protocol (PP) analysis set and/or did not have observed week 1 SBM count because of early withdrawal or other reasons, 174 subjects per treatment group (a total of 522 subjects) were required for this study.
The populations evaluated in study 302 included a modified intent to treat (mITT) population, defined as all randomized subjects who used at least one dose of study medication and recorded at least one diary entry, a PP population, a completer (COMP) population, and a safety population. The primary analysis was conducted in the PP population, which included all mITT subjects without any major protocol violations who were evaluated for at least 4 observation days during the first post-treatment week and had 70–125% compliance with study treatment as determined and verified by study diary data. The COMP population consisted of only those subjects who completed the entire treatment period of the study with a recorded primary endpoint and was used for supportive efficacy analyses. The safety population included all randomized subjects who received at least one dose of double-blinded study medication.
Demographic data were summarized by treatment group and in the overall population using descriptive statistics, including means and medians with standard deviations, minimum and maximum for continuous variables, and counts and percentages for each level of categorical variables.
For the SBM data count, the 90% CI of the ratio between the mean week 1 observed SBM count in the groups receiving the sprinkle and capsule formulations was calculated by back-transforming the 90% CI on the difference in the log-transformed mean counts between groups. A 90% CI of the ratio between the sprinkle and capsule mean at week 1 observed SBM count within 0.80–1.25 was considered to demonstrate bioequivalence. Changes in SBM counts were also evaluated by demographic subgroups including age, sex, race, and weekly SBM frequency rate at baseline (< 1.5 vs ≥ 1.5) to assess the consistency of treatment effect.
For the secondary endpoints, data were summarized by treatment group using descriptive statistics (including 95% CIs). The superiority analysis between lubiprostone formulations and placebo was conducted using analysis of covariance (ANCOVA) models. A van Elteren test stratified by baseline SBM frequency (< 1.5 or ≥ 1.5) and pooled sites was performed for each comparison between each lubiprostone formulation and placebo.
For the safety analysis, the incidence of AEs was summarized by treatment group and overall, and differences in incidence rates between the lubiprostone groups and the placebo group were analyzed using Fisher’s exact test. Changes from baseline and the incidence of abnormalities in clinical laboratory data and vital signs were summarized by treatment group.
Study 304
The sample size chosen for this study was based on precedent set by similar PK studies and was not based on power calculations.
In study 304, the PK study, data were analyzed using an analysis of variance (ANOVA) model with treatment as a fixed effect. The 90% CIs for the mean difference between treatments were constructed for the natural log-scale values of each parameter for lubiprostone. Confidence intervals were based on the least squares means estimation using the mean square error derived from the ANOVA models.
Results
Study Populations
A total of 552 subjects were randomized in study 302, the bioequivalence study, to lubiprostone sprinkle (n = 276), lubiprostone capsule (n = 131), and placebo (n = 145). A total of 514 subjects completed the study (Table 1). Subjects were mostly female (84.4%) and white (52.9%), and were on average 47.6 years of age. The mean BMI of subjects in the trial was 30.6 kg/m2. Baseline and demographic characteristics of subjects in the study were similar among treatment groups (Table 2).
In the PK study, study 304, 35 study subjects (16 men and 19 women) were enrolled in cohort 1 (Table 2). Thirty subjects in this cohort completed the study. Fourteen subjects were enrolled in cohort 2 (six men and eight women), and 13 subjects completed the study (Tables 1, 2). The demographic and baseline characteristics of the subjects are summarized in Table 2. Cohort 1 included a greater percentage of Hispanic subjects than cohort 2.
Bioequivalence (Study 302)
In the PP population, the mean SBM count at week 1 was higher for the capsule treatment group than for the sprinkle treatment group, and the 90% CI of the mean SBM count ratio at week 1 between the sprinkle and capsule groups was 0.688–0.947 (Fig. 2). Results were similar in the mITT population and the COMP population; in no population was the 90% CI within the predefined 0.80–1.25 range.

