
Abstract
Drug-induced lipid accumulation in the liver may induce two clinically relevant conditions, drug-induced steatosis (DIS) and drug-induced steatohepatitis (DISH). The list of drugs that may cause DIS or DISH is long and heterogeneous and includes therapeutically relevant molecules that cannot be easily replaced by less hepatotoxic medicines, therefore making specific strategies necessary for DIS/DISH prevention or treatment. For years, the only available tools to achieve these goals have been antioxidant drugs and free radical scavengers, which counteract drug-induced mitochondrial dysfunction but, unfortunately, have only limited efficacy. In the present review we illustrate how in vitro preclinical research unraveled new key players in the pathogenesis of specific forms of DISH, and how, in a few cases, proof of concept of the beneficial effects of their pharmacological modulation has been obtained in vivo in animal models of this condition. The key issue emerging from these studies is that, in selected cases, liver toxicity depends on mechanisms unrelated to those responsible for the desired, primary pharmacological effects of the toxic drug and, therefore, specific strategies can be designed to overcome steatogenicity without making the drug ineffective. In particular, the hepatotoxic drug could be given in combination with a second molecule intended to selectively antagonize its liver toxicity whilst, ideally, potentiating its desired pharmacological activity. Although most of the evidence that we discuss is from in vitro or animal models and will need to be further explored and validated in humans, it highlights new avenues to be pursued in order to improve the safety of steatogenic drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Pharmacological therapy may lead to steatosis (drug-induced steatosis, DIS) and steatohepatitis (drug-induced steatohepatis, DISH). |
The list of drugs likely causing DIS or DISH is long and heterogeneous, including therapeutically relevant molecules that cannot be easily replaced by less hepatotoxic medicines, making specific strategies necessary for DIS/DISH prevention or treatment. |
For years, the only available tools to achieve these goals have been antioxidant drugs and free radical scavengers, which counteract drug-induced mitochondrial dysfunction but have only limited efficacy. |
Liver toxicity may depend on mechanisms unrelated to those responsible for the desired, primary pharmacological effects. |
Specific strategies can be designed to overcome steatogenicity without making the drug ineffective, even though most of the evidence that we discuss is from in vitro or animal models and will need to be further explored and validated in humans. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.13858256.
Introduction
Prescription drugs are a well-known cause of hepatotoxicity. The estimated prevalence of drug-induced liver injury (DILI) in the general population is about 14–19 cases per 100,000 people [1] but these figures are likely underestimated not only because the pharmacovigilance chain is sometimes unreliable but also because the diagnosis of this condition is difficult and requires a high index of suspicion [2]. Drug-induced steatosis (DIS) and drug-induced steatohepatitis (DISH) are two of the most clinically relevant presentations of DILI. In both these forms of DILI lipids accumulate in liver cells, more typically causing macrovesicular steatosis and sometimes microvesicular steatosis or phospholipidosis [3]. In DISH, in addition to steatosis, prominent inflammatory changes also occur together with hepatocyte degeneration and death [4]. DILI may evolve with the features of a potentially lethal acute disease, or as chronic progressive liver injury, which progresses to cirrhosis [5, 6].
Currently the only way to make the diagnosis of DIS is by the means of imaging studies or, less frequently, histology; whereas, DISH can be identified only if suspected patients undergo liver biopsy. What is more, an exhaustive clinical evaluation and comprehensive causality assessment to rule out other possible causes and determine the association with the suspected drug are required [7]. Even though they are distinct disorders, the slow progressive forms of DIS and DISH are often confused with or misdiagnosed as non-alcoholic fatty liver disease (NAFLD)—which has been recently renamed metabolic associated fatty liver disease (MALFD) [8]- or non-alcoholic steatohepatitis (NASH), which are much more common and, by definition, not caused by drugs. To make matters worse, NAFLD conveys a nearly fourfold increase of DILI risk in obese middle‐aged patients possibly because of similar pathogenetic mechanisms and, therefore, NAFLD and DISH often coexist making it hard to establish a causative role for drugs in determining the liver damage in a specific patient [9].
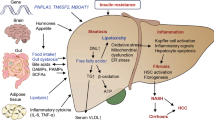
The list of drugs that may cause DIS or DISH (Box 1) is long and heterogeneous and includes therapeutically relevant drugs that cannot be easily replaced by less hepatotoxic medicines [6, 10,11,12,13]. Therefore, the issue of implementing strategies to prevent or mitigate their toxicity for the liver appears of utmost clinical relevance. Unfortunately, the therapeutic armory available to achieve these goals is currently limited to few antioxidant drugs of little clinical efficacy that are administered to counteract the oxidative mechanisms involved in the pathogenesis of DISH [14]. As a matter of fact, DISH has been classically explained as the consequence of the mitochondrial dysfunction induced by the hepatotoxic drug itself or by its metabolites which promote reactive oxygen species (ROS) production, lipid peroxidation, and further mitochondrial damage. These events finally lead to the inhibition of hepatic fatty acid oxidation, to cell damage and death, and the production of inflammatory cytokines (mainly tumor necrosis alpha and interleukin-6), causing inflammation, fibrogenic response, and cell death [15]. Nonetheless, basic research in cellular and animal models showed that the pathophysiology of DIS and DISH is much more complicated since steatogenic drugs may act by multiple mechanisms and induce lipid accumulation in the liver by disturbing lipid and lipoprotein metabolism at several levels [16]. In particular, steatogenic drugs may increase the amount of fatty acid in the hepatocytes, impair lipid incorporation in apolipoproteins, or reduce the secretion of lipoproteins [17] (Fig. 1). Fatty acids in the hepatocytes may increase because of enhanced synthesis, decreased degradation, or enhanced influx from the blood of fatty acids mobilized in the periphery. Fatty acid synthesis is mainly regulated at the level of the gene expression of key enzymes involved in this process. The main transcription factors operating this regulation are carbohydrate responsive element binding protein (ChREBP), which is glucose sensitive, and sterol regulatory element binding protein 1c (SREBP1c), which is regulated by insulin [18]. Fatty acids are degraded through β-oxidation, which mainly takes place in mitochondria and can be, therefore, functionally impaired by drugs exerting toxic effects on these organelles. In addition, gene expression of the enzymes of this metabolic pathway is regulated by peroxisome proliferator-activated receptor alpha (PPARα), which are well-known drug targets. Fatty acids enter the hepatocytes from the blood via CD36 (cluster of differentiation 36), a fatty acid translocase whose activity is regulated by AMP-activated protein kinase (AMPK) and gene expression by liver X receptor (LXR) and pregnane X receptor (PXR), as well as PPARα and PPARγ. Lipids synthesized in the liver are incorporated into nascent lipoproteins through the action of a specific microsomal triglyceride transfer protein (MTP) whose dysfunction will induce lipid accumulation. Whatever the mechanism involved, excess lipids in the liver will determine the formation of lipid droplets that represent a hallmark of steatosis. Evidence has been recently accumulated that these droplets are not just a passive form of storage but physiologically active organelles. Indeed, they regulate liver lipid metabolism and their own fate through a family of proteins known as lipid droplet proteins, which include perilipins and the cell death-inducing DFF45-like effector (CIDE) family proteins Cidea, Cideb, and Cidec/Fsp27. As a matter of fact, alterations in these lipid droplets are now considered a relevant pathogenetic mechanism of steatosis [19].

