
Abstract
Renal tubular acidosis (RTA) occurs when the kidneys are unable to maintain normal acid−base homeostasis because of tubular defects in acid excretion or bicarbonate ion reabsorption. Using illustrative clinical cases, this review describes the main types of RTA observed in clinical practice and provides an overview of their diagnosis and treatment. The three major forms of RTA are distal RTA (type 1; characterized by impaired acid excretion), proximal RTA (type 2; caused by defects in reabsorption of filtered bicarbonate), and hyperkalemic RTA (type 4; caused by abnormal excretion of acid and potassium in the collecting duct). Type 3 RTA is a rare form of the disease with features of both distal and proximal RTA. Accurate diagnosis of RTA plays an important role in optimal patient management. The diagnosis of distal versus proximal RTA involves assessment of urinary acid and bicarbonate secretion, while in hyperkalemic RTA, selective aldosterone deficiency or resistance to its effects is confirmed after exclusion of other causes of hyperkalemia. Treatment options include alkali therapy in patients with distal or proximal RTA and lowering of serum potassium concentrations through dietary modification and potential new pharmacotherapies in patients with hyperkalemic RTA including newer potassium binders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Normal acid–base homeostasis is maintained by the kidneys and respiratory system |
Renal tubular acidosis (RTA) occurs when the kidneys are unable to adequately reclaim filtered bicarbonate or excrete sufficient hydrogen ions because of defects in tubular transport |
Three main types of RTA are common in clinical practice: distal (type 1), proximal (type 2), and hyperkalemic (type 4) RTA |
Differential diagnosis of RTA should consider presenting signs and symptoms, the anion gap, serum biochemistry, and urine pH |
Treatment options depend on the type of RTA diagnosed; alkali therapy may be used in patients with type 1 or 2 RTA, and newer potassium binders may be beneficial for patients with type 4 RTA |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13286027.
Introduction
The maintenance of acid–base homeostasis is a major function of the body, accomplished by intracellular and extracellular buffering mechanisms, exhalation of carbon dioxide (CO2) through the respiratory system, and acid excretion by the kidneys [1]. In healthy individuals ingesting a typical Western diet, metabolism produces acid or alkali components [1], with a daily net acid production of approximately 1 mmol/kg of body weight per day [2]. The amino acids lysine and arginine are metabolized to generate acids, while metabolism of glutamate and aspartate and organic anions, such as acetate and citrate, yield alkali [3]. Metabolism of sulfur-containing amino acids (i.e., methionine and cysteine) yields sulfuric acid, while dietary phosphate yields phosphoric acid [3].
Metabolic acidosis is defined as an excess in non-volatile acid load that results in a primary decrease in plasma bicarbonate (HCO3–) associated with a low plasma pH [1]. Metabolic acidosis can have a normal or high anion gap, where the anion gap refers to difference between the number of cations (i.e., sodium [Na+] and potassium [K+]) and the number of anions (i.e., chloride [Cl–] and HCO3–) [4]. Typically, an anion gap greater than 20 mmol/L may be considered as high; anion gaps greater than 18 mmol/L should be further investigated [4].
Renal tubular acidosis (RTA) develops as a consequence of impaired urinary acidification and is characterized by normal anion gap metabolic acidosis. RTA occurs when the kidneys are unable to adequately reclaim filtered HCO3– or secrete sufficient hydrogen ions (H+) to maintain acid–base homeostasis [5]. This review describes the physiological processes involved in the maintenance of normal acid–base homeostasis and provides an overview of the different types of RTA. The clinical presentation, differential diagnosis, and management options for patients with RTA are also discussed.
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Normal Acid–Base Homeostasis
Kidneys
The kidneys contribute to the maintenance of acid–base homeostasis by reabsorbing HCO3− in the proximal tubule, as well as regenerating HCO3– in the cortical collecting duct [1, 5]. HCO3– reabsorption involves secretion of H+ into the proximal tubule through the Na+/H+ exchanger 3 (NHE3) and H+-adenosine triphosphatase (H+-ATPase) transporters, after which luminal HCO3– reacts with secreted H+ to form CO2 and water, a process that is catalyzed by membrane-bound carbonic anhydrase isoform IV (Fig. 1a) [1, 5, 6]. CO2 is then freely absorbed across the proximal tubular membrane, where cytosolic carbonic anhydrase II catalyzes its reaction with water to form carbonic acid that rapidly breaks down to HCO3–, which is reabsorbed into the blood through the Na+/HCO3– cotransporter (NBCe1), and H+, which is then secreted back into the tubular lumen [5]. HCO3– is also reabsorbed by a similar mechanism in the thick ascending limb of the loop of Henle (Fig. 1b) [6].