Mean (SD) SBM count at week 1 vs baseline in the PP population, study 302. Lubi lubiprostone, PBO placebo, PP per protocol, SBM spontaneous bowel movement, SD standard deviation
Subgroup analyses revealed that sex, age, race, and baseline weekly SBM frequency rate (< 1.5 vs ≥ 1.5) had no impact on the consistency of treatment effect or on treatment differences between study arms. SBM count at week 1 showed consistently higher values for the capsule group than for the sprinkle group in all subsets analyzed (Table 3).
For all secondary endpoints, statistically significant differences were observed in favor of both lubiprostone formulations (sprinkle and capsule) compared with placebo (Table 4). In the PP population, bioequivalence was established for the mean stool consistency score (90% CI [0.843, 0.995]) as well as for the mean change from baseline at week 1 in straining (90% CI [0.962, 1.174]). Similar results were reported for the mITT and COMP populations. An analysis comparing the impact of the sprinkle and capsule formulations on mean change from baseline in stool consistency and the amount of straining at week 1 reported 90% CIs that were outside the predetermined range for bioequivalence.
Pharmacokinetics (Study 304)
Because of limited quantifiable data available for lubiprostone, concentration data and PK parameters of lubiprostone were not summarized. Instead, data on the PK parameters of the M3 metabolite of lubiprostone were analyzed and are presented here.
Overall, M3 exposure (AUC0–t) was approximately + 44% higher and peak concentrations (Cmax) were approximately twofold higher with the sprinkle formulation than with the capsules under fasted conditions (Table 5). When M3 exposure in fed and fasted conditions with the sprinkle formulation were compared, overall M3 exposure was approximately 11% lower and peak concentrations were approximately 59% lower under fed conditions than fasted conditions.
Safety
Average subject exposure to lubiprostone was similar for both formulations in both studies. In general, both lubiprostone formulations were well tolerated, and the overall safety profiles reported in both studies were consistent with the known profile for lubiprostone. In the bioequivalence study (study 302), 7 of 275 subjects (2.5%) in the lubiprostone sprinkle arm and 4 of 130 subjects (3.1%) in the lubiprostone capsule arm discontinued the study because of a treatment-related AE, compared with 2 of 143 subjects (1.4%) in the placebo arm (Table 6). No severe or serious AEs were reported. Most AEs reported with either formulation were GI related. There was a trend toward a lower incidence of the common GI-related AEs, although not statistically significant in most cases, among individuals receiving the sprinkle formulation compared with the capsule formulation. The incidence of vomiting was statistically significantly lower with the sprinkle than with the capsule formulation (0% vs 3.1%; P = 0.0103).
A summary of the TEAEs reported in both cohorts of study 304, the PK study, is shown in Table 6. Overall, four individuals discontinued the study because of an AE following the administration of the sprinkle formulation in cohort 1, and one subject discontinued the study because of orthostatic hypotension (a pre-specified discontinuation criterion) in cohort 2. More AEs were reported following administration of the sprinkle formulation in a fasted state than in a fed state (16 vs 8 events for fasted state vs fed state, respectively), although the percentage of subjects reporting AEs was similar. Because the dose in the PK study (study 304) was supratherapeutic, safety results observed in this study may not be representative of the safety of treatment with lubiprostone at the recommended dose. Moreover, given that this study was conducted in healthy volunteers, AEs related to PD and expected clinical effects of lubiprostone on the GI tract should be expected.
Discussion
In the PD bioequivalence study, both lubiprostone formulations were similar in the observed SBM count at week 1 and were superior to placebo, but the bioequivalence of the sprinkle vs capsule formulations could not be established (based on 90% CI for primary and most secondary endpoints). In the PK study, direct measurement of plasma concentrations of lubiprostone was not possible. However, evaluation of concentrations of the M3 metabolite showed greater systemic exposure with the sprinkle formulation than with the capsule formulation.
Nonetheless, both formulations of lubiprostone were well tolerated, with a profile highly consistent with the known safety of lubiprostone. Most reported AEs were GI in nature. In the bioequivalence study, subjects assigned to the sprinkle formulation reported a lower incidence of GI-related AEs than those on the capsule formulation. Safety data from the PK study, although obtained at supratherapeutic doses and in healthy volunteers rather than patients, also confirmed that lubiprostone administration in a fed state is associated with a reduced incidence of reported AEs.