Basic mechanisms of drug-induced steatosis. The figure shows a pictorial representation of very general mechanisms responsible for drug-induced steatosis (DIS). The left part of the figure shows that, in physiological conditions, the excess of intracellular lipids depends on the amount of free fatty acids (FFA) available for the synthesis of both triglycerides and cholesterol. FFA may either enter the cell from the blood through specific transporters or be synthesized through an intracellular pathway which is highly regulated at the transcriptional level. FFA are degraded mainly through mitochondrial β-oxidation. Specific lipid droplet proteins further control lipid homeostasis and the stability of lipid droplets. The right part of the figure illustrates specific alterations in intracellular lipid homeostasis that may cause DIS: on the one hand FFA degradation through β-oxidation can be decreased and, on the other hand, FFA intracellular concentration may be increased either because of enhanced influx, increased synthesis, or both. Finally, increased expression of specific lipid droplet proteins may be involved
In the present review we will cover some notable examples of drugs acting on some of the pathophysiological steps that we quickly defined above, with the main aim of illustrating how, once that the primary steatogenic mechanism of a drug has been determined, specific strategies can be envisaged to selectively prevent or reduce liver toxicity. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Tamoxifen, Corticosteroids, and Anabolic Agents: If Multiple Intracellular Receptors Are Involved, Just Modulate the Right One to Reduce the Risk of DISH
A heterogeneous group of drugs acting on intracellular receptors, which includes tamoxifen, corticosteroids, and anabolic/androgenic steroids (AAS), may induce DIS and DISH. These drugs have in common their ability to perturb, in different ways, the complex molecular mechanisms which allow steroid hormones to exert diversified effects in different tissues thanks to the ability of their receptors to take multiple conformations and interact with multiple intracellular effectors.
Tamoxifen (TAM) is one of the most important pharmacological tools for the treatment of breast cancer. Unfortunately, in about 2% of cases it causes DISH, which usually occurs by 2 years from the beginning of therapy with a median time to onset between 6 and 22 months [20]. Although DISH does not seem to modify life expectancy of patients with breast cancer [21], and in most cases it regresses by 1.2 years from the end of tamoxifen treatment [22], its progression to cirrhosis has been described in a few patients [23,24,25,26].
TAM is a selective estrogen receptor modulator (SERM), which means that it exerts either agonist or antagonist effects on estrogen receptors (ER) in a tissue-dependent manner, for instance acting as an ER antagonist in the breast and as an agonist in the bone. TAM tissue selectivity depends on its ability to differentially modulate the various ER isoforms interfering with their genomic and non-genomic effects. Genomic effects (i.e., the regulation of gene transcription) are exerted through two different ERs, ERα and ERβ [27, 28], that can be selectively activated or blocked with specific agonists or antagonists [29], whereas non-genomic effects (i.e., the regulation of intracellular signaling pathways including, for instance, the mitogen-activated protein kinase (MAPK) and the phosphoinositide 3-kinase (PI3K)/Akt pathway [30, 31]), seem to be mediated by specific receptors such as orphan G-protein coupled estrogen receptor 30 (GPER1) and ER36, an isoform of ERα that lacks ER transactivation domains and, therefore, cannot take part in genomic regulation [32,33,34]. The evidence that estrogens exert beneficial effects on liver metabolism by improving insulin sensitivity and preventing lipid accumulation and steatohepatitis, both in animal models [35,36,37] and in postmenopausal women under hormonal replacement therapy [38,39,40], suggests that TAM could promote DISH through its estrogen antagonist activity. Early studies pointed to ERα as the ER isoform involved in these effects since it was shown that ERα knockout mice have a reduced insulin sensitivity and show increased tissue fat accumulation [41], whereas selective ERα receptor agonists alleviate hepatic steatosis in male aromatase knockout mice [42]. More recently a role in liver protection from steatosis has been demonstrated also for ERβ. Ponnusamy et al. [43] showed, indeed, that the selective ERβ isoquinolinone agonist LGND2 prevented in mice the accumulation of lipids in the liver induced by diets rich of fat or deficient in methionine and choline. They also demonstrated that this effect depends on the repression of PXR-induced genes and on the induction of farnesoid X receptor (FXR)-regulated genes. The most important implication of these results is that they could provide a rational basis for a new approach aiming to prevent or revert tamoxifen-induced DISH by concomitantly giving a selective ERβ agonist. As a matter of fact, while ERα activity promotes breast cancer growth and survival, and its blockade is essential for tamoxifen anticancer activity, ERβ seems to inhibit breast cancer and its stimulation with agonists such as LGND2 to prevent DISH is not expected to impair tamoxifen antineoplastic effects [44, 45]. Therefore, given the multiplicity of ERs and the divergence of their effects, pharmacological strategies acting on these receptors could be optimized by opportunely combining drugs acting on their different forms. More recently, a similar strategy has been investigated by targeting plasma membrane ERs. More specifically, Gu et al. showed that the ER36 antagonist anordrin prevents tamoxifen-induced DISH in mice [46]. Importantly, anordrin also suppresses the growth of breast and endometrial cancer cells in vitro, suggesting that its combination with tamoxifen should not impair the antineoplastic effect of this drug on breast cancer and should also help reduce the risk of tamoxifen-induced endometrial cancer.
Corticosteroids are another class of drugs acting on intracellular receptors—specifically glucocorticoid receptors (GR)—that may induce hepatic steatosis and, more rarely, DISH [47]. Corticosteroids are precious drugs in clinical practice, being very efficacious in a large group of inflammatory and immunological disorders, but their toxicity is a reason for concern, especially in the case of long-term use at high doses. Remarkably, metabolic alterations, including insulin resistance, are among the most common corticosteroid unwanted effects during chronic therapy of diseases such as asthma, systemic lupus erythematosus, rheumatoid arthritis, psoriasis, inflammatory bowel disease, and nephritic syndrome [48]. Both preclinical data in cellular and in animal models and clinical studies in humans strongly support the role of corticosteoids in the pathogenesis of NAFLD [49]. Nonetheless, a low prevalence of hepatic steatosis has been observed in patients with Cushing’s syndrome despite the marked insulin resistance observed in this condition possibly because, in these patients, cortisol excess inhibits IL-6-dependent low-grade chronic inflammation [50,51,52].
Several mechanisms cooperate to cause corticosteroid-induced hepatic steatosis by exerting direct effects in the liver or indirect effects in the brain, where they affect behavior and appetite, or in the adipose tissue, where they influence insulin sensitivity and lipolysis (Box 2). Similar to what we described for ERs, GRs also exert a diversified series of pharmacological effects because they may interact with different coactivators and corepressors by taking multiple conformations, which could be selectively stabilized or repressed by selective receptor modulators [53]. Several GR receptor modulators have been developed and some of them, such as mifepristone, also have approved clinical indications. A proof of concept that also the steatogenic effects of corticosteroids could be selectively modulated has been obtained with CORT118335, a selective GR modulator in clinical development for olanzapine-induced weight gain (clinical trial NCT03818256) [54]. To be more specific, when compared with corticosterone, CORT118335 loses glucocorticoid ability to promote hepatic cholesterol and triglyceride accumulation via increased free fatty acid accumulation and enhanced de novo synthesis, but maintains glucocorticoid ability to induce very-low-density lipoprotein (VLDL) synthesis [55]. Transcriptome analysis showed that, unlike corticosterone, CORT118335 did not enhance the transcription of genes involved in the uptake of free fatty acids or in the synthesis of cholesterol. Because of this pharmacological profile, not only CORT118335 does not promote steatosis but it is also expected to exert antisteatogenic effects. As matter of fact, Koorneef et al. [55] showed that CORT118335 prevents and reverts liver steatosis in mice fed with high-fat diet. Remarkably, CORT118335’s favorable effects are not exerted only in the liver but this compound also decreases fat mass expansion in the adipose tissue [56, 57]; moreover, by acting in the brain, it dampens endocrine and behavioral response to stress [58]. On the basis of the encouraging results of the preclinical studies, a clinical trial (NCT03823703) has been started to investigate the effect of CORT118335 on NASH and its results are expected in April 2022. CORT118335 does not seem to possess a strong anti-inflammatory activity and, therefore, it is unlikely that it could represent a less hepatotoxic alternative to the classical steatogenic corticosteroids. However, the case of CORT118335 represents an interesting proof of the concept that GR can be modulated to remove their steatogenic potential. Moreover, it remains to be investigated whether if given in combination with anti-inflammatory glucocorticoids it could reduce their hepatotoxic potential.