(These figures were published in Comprehensive Clinical Nephrology: 5th Edition, Palmer BF, Normal acid–base balance, pp. 142–148, Copyright Elsevier (2014) [6])
Schematic diagrams illustrating bicarbonate (HCO3–) reabsorption and regeneration in the kidney. a HCO3– reabsorption in the proximal tubule. Hydrogen ions (H+) are secreted into the lumen via apical sodium (Na+)/H+ exchanger 3 (NHE3) and H+-ATPase transporters. Apical carbonic anhydrase (CA) IV catalyzes the reaction between H+ and HCO3–, which forms H2CO3 that rapidly dissociates to water and carbon dioxide (CO2). CO2 diffuses back across the apical membrane, where CA-II catalyzes its reaction with intracellular hydroxide ions (OH–) to form H+ and HCO3–. HCO3– is transported across the basolateral membrane by the Na+/HCO3–/CO32– cotransporter (NBCe1). b HCO3– reabsorption in the thick ascending limb. As in the proximal tubule, H+ is secreted into the lumen via NHE3, where it reacts with HCO3– to release CO2 that diffuses back across the apical membrane. HCO3– is transported across the basolateral membrane by NBCe1 and the kidney anion exchanger (AE1). c H+ secretion by α-intercalated cells in cortical collecting duct (CCD). H+ is secreted into the lumen by H+/K+-ATPase and vacuolar (v) H+-ATPase transporters on the apical membrane. Intracellular OH– generated by H+ secretion reacts with CO2 via CA-II to form HCO3–, which is removed by basolateral AE1. The resulting intracellular chloride (Cl–) exits via conductance channels in the basolateral membrane. Luminal K+ transported into the cell via H+/K+-ATPase can exit via channels in the apical or basolateral membrane, depending on K+ balance. d HCO3– secretion by β-intercalated cells in CCD. H+-ATPase transports H+ across the basolateral membrane. Intracellular OH– generated by H+ secretion reacts with CO2 via CA-II to form HCO3–, which is transported into the lumen by the apical Cl–/HCO3– exchanger pendrin. Intracellular Cl– exits via conductance channels in the basolateral membrane
In the collecting duct, α-intercalated cells secrete H+ by an apical H+-ATPase. This process generates HCO3–, which is transported into the blood by the basolateral Cl–/HCO3– exchanger kidney anion exchanger 1 (AE1). β-Intercalated cells secrete HCO3– into the lumen of the cortical collecting duct in exchange for Cl– through the apical Cl–/HCO3– exchanger pendrin [5]. HCO3– secretion is driven by H+ secretion across the basolateral surface of the cell via an H+-ATPase (Fig. 1c, d) [6].
In the cortical collecting duct, H+ secretion by apical H+-ATPase in α-intercalated cells is regulated by apical epithelial Na+ channels (ENaC) in principal cells, as well as aldosterone, angiotensin II, and the calcium-sensing receptor [5]. Specifically, reabsorption of Na+ through ENaC creates a lumen-negative charge secondarily increasing the rate of H+ secretion. Secreted H+ is buffered by ammonia (NH3) and titratable acids, such as phosphate. NH3 is produced as the result of glutamine metabolism and secreted into the proximal tubule lumen, where it combines with H+ to form ammonium (NH4+; Fig. 2a) [1, 5, 6]. Following reabsorption of NH4+ in the medullary thick ascending limb by substituting K+ for NH4+ on apical Na+/K+/2Cl– cotransporters (Fig. 2b), NH3 is secreted into the collecting duct, whereupon it combines with H+ secreted from α-intercalated cells to form excreted NH4+ [1, 5].