In general, in the bioequivalence study conducted at therapeutic doses, the sprinkle formulation appeared to behave as a less-intense version of lubiprostone capsules, meaning that the sprinkle formulation provided less potency in efficacy and was associated with fewer AEs. These results are noteworthy given the potentially higher concentrations of the M3 metabolite of lubiprostone reported in the PK study.
It appears reasonable, on the basis of (1) the different PK profile, (2) the lower effect size of the sprinkle formulation, and (3) the lower incidence of AEs but overall highly similar safety profile of the sprinkle formulation, that the release profile of lubiprostone from the sprinkle formulation was not similar enough to the one from the capsule formulation to trigger the same biological response. Results obtained with the sprinkle formulation are comparable to those observed with a lower dose of lubiprostone capsule formulation (i.e., lower than 24 μg BID). In a phase 2b study of multiple doses of orally administered lubiprostone (24, 48, or 72 μg/day) in patients with chronic constipation, SBM frequencies at week 1 were significantly higher in patients taking lubiprostone 48 or 72 μg/day compared with placebo (P ≤ 0.003), but not in the 24-μg group. However, by week 2, all three lubiprostone doses yielded significantly higher SBM rates vs placebo (P ≤ 0.020). Significantly larger proportions of patients taking lubiprostone 48 and 72 μg/day also experienced an SBM on the first treatment day (P ≤ 0.009) [16].
These studies have several limitations. The bioequivalence study was limited because the planned 1:1:1 randomization schedule was not appropriately balanced, resulting in higher-than-expected numbers of subjects assigned to the lubiprostone sprinkle formulation. The study did not analyze the distribution of parous/non-parous women, given that obstetrics trauma may affect rectal contractile function, and incomplete response to constipation treatment. The PK and bioavailability study was limited by its small size. Additionally, medications that were discontinued before randomization may have provided action that lasted longer than the screening period (14 days), and therefore the improvement in the stool frequency found during the study could have potentially resulted from a continued effect of these medications. Given these results and related observations, the lubiprostone sprinkle and capsule formulations do not appear to be truly bioequivalent. Additional research and modifications to the pharmacological profile of the lubiprostone sprinkle formulations (with uncertain outcome) would be needed before it might be studied clinically again. Regardless, the findings reported here have an impact for patients with difficulty swallowing, which could be related to neurologic disorders, motility disorders, anatomic disorders, neoplasm, or people who simply cannot swallow pills or capsules.
Conclusions
Two randomized studies were conducted to evaluate whether sprinkled lubiprostone formulation, designed as an alternative to swallowing capsules, showed biological equivalence to the currently available capsules. Although the sprinkled lubiprostone formulation caused improvement in bowel function, it did not appear to be truly biologically equivalent.
Change history
24 May 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12325-021-01776-w
References
Drossman DA. Functional gastrointestinal disorders: history, pathophysiology, clinical features, and Rome IV. Gastroenterology. 2016;150(1262–79):e1262.
Suares NC, Ford AC. Prevalence of, and risk factors for, chronic idiopathic constipation in the community: systematic review and meta-analysis. Am J Gastroenterol. 2011;106:1582–91 (quiz 1581, 1592).
McCrea GL, Miaskowski C, Stotts NA, Macera L, Varma MG. A review of the literature on gender and age differences in the prevalence and characteristics of constipation in North America. J Pain Symptom Manag. 2009;37:737–45.
Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol. 2011;25:3–18.
Dennison C, Prasad M, Lloyd A, Bhattacharyya SK, Dhawan R, Coyne K. The health-related quality of life and economic burden of constipation. Pharmacoeconomics. 2005;23:461–76.
Sanchez MI, Bercik P. Epidemiology and burden of chronic constipation. Can J Gastroenterol. 2011;25(Suppl B):11B-15B.
Lacy BE, Levy LC. Lubiprostone: a novel treatment for chronic constipation. Clin Interv Aging. 2008;3:357–64.
Barish CF, Drossman D, Johanson JF, Ueno R. Efficacy and safety of lubiprostone in patients with chronic constipation. Dig Dis Sci. 2010;55:1090–7.
Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: lubiprostone in patients with constipation-associated irritable bowel syndrome—results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther. 2009;29:329–41.
Jamal MM, Adams AB, Jansen JP, Webster LR. A randomized, placebo-controlled trial of lubiprostone for opioid-induced constipation in chronic noncancer pain. Am J Gastroenterol. 2015;110:725–32.
Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol. 2008;103:170–7.
Liu F, Ranmal S, Batchelor HK, et al. Patient-centred pharmaceutical design to improve acceptability of medicines: similarities and differences in paediatric and geriatric populations. Drugs. 2014;74:1871–89.
Drumond N, van Riet-Nales DA, Karapinar-Carkit F, Stegemann S. Patients’ appropriateness, acceptability, usability and preferences for pharmaceutical preparations: results from a literature review on clinical evidence. Int J Pharm. 2017;521:294–305.
Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–4.
US Food and Drug Administration. Draft guidance on lubiprostone. Recommended Aug 2010; revised July 2015. https://www.accessdata.fda.gov/drugsatfda_docs/psg/Lubiprostone_draft_Oral%20cap_RLD%2021908_RC07-15.pdf. Accessed 19 Feb 2021.
Johanson JF, Ueno R. Lubiprostone, a locally acting chloride channel activator, in adult patients with chronic constipation: a double-blind, placebo-controlled, dose-ranging study to evaluate efficacy and safety. Aliment Pharmacol Ther. 2007;25:1351–61.
Acknowledgements
We thank the participants of the studies. The authors also acknowledge the valued contribution from Dr. Richard Krause, who has passed away.
Funding
This study is supported by Mallinckrodt Pharmaceuticals, Bedminster, NJ, USA, and Takeda Development Center Americas, Inc., Deerfield, IL, USA. The study sponsors also supported the journal’s Rapid Service fee.
Medical Writing Assistance
Medical writing support was provided by Nikhilesh Sanyal, PhD, of Syneos Health Medical Communications, LLC, and was supported by Mallinckrodt Pharmaceuticals and Takeda Development Center Americas, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All authors contributed to data analysis, drafting or revising the article, providing final approval of the version to be published, and agree to be accountable for all aspects of the work.
Compliance with Ethics Guidelines
Studies reported here were conducted in accordance with Good Clinical Practice (GCP) standards and applicable country and/or local statutes and regulations regarding ethical committee review, informed consent, and the protection of human subjects participating in biomedical research. The study protocols were approved by the participating centers’ institutional review boards (IntegReview IRB, Austin, TX) and written informed consent was obtained from each subject prior to initiation of any study-related procedures.
Code Availability
SAS® software (SAS Institute, Cary, NC) was used to generate the randomization code and stratification for subjects to treatment groups at each investigational site.
Disclosures
Angel Chen: employee of Sucampo Pharmaceuticals Inc. during conduct of the study. Peter Lichtlen: employee and stock option holder of Sucampo Pharmaceuticals Inc. during conduct of the study. Taryn Losch-Beridon: employee and stock option holder of Sucampo Pharmaceuticals Inc. during conduct of the study. Shadreck Mareya: employee and stock option holder of Sucampo Pharmaceuticals Inc. during conduct of the study. Atoya Adams, Charles Barish, Patrick Dennis, Richard Krause and Jeffrey Schneider have nothing to disclose. Angel Chen, Peter Lichtlen, Taryn Losch-Beridon, and Shadreck Mareya were employed at Sucampo Pharmaceuticals at the time that the work for this manuscript was completed. Richard Krause is deceased.
Data Availability
The datasets generated during and/or analyzed during the current studies are available from Mallinckrodt Pharmaceuticals upon reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original online version of this article was revised due to retrospective open access.
Angel Chen, Peter Lichtlen, Taryn Losch-Beridon, and Shadreck Mareya were employed at Sucampo Pharmaceuticals at the time that the work for this manuscript was completed.
Richard Krause is deceased.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Adams, A., Barish, C., Chen, A. et al. Capsule and Sprinkle Formulations of Lubiprostone Are Not Biologically Similar in Patients with Functional Constipation. Adv Ther 38, 2936–2952 (2021). https://doi.org/10.1007/s12325-021-01707-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01707-9