The hepatotoxic potential of AAS has been known for a long time and has been classically related to the occurrence of hepatocellular necrosis and intrahepatic cholestasis with jaundice [59]. The ability of these drugs to induce DIS and DISH, surprisingly not observed in the first case series, has been described only recently [60]. In 2011, research conducted in Brazil showed fatty liver disease in 12.6% of 95 AAS asymptomatic users who, however, unlike the typical patients with NAFLD, did not have insulin resistance [61]. Similar results were obtained in a second study on 182 asymptomatic bodybuilders using AAS [62]. To make matters worse and more complicated, there is clear evidence that pre-existing NAFLD may make adolescents more vulnerable to hepatotoxicity from AAS and likely presenting fatty liver [63]. How AAS induce DIS and DISH is still unclear, and several mechanisms have been proposed including the increase in hepatic lysosomal hydrolases, a decrease in principal components of the microsomal drug-metabolizing system, and a functional impairment of the mitochondrial respiratory chain complexes, which, notably, may occur without alteration of laboratory serum liver tests [64]. In the perspective of our discussion about pharmacological actions exerted on steroid receptors it is worth recalling here that androgens may directly affect lipid metabolism by acting in the liver [65]. In physiological conditions, testosterone, unlike AAS, seems to prevent steatogenesis as suggested by the evidence that the prevalence of NAFLD is increased in hypogonadal men [65]. Opposite effects have been observed in women and evidence has been reported that androgens have a role in causing NAFLD in the context of polycystic ovary syndrome [66], suggesting that the effects of androgens in the liver are highly dimorphic [67, 68]. Depending on the tissue considered, testosterone may exert its effect as such or after having been converted by the enzyme 5α-reductase (5AR) into its reduced form dihydrotestosterone (DHT). The evidence that hepatic steatosis develops in 5AR knockout mice [69] and in humans taking drugs that inhibit this enzyme [70] strongly suggests that most of the androgenic effects in the liver are exerted by DHT, even though it cannot be excluded that part of these effects depend on the concomitant alteration of the metabolism of glucocorticoids, which are an additional 5AR substrate [71, 72]. It is currently unclear whether anabolic androgens could affect DHT effects in the liver and how much such an effect could impact on the steatogenic potential of these compounds. It is, however, worth noticing that many of the AAS are resistant to 5AR and, at the same time, they also decrease endogenous testosterone release (and, consequently, DHT) by exerting a feedback action on the hypothalamus–pituitary axis, hence shifting the androgenic effects towards a predominantly anabolic action.
Atypical Antipsychotics: Hit at the Core of the Machinery Regulating Lipogenic Gene Expression
The development of atypical antipsychotics (AAPs) represented a major advancement for the treatment of psychosis because of their higher efficacy against negative symptoms and lower prevalence of extrapyramidal unwanted effects in comparison with typical antipsychotics. Unfortunately, AAPs also possess an intrinsic high metabolic toxicity, and their use has been linked to the appearance of metabolic syndrome in 15–70% of patients [73]. It has been estimated that about 25% of the patients receiving AAPs will develop NAFLD with a FLI (fatty liver index) score ≥ 60 by 3 years from the beginning of the treatment [73] and rare cases of progression to cirrhosis and hepatocellular carcinoma have been reported as well [74]. For a long time, the metabolic toxicity of AAPs has been explained as the consequence of the blockade of central receptors controlling feeding behavior, mainly of the 5-HT2 and specifically 5-HT2C serotoninergic and H1 histaminergic type, hence suggesting that better-tolerated drugs could be obtained by finely tuning their receptor specificity [75,76,77]. More recently, a novel mechanism has been elucidated which could open the way to new potential strategies for preventing AAP-induced DISH. Fernø et al. first observed that in glial cells antipsychotic drugs, namely clozapine and haloperidol, increase the synthesis of lipids by inducing the transcription of lipogenic genes including HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase), HMGCS1 (3-hydroxy-3-methylglutaryl-coenzyme A synthase 1), FASN (fatty acid synthase), and SCD (stearoyl-CoA desaturase) [78]. Similar results were later obtained also, in primary cultures of hepatocytes and, in vivo, in the liver of mice treated with risperidone [79, 80]. The exact mechanism by which antipsychotic drugs induce lipogenic gene transcription is still matter of discussion but it seems to involve the endoplasmic reticulum multiprotein complex composed of SREBPs, and their regulators: Srebp cleavage-activating proteins (SCAPs), insulin-induced gene 1 (INSIG1), and progesterone receptor membrane component 1 (PGRMC1) (Fig. 2). SREBPs are atypical transcription factors since they are synthesized as integral endoplasmic reticulum proteins which need to be proteolytically cleaved to be converted in active forms which, then, translocate to the nucleus to regulate gene transcription [81]. Three main forms of SREBPs have been identified: SREBP1a and SREBP1c (both encoded by the SREBF1 gene), which mainly control the expression of genes involved in the synthesis of triglycerides, and SREBP2 (encoded by SREBF2 gene), which regulates cholesterol synthesis. In normal conditions, SREBPs are retained in endoplasmic reticulum membranes because their chaperone SCAP is bound to INSIG proteins; when the concentrations of sterols or free fatty acid increase in the cytoplasm, SCAP is released from the INSIG proteins and may promote SREBP translocation to the Golgi where these transcription factors are cleaved to the active forms. PGRMC1 binds to INSIG1 and SCAP and cooperates in preventing SREBP translocation to the Golgi as also indicated by the activation of SREBP-dependent gene transcription and the appearance of hepatic steatosis in PGRMC1 knockout mice [82]. Several lines of evidence support the hypothesis that AAPs may exert their detrimental metabolic effects through the activation of the SREBP machinery. First, it has been demonstrated that, in continuous human hepatocyte cell lines, SREBP1 and SREBP2 are cleaved and activated upon exposure to these drugs [83]. Second, AAPs increase SREBP1 and SREBP2 and reduce INSIG2 and PGRMC1 expression in the liver. Third, polymorphisms of INSIG2 have been linked to the risk of AAP-induced detrimental metabolic effects [84, 85]. Finally, AAP-induced lipogenesis in adipocytes in vitro may be reverted by the overexpression of INSIG2 [86]. The exact mechanism by which AAPs may activate SREBP remains unclear. However, evidence has been reported that because of their amphiphilic structure these drugs could bind sterols in the cytoplasm and prevent their translocation to the endoplasmic reticulum, hence indirectly activating SREBPs [87]. The hypothesis that SREBP activation could be involved in causing AAP-induced metabolic effects suggests that these detrimental effects could be prevented or mitigated by therapeutic strategies targeting the SREBP machinery. A proof of concept of this principle has been recently provided by Cai et al. who showed that the steroid receptor antagonist mifepristone increases the expression of PGRMC1 (and INSIG2) in the liver and prevented AAP-induced increase in serum triacylglycerols, total cholesterol, and free fatty acids [88] (Fig. 2). These findings provided a potential explanation for previous observations that both in experimental animals and in humans mifepristone may reduce and prevent the unwanted metabolic effects of AAPs [89,90,91]. In conclusion, strategies preventing SREBP activation could be effective in preventing the undesired metabolic effects of APPs. However, before planning controlled studies to assess the efficacy of mifepristone or similar compounds in humans, it will be important to understand better whether the increase in SREBP-target gene expression in glial cells is required for the antipsychotic effects of these drugs—as originally proposed by Fernø et al. [78]—or not.

Mechanisms of DIS caused by atypical antipsychotic drugs and of its improvement by mifepristone. The left part of the figure shows the mechanism responsible for DIS induced by atypical antipsychotics (AAPs). AAPs increase the expression of sterol response element binding proteins (SREBPs), the master regulators of lipogenic gene transcription, at the same time also reducing the expression of insulin induced gene 1 (INSIG1) and progesterone receptor membrane component 1 (PGRMC1), two proteins which prevent SREBP activation by causing its retention in the endoplasmic reticulum as a complex bound to SREBP cleavage-activating proteins (SCAPs). In addition, AAPs bind to intracellular lipids, hence preventing their interaction with SCAPs and INSIG1, which is the signal causing the retention of the SCAP/SREBP complex in the endoplasmic reticulum. The right part of the figure illustrates how mifepristone could prevent or improve AAP-induced DIS. This progesterone and glucocorticoid antagonist increases the expression of INSIG2 and PGMRC1, hence counterbalancing the inhibitory effects of AAPs and preserving the inhibitory activity of these proteins on SREBP activation and, consequently, lipogenesis and DIS development
Amiodarone: Relief for a Stressed Endoplasmic Reticulum
The class III antiarrhythmic drug amiodarone is known for its hepatotoxic potential since it induces an asymptomatic increase in transaminases in about 25% of patients, hepatitis in 1–3% of cases. Moreover, amiodarone is one of the most prevalent causes of DISH, which occurs in 1–3% of patients taking this drug chronically [1, 92, 93]. The latency of amiodarone-induced DISH may be quite short, and cases have been observed after about 2 months of treatment [94]. Unlike other steatogenic drugs, amiodarone-induced hepatic damage may progress after the drug has been discontinued and this could depend on the long-term persistence of the drug that is concentrated in the liver where it may reach concentrations 500 times higher than in blood [95]. Especially worrying is the evidence that amiodarone-induced DISH may evolve into cirrhosis, although this occurs quite rarely.