(Adapted with permission from Palmer 2014 [6])
Schematic diagrams illustrating ammonia (NH3) production and transport in the kidney. a NH3 production in the proximal tubular cells. After glutamine uptake via sodium (Na+)-coupled neutral amino acid transporter 3 (SNAT3), mitochondrial glutamine metabolism results in production of ammonium (NH4+). NH3 passively diffuses across apical membrane and hydrogen (H+) is transported via apical Na+/H+ exchanger 3 (NHE3) and H+-ATPase; NH3 and H+ combine in the lumen to form NH4+. b NH3 transport in the thick ascending limb. Lumen-positive voltage drives passive paracellular transport of NH4+ from the lumen into the blood. By substituting for potassium (K+), NH4+ is also transported into the cell via the Na+/K+/2Cl– transporter and the apical membrane K+ channel (ROMK). The basolateral Na+/bicarbonate (HCO3–) cotransporter (NBCn2) may play a role in maintaining cellular pH. NH4+ crosses the basolateral membrane into the blood via NHE4
Respiratory System
Acid–base homeostasis is also maintained by the brainstem and lungs. Alveolar ventilation increases in response to a decrease in cerebral interstitial pH (or vice versa) [1]. A small decrease in plasma pH stimulates alveolar ventilation and lowers arterial partial pressure of CO2 (pCO2) under normal conditions, which minimizes the drop in plasma pH [1].
Classification of RTA
RTA can occur as the result of one or more defects in tubular transport of H+ or HCO3− [7]. It can occur in patients with a normal or only mildly decreased glomerular filtration rate (GFR), as well as those with chronic kidney disease (CKD) [7, 8]. RTA is a cause of hyperchloremic normal anion gap acidosis, in which the loss of serum HCO3– leads to retention of Cl– so that the anion gap remains normal [9].
Three types of RTA are commonly observed in clinical practice (Fig. 3; Table 1). Types 1 and 2 RTA are caused by defects in the distal and proximal tubule, respectively, and type 4 RTA is characterized by abnormal excretion of acid and K+ in the collecting duct, which leads to hyperkalemic acidosis. Type 3 RTA is rare and has features of both distal and proximal RTA (because of the rarity of this condition, it will not be discussed further) [5]. Here, we use illustrative cases to describe the clinical characteristics of the three main forms of RTA.

A schematic diagram illustrating the underlying kidney tubule defects causing the different types of renal tubular acidosis (RTA). Distal (type 1) RTA is caused by either impaired hydrogen (H+) secretion by vacuolar (v) H+-ATPase or H+/K+-ATPase or increased H+ permeability of luminal membrane by α-intercalated cells of the collecting duct, which leads to a reduction in net H+ secretion. Proximal (type 2) RTA is caused by defects in bicarbonate (HCO3–) reabsorption in the proximal tubule, due to either impaired HCO3– transport across the basolateral membrane or inhibition of carbonic anhydrase (CA) activity. Hyperkalemic (type 4) RTA is caused by aldosterone deficiency or resistance, which leads to reduced Na+ (sodium) reabsorption by principal cells of the collecting duct and decreased transepithelial voltage, leading to diminished H+ secretion by α-intercalated cells and K+ secretion by principal cells. AE1 kidney anion exchanger, ENaC epithelial Na+ channel, MR mineralocorticoid receptor, NHE3 Na+/H+ exchanger 3, ROMK apical membrane K+ channel
Distal RTA (Type 1)
Clinical Case Presentation 1 A 34-year-old woman presents with diffuse muscle weakness over several days. She also reports dry eyes and dry mouth over the last several months. Laboratory analysis shows the following: Na+ 141 mmol/L, K+ 2.5 mmol/L, Cl– 112 mmol/L, and HCO3– 16 mmol/L, and an arterial blood gas test shows pH 7.32 and pCO2 31 mmHg. Serum creatinine is 1.4 mg/dL and urine pH is 6.8. Ultrasound imaging shows normal sized kidneys with evidence of numerous calcifications distributed throughout both kidneys. The patient is diagnosed with type 1 RTA, thought to be secondary to underlying Sjögren’s syndrome. |
Patients with distal (type 1) RTA are unable to acidify their urine despite severe metabolic acidosis. Distal RTA is characterized by impaired urinary acid secretion and evidence of kidney K+ wasting with either normal or minimally reduced GFR and persistently alkaline urine pH (> 5.3; Table 1) [10]. Patients with distal RTA often present with symptoms relating to hypokalemia, such as polydipsia and polyuria due to impaired urinary concentration ability, as well as muscle weakness, as seen in Clinical Case Presentation 1. This patient also presented with nephrolithiasis; distal RTA is a risk factor for nephrolithiasis and nephrocalcinosis due to hypercalciuria and hypocitraturia, combined with a persistently alkaline urine pH [5, 11]. Children with distal RTA also commonly present with failure to thrive, growth retardation, and rickets [11, 12].