Classical studies performed on isolated mitochondria established that amiodarone and its metabolite desmethylamiodarone are directly toxic for these organelles [96, 97]. These compounds inhibit the respiratory chain and, independently from the blockade of respiration, they also decrease fatty acid β-oxidation [96, 98]. Although mitochondrial permeabilization and cytochrome c leakage, which is a potent apoptosis-inducing signal, were observed in vitro upon amiodarone exposure [99], the events downstream of mitochondrial toxicity and how they could lead to DISH have been poorly understood for many years. Recently, Erez et al. reported convincing evidence that endoplasmic reticulum stress could be involved, and their findings could open the way to new targeted approaches for the prevention or treatment of amiodarone-induced DISH [100] (Fig. 3). Specifically, they showed that, both in immortalized hepatocytes in vitro and in the liver of mice in vivo, amiodarone potently activates the endoplasmic reticulum stress response as demonstrated by the increased expression of spliced X-box binding protein 1 (sXBP1), of its direct target endoplasmic reticulum–localized DnaJ 4 (Erdj4), of CCAAT-enhancer-binding protein homologous protein (CHOP), and activating transcription factor 4 (Atf4) [100]. A plausible mechanism responsible for endoplasmic reticulum stress induction was the decrease in Ca2+ concentration in the endoplasmic reticulum caused by amiodarone through the inhibition of mitochondrial respiration and, consequently, the decrease in intracellular ATP, which is essential for the pumping activity of the smooth endoplasmic reticulum Ca2+ pump (SERCA). Chop induction contributes to lipid accumulation by promoting the expression of the lipid droplet proteins cell death activator (Cidea), cell death inducing DFFA like effector C (Cidec), and perilipin-2. The crucial role of Chop in amiodarone-induced endoplasmic reticulum stress is supported by the evidence that Dit3−/− mice, which lack Chop, are less susceptible to amiodarone-induced steatosis. Therefore, a likely mechanism of amiodarone-induced DISH could be that on the one hand amiodarone inhibits mitochondrial β-oxidation and fatty acid degradation, whereas, on the other hand, it promotes lipid droplet formation. Note that endoplasmic reticulum stress is also a well-known apoptosis-inducing mechanism and its involvement in amiodarone-induced cell death has been documented as well [101]. The most important consequence of these findings is that if amiodarone-induced DISH so critically relies on endoplasmic reticulum stress, it theoretically could be prevented or alleviated by relieving endoplasmic reticulum stress. As a matter of fact, 1-(3,4-dihydroxyphenyl)-2-thiocyanate-ethanone (Bix), a chemical which potently induces binding immunoglobulin protein (Bip) and, therefore, decreases endoplasmic reticulum stress by promoting protein folding [102], reduced lipid accumulation in the hepatocytes and liver damage in mice treated with amiodarone (Fig. 3).

Mechanisms of DIS caused by amiodarone and of its improvement by thiocyanic acid 2-(3,4-dihydroxyphenyl)-2-oxoethyl ester (Bix). The left part of the figure shows the mechanism responsible for DIS induced by amiodarone. This antiarrhythmic drug induces endoplasmic reticulum stress by inhibiting mitochondrial respiration, hence causing a decrease in ATP synthesis and in ATP-dependent Ca2+ accumulation in the endoplasmic reticulum. As a consequence of endoplasmic reticulum stress, the expression of Chop is induced which, in turn, enhances the expression of the droplet proteins CIDEA, CIDEC, and perilipin-2, finally leading to intracellular lipid droplet accumulation. The right part of the figure illustrates how Bix could prevent or improve amiodarone-induced DIS. This compound is a specific inducer of the gene expression of binding immunoglobulin protein (Bip), a protein which reduces endoplasmic reticulum stress acting as a protein chaperone. Because of reduced endoplasmic reticulum stress, fewer droplet proteins are expressed and DIS is prevented or reduced
Methotrexate: If You Cannot Make it by Yourself Ask a Friend for Help
Methotrexate (MTX) is a folic acid antagonist with antiproliferative and immunosuppressant activity that even nowadays, in the biological therapy era, is still used for the long-term treatment of autoimmune diseases such as psoriasis, rheumatoid arthritis, and bowel inflammatory diseases. The hepatotoxic potential of this drug was observed many years ago and early reports showed the frequent occurrence of steatosis with pathological features very similar to those of alcoholic steatosis in patients chronically taking low dose MTX. It was also observed that about 20% of patients developed cirrhosis and, therefore, stringent guideline recommendations were issued for the early identification and the prevention of this drug-induced complication also by routinely performing liver biopsy before and during therapy [103]. Although more recent studies revised down the estimate of the prevalence of MTX-induced cirrhosis [104] and there was a general relaxation in performing liver biopsies in MTX-treated patients [105], MTX still remains one of the major causes of DISH. The mechanism responsible for MTX-induced DISH have been only partially elucidated but available data suggest that MTX-induced free radical generation could be involved. As a matter of fact, MTX reduces oxygen uptake and decreases oxidative phosphorylation in isolated mitochondria [106]; in addition, it inhibits several mitochondrial enzymes including 2-oxoglutarate, isocitrate, malate, and pyruvate dehydrogenases [107]. MTX-induced free radical generation depletes hepatic glutathione, promotes apoptotic cell death, and activates the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) [108] and the synthesis of cytokines, mainly tumor necrosis factor alpha (TNFα), which maintain a strong inflammatory response [109]. Consequently, it has been suggested that MTX-induced DISH could be ameliorated by TNFα pharmacological inhibition. This hypothesis has been confirmed in animal models of this disease. In particular, Cure et al. showed that the anti-TNFα monoclonal antibody infliximab reduced the severity of liver damage induced in rats by MTX administration; nitric oxide (NO) synthesis was decreased as well, whereas the expression of arginase and carbamoyl phosphate synthetase, two enzymes which decrease tissue levels of nitric oxide and, therefore, exert anti-inflammatory effects, were increased [110]. Hafez et al. reported similar results in rats treated with etanercept, another widely used anti-TNFα drug consisting in an IgG1 Fc fragment fused with TNFα receptors, which were protected from MTX-induced hepato- and nephrotoxicity [111]. Hafez et al. also reported evidence that this protective effect could be partially dependent on a decrease in the expression of inducible nitric oxide synthase (iNOS) and be replicated by the iNOS inhibitor aminoguanidine. The evidence that anti-TNFα drugs may reduce MTX-induced hepatotoxicity is potentially of great clinical interest because these drugs are often associated with MTX to potentiate immunosuppressant activity. Clinical study will be necessary to substantiate the potential beneficial effect of combining these drugs in humans, also considering that a note of caution derives from published (though controversial) data suggesting that anti-TNFα agents themselves could be hepatotoxic [112, 113]. Nonetheless, the preclinical evidence we reviewed provides an elegant example of the concept that DISH could be prevented by choosing appropriate drug combinations that also give definite advantages for the primary goal of the therapy.
Valproic Acid: Don’t Break Me Down!
Valproic acid (VPA) is a drug commonly used by physicians to manage seizures and psychiatric disorders. The first warning about the risks of fatty liver with VPA came from a pediatric study performed in a group of 100 children, but thereafter, this idiosyncratic reaction was more and more often reported also in other age groups [114, 115] and nowadays it is clear that about 60% of patients treated with this drug will develop DIS or DISH [116]. The genesis of DIS in patients taking VPA is probably multifactorial and different mechanisms have been proposed that could possibly coexist and synergize (Box 3). It has been suggested that the high prevalence of DIS/DISH in patients taking valproate could be the consequence of the increase in body weight caused by this drug. This hypothesis is supported by the evidence that the association between fatty liver and VPA is more relevant in patients who are already overweight/obese or present features of metabolic syndrome when they start VPA treatment [116,117,118]. However, recent studies unveiled specific, direct detrimental effects exerted by valproate in the hepatocytes. It has been shown, indeed, that this drug causes an increase in free radical generation, which could explain the histopathological features of microvesicular hepatosteatosis, commonly observed in VPA toxicity, that are suggestive of an impairment of mitochondrial β-oxidation with a concomitant increase in peroxisomal β-oxidation [119]. Effects of the drug, which is a well-known inhibitor of histone deacetylation, on the epigenetic regulation of genes involved in intracellular transport of fatty acids and in β-oxidation including PPARγ, and PPARα, aryl hydrocarbon receptor (AHR), and fatty acid translocase CD36 may partly account for valproate’s steatogenic activity [120]. Increased oxidative stress is an important additional factor contributing to liver toxicity [121, 122]. Most of the oxidative damage does not seem to be primarily mitochondrial in origin and is instead contributed by a cytochrome-dependent valproate metabolism through CYP2E1, a cytochrome form that has been implicated in the pathogenesis of multiple forms of NAFLD [123] (Fig. 4). Ma et al. recently showed that the exposure to valproate causes an increase in the expression and activity of CYP2E1 both in vitro in cultured human hepatocytes and in vivo in male C57B/6J mice [124]. Importantly, the pharmacological inhibition of CYP2E1 with diallyl sulfide, a natural compound contained in garlic, attenuated valproate-induced free radical generation and steatogenic effects both in vitro and in vivo; this favorable effect was accompanied and probably determined by the reduction of valproate-induced expression of CD36 and of diacylglycerol acyltransferase 2 [124], exemplifying an additional rational strategy to limit or reverse DIS and DISH, i.e., the pharmacological inhibition of selective drug-metabolizing pathways (Fig. 4).

Mechanisms of DIS caused by valproate and of its improvement by diallyl sulfide. The left part of the figure shows one of the mechanisms proposed to explain valproate-induced DIS, which is presumably a multifactorial process (see text for more details). This antiepileptic drug induces the expression in the hepatocytes of CYP2E1, a specific cytochrome P450 isoform whose enhanced activity causes free radical accumulation. Consequently, the expression increases of the FFA transporter CD36 and of diacylglycerol acyltransferase 2 (DGAT2), a key enzyme in the synthesis of triglycerides. The right part of the figure illustrates how diallyl sulfide could prevent or improve valproate-induced DIS. This compound is an inhibitor of CYP2E1, which, by preventing enzyme activation and the consequent free radical generation, may also prevent CD36 and DGAT2 induction and lipid accumulation as well
Concluding Remarks
The opinion of the pharmacologist: DIS and DISH are good examples of drug-induced toxicities that complicate the use of clinically relevant medicines since they are difficult to detect, are frequently unrecognized, and may ultimately cause “unexpected” serious medical consequences for the patients. The representative cases that we illustrated in the present review exemplify how a better knowledge of the toxicodynamic mechanisms responsible for potentially serious adverse drug reactions may help in rationally designing strategies aiming to overcome these toxicities and ultimately improve the drug safety profile. Some of the mechanisms we discussed seem to be quite specific for selected drugs and, therefore, they could represent a substantial change of perspective in the treatment or prevention of DIS and DISH that were classically focused on not specifically targeting free radical generation and the consequent oxidative damage. Most of the evidence that we discussed is from in vitro or animal models and, therefore, will need to be further explored and validated in humans.
The opinion of the hepatologist: In clinical practice identifying DIS and DISH may be quite difficult. In this regard, excluding with sufficient precision other possible causes according to the pattern of liver damage (the main role of liver biopsy) is central to a correct diagnosis. What is more, diagnostic scales, such as Roussel Uclaf Causality Assessment Method, based on the international DILI consensus criteria, or Digestive Disease Week Japan scale or Maria & Victorino scale, should be adopted [125]. Considering these difficulties, substantial efforts should be directed to the precocious identification of potentially hepatotoxic drug(s) in the therapy of single patients to implement specific surveillance programs with the final aim of improving the prognosis of DILI. In this context it would be extremely valuable to have the support of additional pharmacological strategies, such as those that we discussed in this review, that could help in preventing or treating DIS and DISH.
Change history
04 June 2021
The original article was revised due to update in funding note
References
Chalasani N, Bonkovsky HL, Fontana R, et al. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN Prospective Study. Gastroenterology. 2015;148(1340–52):e7.
Tarantino G, Di Minno MN, Capone D. Drug-induced liver injury: is it somehow foreseeable? World J Gastroenterol. 2009;15:2817–33.
Bessone F, Dirchwolf M, Rodil MA, Razori MV, Roma MG. Review article: drug-induced liver injury in the context of nonalcoholic fatty liver disease - a physiopathological and clinical integrated view. Aliment Pharmacol Ther. 2018;48:892–913.
Aly FZ, Kleiner DE. Update on fatty liver disease and steatohepatitis. Adv Anat Pathol. 2011;18:294–300.
Andrade RJ, Robles-Díaz M. Diagnostic and prognostic assessment of suspected drug-induced liver injury in clinical practice. Liver Int. 2020;40:6–17.
Kullak-Ublick GA, Andrade RJ, Merz M, et al. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut. 2017;66:1154–64.
Pavlik L, Regev A, Ardayfio PA, Chalasani NP. Drug-induced steatosis and steatohepatitis: the search for novel serum biomarkers among potential biomarkers for non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Drug Saf. 2019;42:701–11.
Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol. 2020;17:387–8.
Tarantino G, Conca P, Basile V, et al. A prospective study of acute drug-induced liver injury in patients suffering from non-alcoholic fatty liver disease. Hepatol Res. 2007;37:410–5.
Farrell GC. Drugs and steatohepatitis. Semin Liver Dis. 2002;22:185–94.
Park WB, Kim W, Lee KL, et al. Antituberculosis drug-induced liver injury in chronic hepatitis and cirrhosis. J Infect. 2010;61:323–9.
Patel V, Sanyal AJ. Drug-induced steatohepatitis. Clin Liver Dis. 2013;17:533–46.
Rabinowich L, Shibolet O. Drug induced steatohepatitis: an uncommon culprit of a common disease. Biomed Res Int. 2015;2015:168905.
Ramachandran A, Visschers RGJ, Duan L, Akakpo JY, Jaeschke H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: current understanding and future perspectives. J Clin Transl Res. 2018;4:75–100.
Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69.
Miele L, Liguori A, Marrone G, et al. Fatty liver and drugs: the two sides of the same coin. Eur Rev Med Pharmacol Sci. 2017;21(1 Suppl):86–94.
Dash A, Figler RA, Sanyal AJ, Wamhoff BR. Drug-induced steatohepatitis. Expert Opin Drug Metab Toxicol. 2017;13:193–204.
Dentin R, Girard J, Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie. 2005;87(1):81–6.
Carr RM, Ahima RS. Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res. 2016;340:187–92.
Saphner T, Triest-Robertson S, Li H, Holzman P. The association of nonalcoholic steatohepatitis and tamoxifen in patients with breast cancer. Cancer. 2009;115:3189–95.
Wang C, Zhou Y, Huang W, et al. The impact of pre-existed and SERM-induced non-alcoholic fatty liver disease on breast cancer survival: a meta-analysis. J Cancer. 2020;11:4597–604.
Nishino M, Hayakawa K, Nakamura Y, Morimoto T, Mukaihara S. Effects of tamoxifen on hepatic fat content and the development of hepatic steatosis in patients with breast cancer: high frequency of involvement and rapid reversal after completion of tamoxifen therapy. AJR Am J Roentgenol. 2003;180:129–34.
Dray X, Tainturier MH, De La Lande P, Marty O, Mallet L. Cirrhose avec stéatohépatite non alcoolique: rôle du tamoxifène [Cirrhosis with non alcoholic steatohepatitis: role of tamoxifen]. Gastroenterol Clin Biol. 2000;24:1122–3.