Distal RTA is caused by impaired distal acidification of the urine due to a reduction in net H+ secretion in the distal nephron, which results in impaired HCO3– regeneration. This defect may be due to impaired H+ secretion through decreased net activity of H+-ATPase or H+/K+-ATPase (i.e., secretory defect) or increased H+ permeability of the luminal membrane (i.e., gradient defect) [10, 13]. Patients with distal RTA typically develop hyperchloremic, hypokalemic metabolic acidosis with a normal anion gap [11, 14]. In general, most patients develop hypokalemia as decreased distal secretion of H+ via H+-ATPase or H+/K+-ATPase ultimately causes decreased K+ reabsorption (i.e., renal K+ wasting) [13, 15]. If H+/K+-ATPase is primarily affected, then less K+ will be reabsorbed. A likely greater mechanism underlying renal K+ wasting is the effect of systemic acidosis on proximal tubular function due to defects in vacuolar H+-ATPase. Acidemia reduces net proximal fluid reabsorption, which leads to volume contraction and activation of the renin–angiotensin–aldosterone system (RAAS), resulting in decreased proximal tubular Na+ reabsorption and increased distal delivery of Na+ to the cortical collecting duct. These processes, along with increased aldosterone secretion, lead to increased K+ secretion [13].
Impaired Na+ reabsorption in the cortical collecting duct due to loss of H+-ATPase function also leads to increased Na+ delivery and urine flow to the medullary collecting duct [10]. Increased distal Na+ delivery stimulates expression of ENaC to promote Na+ reabsorption, and the resultant increase in ENaC-dependent Na+ reabsorption stimulates K+ channel-dependent K+ secretion [10]. Increased urinary flow activates large conductance big K+ channels, which further promote K+ secretion and contribute to hypokalemia development [16]. In contrast, renal Na+ wasting is suspected to be caused by natriuresis due to increased levels of brain natriuretic peptide or reduced sympathetic tone in the kidneys [17].
On the basis of the underlying defect, distal RTA may be classified as hereditary (primary) or acquired (secondary) [5]. For example, primary distal RTA may be caused by autosomal recessive mutations in A4 (ATP6V0A4) and B1 (ATP6B1) subunit genes of vacuolar H+-ATPase [18,19,20]. Mutations in the AE1 gene encoding the basolateral Cl–/HCO3– exchanger protein of α-intercalated cells are associated with autosomal dominant primary distal RTA [21]. Acquired distal RTA may occur as a result of some medications, including amphotericin B [22], lithium [23], and high doses of ibuprofen [24]. Secondary distal RTA often develops in association with systemic diseases [7], such as Sjögren’s syndrome (as in Clinical Case Presentation 1) [25, 26], systemic lupus erythematosus [27], or primary sclerosing cholangitis [28], and inherited conditions, such as sickle cell anemia [29] and Wilson’s disease [30].
NH3 synthesis is normally increased in response to low pH and low serum K+ concentrations [5, 7]; however, the inability to acidify luminal fluid in distal RTA causes decreased NH3 secretion in the proximal tubule [7], which may lead to the development of elevated serum NH3 concentrations (hyperammonemia) in some cases [11, 31]. Furthermore, medullary transfer of NH4+ may be impaired because of renal interstitial disease, which often occurs in association with the patient’s underlying condition or as a result of hypokalemia (i.e., kaliopenic nephropathy) or nephrocalcinosis [7].