Kotiloglu G, Aki ZS, Ozyilkan O, Kutlay L. Tamoxifen-induced cirrhotic process. Breast J. 2001;7:442–3.
Oien KA, Moffat D, Curry GW, et al. Cirrhosis with steatohepatitis after adjuvant tamoxifen. Lancet. 1999;353:36–7.
Roy S, Ghosh J, Shetty N, Menon MB, Ramaswamy A, Gupta S. Tamoxifen and fulvestrant induced steatohepatitis with cirrhosis: a rare case report. South Asian J Cancer. 2019;8:225.
Dey P, Barros RP, Warner M, Ström A, Gustafsson JÅ. Insight into the mechanisms of action of estrogen receptor β in the breast, prostate, colon, and CNS. J Mol Endocrinol. 2013;51:T61-74.
Enmark E, Pelto-Huikko M, Grandien K, et al. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258–65.
Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids. 2014;90:13–29.
Segars JH, Driggers PH. Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol Metab. 2002;13:349–54.
Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab. 2002;13:422–7.
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30.
Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α 66. Biochem Biophys Res Commun. 2005;336:1023–7.
Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-α, hER-α36: transduction of estrogen and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. 2006;103:9063–8.
Kumagai S, Holmang A, Bjorntorp P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta Physiol Scand. 1993;149:91–7.
Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–17.
Wagner JD, Thomas MJ, Williams JK, Zhang L, Greaves KA, Cefalu WT. Insulin sensitivity and cardiovascular risk factors in ovariectomized monkeys with estradiol alone or combined with nomegestrol acetate. J Clin Endocrinol Metab. 1998;83:896–901.
Margolis KL, Bonds DE, Rodabough RJ, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. 2004;47:1175–87.
McKenzie J, Fisher BM, Jaap AJ, Stanley A, Paterson K, Sattar N. Effects of HRT on liver enzyme levels in women with type 2 diabetes: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2006;65:40–4.
Venetsanaki V, Polyzos SA. Menopause and non-alcoholic fatty liver disease: a review focusing on therapeutic perspectives. Curr Vasc Pharmacol. 2019;17:546–55.
Ribas V, Nguyen MT, Henstridge DC, et al. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am J Physiol Endocrinol Metab. 2010;298:E304–19.
Chow JD, Jones ME, Prelle K, Simpson ER, Boon WC. A selective estrogen receptor α agonist ameliorates hepatic steatosis in the male aromatase knockout mouse. J Endocrinol. 2011;210:323–34.
Ponnusamy S, Tran QT, Thiyagarajan T, Miller DD, Bridges D, Narayanan R. An estrogen receptor β-selective agonist inhibits non-alcoholic steatohepatitis in preclinical models by regulating bile acid and xenobiotic receptors. Exp Biol Med (Maywood). 2017;242:606–16.
Song P, Li Y, Dong Y, et al. Estrogen receptor β inhibits breast cancer cells migration and invasion through CLDN6-mediated autophagy. J Exp Clin Cancer Res. 2019;38:354.
Zhou Y, Liu X. The role of estrogen receptor beta in breast cancer. Biomark Res. 2020;8:39.
Gu W, Xu W, Sun X, et al. Anordrin eliminates tamoxifen side effects without changing its antitumor activity. Sci Rep. 2017;7:43940.
Shimizu H, Shimizu T, Takahashi D, et al. Corticosteroid dose increase is a risk factor for nonalcoholic fatty liver disease and contralateral osteonecrosis of the femoral head: a case report. BMC Musculoskelet Disord. 2019;20:88.
Zhou PZ, Zhu YM, Zou GH, et al. Relationship between glucocorticoids and insulin resistance in healthy individuals. Med Sci Monit. 2016;22:1887–94.
Woods CP, Hazlehurst JM, Tomlinson JW. Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol. 2015;154:94–103.
Nosadini R, Del Prato S, Tiengo A, et al. Insulin resistance in Cushing’s syndrome. J Clin Endocrinol Metab. 1983;57:529–36.
Rockall AG, Sohaib SA, Evans D, et al. Hepatic steatosis in Cushing’s syndrome: a radiological assessment using computed tomography. Eur J Endocrinol. 2003;149:543–8.
Tarantino G, Finelli C. Pathogenesis of hepatic steatosis: the link between hypercortisolism and non-alcoholic fatty liver disease. World J Gastroenterol. 2013;19:6735–43.
Meijer OC, Koorneef LL, Kroon J. Glucocorticoid receptor modulators. Ann Endocrinol (Paris). 2018;79(3):107–11.
Atucha E, Zalachoras I, van den Heuvel JK, et al. A mixed glucocorticoid/mineralocorticoid selective modulator with dominant antagonism in the male rat brain. Endocrinology. 2015;156(11):4105–14.
Koorneef LL, van den Heuvel JK, Kroon J, et al. Selective glucocorticoid receptor modulation prevents and reverses nonalcoholic fatty liver disease in male mice. Endocrinology. 2018;159(12):3925–36.
Mammi C, Marzolla V, Armani A, et al. A novel combined glucocorticoid-mineralocorticoid receptor selective modulator markedly prevents weight gain and fat mass expansion in mice fed a high-fat diet. Int J Obes (Lond). 2016;40(6):964–72.
Nguyen ET, Berman S, Streicher J, et al. Effects of combined glucocorticoid/mineralocorticoid receptor modulation (CORT118335) on energy balance, adiposity, and lipid metabolism in male rats. Am J Physiol Endocrinol Metab. 2019;317(2):E337–49.
Nguyen ET, Caldwell JL, Streicher J, et al. Differential effects of imipramine and CORT118335 (glucocorticoid receptor modulator/mineralocorticoid receptor antagonist) on brain-endocrine stress responses and depression-like behavior in female rats. Behav Brain Res. 2018;336:99–110.
Robles-Diaz M, Gonzalez-Jimenez A, Medina-Caliz I, et al. Distinct phenotype of hepatotoxicity associated with illicit use of anabolic androgenic steroids. Aliment Pharmacol Ther. 2015;41:116–25.
Stimac D, Milić S, Dintinjana RD, Kovac D, Ristić S. Androgenic/anabolic steroid-induced toxic hepatitis. J Clin Gastroenterol. 2002;35:350–2.
Schwingel PA, Cotrim HP, Salles BR, et al. Anabolic-androgenic steroids: a possible new risk factor of toxicant-associated fatty liver disease. Liver Int. 2011;31:348–53.
García-Cortés M, Robles-Díaz M, Ortega-Alonso A, Medina-Caliz I, Andrade RJ. Hepatotoxicity by dietary supplements: a tabular listing and clinical characteristics. Int J Mol Sci. 2016;17:537.
Awai HI, Yu EL, Ellis LS, Schwimmer JB. Liver toxicity of anabolic androgenic steroid use in an adolescent with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2014;59:e32–3.
Neri M, Bello S, Bonsignore A, et al. Anabolic androgenic steroids abuse and liver toxicity. Mini Rev Med Chem. 2011;11:430–7.
Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol. 2015;2015:294278.
Cai J, Wu CH, Zhang Y, et al. High-free androgen index is associated with increased risk of non-alcoholic fatty liver disease in women with polycystic ovary syndrome, independent of obesity and insulin resistance. Int J Obes (Lond). 2017;41(9):1341–7.
Schiffer L, Kempegowda P, Arlt W, O’Reilly MW. Mechanisms in endocrinology: the sexually dimorphic role of androgens in human metabolic disease. Eur J Endocrinol. 2017;177(3):R125–43.
Ballestri S, Nascimbeni F, Baldelli E, Marrazzo A, Romagnoli D, Lonardo A. NAFLD as a sexual dimorphic disease: role of gender and reproductive status in the development and progression of nonalcoholic fatty liver disease and inherent cardiovascular risk. Adv Ther. 2017;34(6):1291–326.
Dowman JK, Hopkins LJ, Reynolds GM, et al. Loss of 5α-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice. Endocrinology. 2013;154(12):4536–47.
Hazlehurst JM, Oprescu AI, Nikolaou N, et al. Dual-5α-reductase inhibition promotes hepatic lipid accumulation in man. J Clin Endocrinol Metab. 2016;101(1):103–13.
Nasiri M, Nikolaou N, Parajes S, et al. 5α-Reductase type 2 regulates glucocorticoid action and metabolic phenotype in human hepatocytes. Endocrinology. 2015;156(8):2863–71.
Othonos N, Marjot T, Woods C, et al. Co-administration of 5α-reductase Inhibitors worsens the adverse metabolic effects of prescribed glucocorticoids. J Clin Endocrinol Metab. 2020;105(9):e3316–28.
Xu H, Zhuang X. Atypical antipsychotics-induced metabolic syndrome and nonalcoholic fatty liver disease: a critical review. Neuropsychiatr Dis Treat. 2019;15:2087–99.
Tan Y, Sun L, Zhou X. Olanzapine-induced cirrhosis and hepatocellular carcinoma by hepatotoxicity in an elder chinese woman: case report and review of the literature. Biomed J Sci Tech Res. 2019;19:14355–7.
Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH. From the cover: antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc Natl Acad Sci USA. 2007;104:3456–9.
Panariello F, De Luca V, de Bartolomeis A. Weight gain, schizophrenia and antipsychotics: new findings from animal model and pharmacogenomic studies. Schizophr Res Treat. 2011;2011:459284.
Reynolds GP, Hill MJ, Kirk SL. The 5-HT2C receptor and antipsychotic induced weight gain - mechanisms and genetics. J Psychopharmacol. 2006;20(4 Suppl):15–8.
Fernø J, Raeder MB, Vik-Mo AO, et al. Antipsychotic drugs activate SREBP-regulated expression of lipid biosynthetic genes in cultured human glioma cells: a novel mechanism of action? Pharmacogenom J. 2005;5:298–304.
Lauressergues E, Staels B, Valeille K, et al. Antipsychotic drug action on SREBPs-related lipogenesis and cholesterogenesis in primary rat hepatocytes. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:427–39.
Lauressergues E, Martin F, Helleboid A, et al. Overweight induced by chronic risperidone exposure is correlated with overexpression of the SREBP-1c and FAS genes in mouse liver. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:423–36.
Ye J, DeBose-Boyd RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol. 2011;3:a004754.
Lee SR, Kwon SW, Kaya P, et al. Loss of progesterone receptor membrane component 1 promotes hepatic steatosis via the induced de novo lipogenesis. Sci Rep. 2018;8:15711.
Raeder MB, Fernø J, Vik-Mo AO, Steen VM. SREBP activation by antipsychotic- and antidepressant-drugs in cultured human liver cells: relevance for metabolic side-effects? Mol Cell Biochem. 2006;289:167–73.
Le Hellard S, Theisen FM, Haberhausen M, et al. Association between the insulin-induced gene 2 (INSIG2) and weight gain in a German sample of antipsychotic-treated schizophrenic patients: perturbation of SREBP-controlled lipogenesis in drug-related metabolic adverse effects? Mol Psychiatry. 2009;14:308–17.
Liou YJ, Bai YM, Lin E, et al. Gene-gene interactions of the INSIG1 and INSIG2 in metabolic syndrome in schizophrenic patients treated with atypical antipsychotics. Pharmacogenom J. 2012;12:54–61.
Chen CC, Hsu LW, Huang KT, Goto S, Chen CL, Nakano T. Overexpression of Insig-2 inhibits atypical antipsychotic-induced adipogenic differentiation and lipid biosynthesis in adipose-derived stem cells. Sci Rep. 2017;7:10901.
Kristiana I, Sharpe LJ, Catts VS, Lutze-Mann LH, Brown AJ. Antipsychotic drugs upregulate lipogenic gene expression by disrupting intracellular trafficking of lipoprotein-derived cholesterol. Pharmacogenom J. 2010;10:396–407.
Cai HL, Tan QY, Jiang P, et al. A potential mechanism underlying atypical antipsychotics-induced lipid disturbances. Transl Psychiatry. 2015;5:e661.
Beebe KL, Block T, Debattista C, Blasey C, Belanoff JK. The efficacy of mifepristone in the reduction and prevention of olanzapine-induced weight gain in rats. Behav Brain Res. 2006;171:225–9.
Gross C, Blasey CM, Roe RL, Allen K, Block TS, Belanoff JK. Mifepristone treatment of olanzapine-induced weight gain in healthy men. Adv Ther. 2009;26:959–69.
Gross C, Blasey CM, Roe RL, Belanoff JK. Mifepristone reduces weight gain and improves metabolic abnormalities associated with risperidone treatment in normal men. Obesity. 2010;18:2295–300.
Babatin M, Lee SS, Pollak PT. Amiodarone hepatotoxicity. Curr Vasc Pharmacol. 2008;6:228–36.
Lewis JH, Ranard RC, Caruso A, et al. Amiodarone hepatotoxicity: prevalence and clinicopathologic correlations among 104 patients. Hepatology. 1989;9:679–85.
Rigas B, Rosenfeld LE, Barwick KW, et al. Amiodarone hepatotoxicity. A clinicopathologic study of five patients. Ann Intern Med. 1986;104:348–51.
Brien JF, Jimmo S, Brennan FJ, Ford SE, Armstrong PW. Distribution of amiodarone and its metabolite, desethylamiodarone, in human tissues. Can J Physiol Pharmacol. 1987;65:360–4.
Fromenty B, Fisch C, Berson A, Letteron P, Larrey D, Pessayre D. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J Pharmacol Exp Ther. 1990;255:1377–84.
Fromenty B, Fisch C, Labbe G, et al. Amiodarone inhibits the mitochondrial beta-oxidation of fatty acids and produces microvesicular steatosis of the liver in mice. J Pharmacol Exp Ther. 1990;255:1371–6.
Spaniol M, Bracher R, Ha HR, Follath F, Krähenbühl S. Toxicity of amiodarone and amiodarone analogues on isolated rat liver mitochondria. J Hepatol. 2001;35:628–36.
Kaufmann P, Török M, Hänni A, Roberts P, Gasser R, Krähenbühl S. Mechanisms of benzarone and benzbromarone-induced hepatic toxicity. Hepatology. 2005;41:925–35.
Erez N, Hubel E, Avraham R, et al. Hepatic amiodarone lipotoxicity is ameliorated by genetic and pharmacological inhibition of endoplasmatic reticulum stress. Toxicol Sci. 2017;159:402–12.
Wandrer F, Frangež Ž, Liebig S, et al. Autophagy alleviates amiodarone-induced hepatotoxicity. Arch Toxicol. 2020;94:3527–39.
Kudo T, Kanemoto S, Hara H, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15:364–75.
Lewis JH, Schiff E. Methotrexate-induced chronic liver injury: guidelines for detection and prevention. The ACG Committee on FDA-related matters. American College of Gastroenterology. Am J Gastroenterol. 1988;83:1337–45.
Dawwas MF, Aithal GP. End-stage methotrexate-related liver disease is rare and associated with features of the metabolic syndrome. Aliment Pharmacol Ther. 2014;40:938–48.
Collin B, Srinathan SK, Finch TM. Methotrexate: prescribing and monitoring practices among the consultant membership of the British Association of Dermatologists. Br J Dermatol. 2008;158:793–800.
Yamamoto N, Oliveira MB, Campello Ade P, Lopes LC, Klüppel ML. Methotrexate: studies on the cellular metabolism. I/ Effect on mitochondrial oxygen uptake and oxidative phosphorylation. Cell Biochem Funct. 1988;6:61–6.
Caetano NN, Campello AP, Carnieri EG, Kluppel ML, Oliveira MB. Effect of methotrexate (MTX) on NAD(P)+ dehydrogenases of HeLa cells: malic enzyme, 2-oxoglutarate and isocitrate dehydrogenases. Cell Biochem Funct. 1997;15:259–64.
Mukherjee S, Ghosh S, Choudhury S, et al. Pomegranate reverses methotrexate-induced oxidative stress and apoptosis in hepatocytes by modulating Nrf2-NF-κB pathways. J Nutr Biochem. 2013;24:2040–50.
Cetiner M, Sener G, Sehirli AO, et al. Taurine protects against methotrexate-induced toxicity and inhibits leukocyte death. Toxicol Appl Pharmacol. 2005;209:39–50.
Cure E, Kirbas A, Tumkaya L, et al. Protective effect of infliximab on methotrexate-induced liver injury in rats: unexpected drug interaction. J Cancer Res Ther. 2015;11:164–9.
Hafez HM, Ibrahim MA, Ibrahim SA, Amin EF, Goma W, Abdelrahman AM. Potential protective effect of etanercept and aminoguanidine in methotrexate-induced hepatotoxicity and nephrotoxicity in rats. Eur J Pharmacol. 2015;768:1–12.
Likhitsup A, Dundulis J, Ansari S, et al. High prevalence of non-alcoholic fatty liver disease in patients with inflammatory bowel disease receiving anti-tumor necrosis factor therapy. Ann Gastroenterol. 2019;32:463–8.
Tang KT, Dufour JF, Chen PH, Hernaez R, Hutfless S. Antitumour necrosis factor-α agents and development of new-onset cirrhosis or non-alcoholic fatty liver disease: a retrospective cohort. BMJ Open Gastroenterol. 2020;7:e000349.
Egger J, Brett EM. Effects of sodium valproate in 100 children with special reference to weight. Br Med J (Clin Res Ed). 1981;283:577–81.
Mnif L, Sellami R, Masmoudi J. Valproic acid and hepatic steatosis: a possible link? About a Case Report Psychopharmacol Bull. 2016;46:59–62.
Luef GJ, Lechleitner M, Bauer G, Trinka E, Hengster P. Valproic acid modulates islet cell insulin secretion: a possible mechanism of weight gain in epilepsy patients. Epilepsy Res. 2003;55:53–8.
de Santa María OSR, Santiago-Fernández C, Cano-Del Pozo M, et al. Tratamiento con valproato y esteatosis hepática [Treatment with valproate and hepatic steatosis]. Rev Neurol. 2005;41:766–7.
Verrotti A, Di Marco G, la Torre R, Pelliccia P, Chiarelli F. Nonalcoholic fatty liver disease during valproate therapy. Eur J Pediatr. 2009;168:1391–4.
Begriche K, Massart J, Robin MA, Borgne-Sanchez A, Fromenty B. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol. 2011;54:773–94.
van Breda SGJ, Claessen SMH, van Herwijnen M, et al. JCS. Integrative omics data analyses of repeated dose toxicity of valproic acid in vitro reveal new mechanisms of steatosis induction. Toxicology. 2018;393:160–70.
Gai Z, Krajnc E, Samodelov SL, Visentin M, Kullak-Ublick GA. Obeticholic acid ameliorates valproic acid-induced hepatic steatosis and oxidative stress. Mol Pharmacol. 2020;97(5):314–23.
Tong V, Teng XW, Chang TK, Abbott FS. Valproic acid I: time course of lipid peroxidation biomarkers, liver toxicity, and valproic acid metabolite levels in rats. Toxicol Sci. 2005;86(2):427–35.
Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018;75(18):3313–27.
Ma L, Wang Y, Chen X, Zhao L, Guo Y. Involvement of CYP2E1-ROS-CD36/DGAT2 axis in the pathogenesis of VPA-induced hepatic steatosis in vivo and in vitro. Toxicology. 2020;445:152585.
García-Cortés M, Stephens C, Lucena MI, Fernández-Castañer A, Andrade RJ. Causality assessment methods in drug induced liver injury: strengths and weaknesses. J Hepatol. 2011;55(3):683–91.
Baxter JD, Forsham PH. Tissue effects of glucocorticoids. Am J Med. 1972;53:573–89.
Gathercole LL, Morgan SA, Bujalska IJ, Hauton D, Stewart PM, Tomlinson JW. Regulation of lipogenesis by glucocorticoids and insulin in human adipose tissue. PLoS One. 2011;6:e26223.
Peckett AJ, Wright DC, Riddell MC. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism. 2011;60:1500–10.
Macfarlane DP, Forbes S, Walker BR. Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J Endocrinol. 2008;197:189–204.
Bagdade JD, Yee E, Albers J, Pykalisto OJ. Glucocorticoids and triglyceride transport: effects on triglyceride secretion rates, lipoprotein lipase, and plasma lipoproteins in the rat. Metabolism. 1976;25:533–42.
Lettéron P, Brahimi-Bourouina N, Robin MA, Moreau A, Feldmann G, Pessayre D. Glucocorticoids inhibit mitochondrial matrix acyl-CoA dehydrogenases and fatty acid beta-oxidation. Am J Physiol. 1997;272(5 Pt 1):G1141–50.
Leclerc N, Noh T, Khokhar A, Smith E, Frenkel B. Glucocorticoids inhibit osteocalcin transcription in osteoblasts by suppressing Egr2/Krox20-binding enhancer. Arthritis Rheum. 2005;52:929–39.
Ahmed A, Rabbitt E, Brady T, et al. A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease. PLoS One. 2012;7:e29531.
Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin North Am. 2014;43:75–102.
Wang JC, Gray NE, Kuo T, Harris CA. Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci. 2012;2:19.
Tataranni PA, Larson DE, Snitker S, Young JB, Flatt JP, Ravussin E. Effects of glucocorticoids on energy metabolism and food intake in humans. Am J Physiol. 1996;271:E317–25.
Dunford EC, Riddell MC. The metabolic implications of glucocorticoids in a high-fat diet setting and the counter-effects of exercise. Metabolites. 2016;6:44.
Uddén J, Björntorp P, Arner P, Barkeling B, Meurling L, Rössner S. Effects of glucocorticoids on leptin levels and eating behaviour in women. J Intern Med. 2003;253:225–31.
Dieguez C, Mallo F, Señaris R, et al. Role of glucocorticoids in the neuroregulation of growth hormone secretion. J Pediatr Endocrinol Metab. 1996;9(Suppl 3):255–60.
Pylvänen V, Knip M, Pakarinen A, Kotila M, Turkka J, Isojärvi JI. Serum insulin and leptin levels in valproate-associated obesity. Epilepsia. 2002;43:514–7.
Verrotti A, Basciani F, De Simone M, Trotta D, Morgese G, Chiarelli F. Insulin resistance in epileptic girls who gain weight after therapy with valproic acid. J Child Neurol. 2002;17:265–8.
Wong HY, Chu TS, Lai JC, et al. Sodium valproate inhibits glucose transport and exacerbates Glut1-deficiency in vitro. J Cell Biochem. 2005;96:775–85.
Qiao L, Schaack J, Shao J. Suppression of adiponectin gene expression by histone deacetylase inhibitor valproic acid. Endocrinology. 2006;147:865–74.
Verrotti A, Basciani F, Morresi S, de Martino M, Morgese G, Chiarelli F. Serum leptin changes in epileptic patients who gain weight after therapy with valproic acid. Neurology. 1999;53:230–2.
Gungor S, Yücel G, Akinci A, Tabel Y, Ozerol IH, Yologlu S. The role of ghrelin in weight gain and growth in epileptic children using valproate. J Child Neurol. 2007;22:1384–8.
Aycicek A, Iscan A. The effects of carbamazepine, valproic acid and phenobarbital on the oxidative and antioxidative balance in epileptic children. Eur Neurol. 2007;57:65–9.
Aires CC, Ijlst L, Stet F, et al. Inhibition of hepatic carnitine palmitoyl-transferase I (CPT IA) by valproyl-CoA as a possible mechanism of valproate-induced steatosis. Biochem Pharmacol. 2010;79:792–9.
Tong V, Teng XW, Chang TK, Abbott FS. Valproic acid II: effects on oxidative stress, mitochondrial membrane potential, and cytotoxicity in glutathione-depleted rat hepatocytes. Toxicol Sci. 2005;86:436–43.
Acknowledgements
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Mauro Cataldi, Vincenzo Citro, Chiara Resnati and Federica Manco, have nothing to disclose. Giovanni Tarantino is currently serving as Section Editor in Advances in Therapy but has nothing further to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cataldi, M., Citro, V., Resnati, C. et al. New Avenues for Treatment and Prevention of Drug-Induced Steatosis and Steatohepatitis: Much More Than Antioxidants. Adv Ther 38, 2094–2113 (2021). https://doi.org/10.1007/s12325-021-01669-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01669-y