Distal RTA is often associated with recurrent nephrolithiasis, nephrocalcinosis, and bone disease [32]. H+ retention also leads to decreases in renal calcium reabsorption and increases in calcium release from bones, which causes hypercalciuria [32]. Citrate reabsorption in the proximal tubules is elevated in distal RTA as a result of increased Na+-dependent dicarboxylate transporter 1 activity, and this leads to hypocitraturia [11, 32]. Hypercalciuria, hypocitraturia, and alkaline urinary pH all promote calcium phosphate precipitation [11, 32].
Incomplete distal RTA has many clinical characteristics in common with distal RTA, including hypocitraturia and alkaline urine pH, and may also be associated with nephrolithiasis and nephrocalcinosis [32]. However, in contrast to overt distal RTA, patients with incomplete distal RTA often have normal serum HCO3– concentrations and NH3 secretion is typically normal or occasionally increased [5, 32].
Clinical Case Presentation 1 highlights some important learning points. Musculoskeletal symptoms are a common clinical manifestation of RTA and can be alleviated with the correction of hypokalemia. Secondly, patients who present with kidney stones should be screened for RTA and incomplete distal RTA should be considered in any patients with nephrocalcinosis and a persistent urine pH of at least 5.5 in the absence of urinary tract infection and normal or near-normal serum HCO3– concentrations. Early treatment of distal RTA also improves nephrocalcinosis symptoms and may prevent recurrence of kidney stones.
Proximal RTA (Type 2)
Clinical Case Presentation 2 A 65-year-old man presents with back pain and unexplained anemia. Upon evaluation, the following laboratory values are obtained: Na+ 139 mmol/L, K+ 3.4 mmol/L, HCO3– 18 mmol/L, Cl– 108 mmol/L, glucose 104 mg/dL, creatinine 2.2 mg/dL, and total protein 10.1 g/dL. An arterial blood gas test shows pH 7.35 and pCO2 33 mmHg. The urinalysis shows 1+ glucose and a urine pH of 5.5. The patient is diagnosed with multiple myeloma complicated by evidence of proximal RTA in association with generalized dysfunction of the proximal tubule (Fanconi syndrome). |
Isolated proximal type 2 RTA is characterized by defects in the reabsorption of filtered HCO3– in the proximal tubule without defects in the transport of other solutes (Table 1) [33, 34]. The threshold serum concentration for HCO3– reabsorption (normally approximately 25 mmol/L) is reduced, leading to delivery of larger quantities of filtered HCO3– to the distal nephron (which has a low capacity for HCO3– reabsorption) and urinary HCO3– wastage [33, 34]. Reductions in serum HCO3– cause acidosis; however, the urine pH remains alkaline because of the presence of urinary HCO3− [34]. When serum HCO3– concentrations decrease below the lower threshold (16–20 mmol/L), a new steady state is reached, whereby all filtered HCO3– is reabsorbed. At this point, the urine contains no HCO3– and is maximally acidic [33].
A diagnosis of proximal RTA may be suspected in patients who present with hypokalemia, normal anion gap metabolic acidosis, and acidic urine (pH < 5.5) [7]. Other signs of proximal tubular dysfunction, including hypophosphatemia, hypouricemia, euglycemic glycosuria, and proteinuria, as observed in Clinical Case Presentation 2, are also consistent with a proximal RTA diagnosis and are reflective of generalized proximal tubular dysfunction [7]. Fractional urinary HCO3– excretion, which is normally less than 5% of filtered HCO3–, is usually approximately 15% in patients with proximal RTA [35]. Isolated proximal RTA, that is decreased HCO3– reabsorption without abnormalities in the transport of other solutes, is rare [34]. The etiology of isolated proximal RTA may be inherited (autosomal recessive mutations in SLC4A4, the gene encoding electrogenic NBCe1 [36]) or acquired (e.g., due to carbonic anhydrase inhibitors [34]). Patients with isolated proximal RTA typically present with growth retardation in early childhood [37].
In patients with proximal RTA, hypokalemia develops as a result of the loss of proximal HCO3– reabsorption [7]. Increased urinary excretion of HCO3– causes a decrease in intravascular volume, which leads to RAAS stimulation. Impaired proximal HCO3– reabsorption also causes increased distal Na+ delivery. The increase in RAAS activity results in elevated aldosterone levels that, combined with elevated distal Na+ concentrations, cause an increase in urinary K+ excretion (i.e., K+ wasting) that leads to hypokalemia [7].
Proximal RTA may occur as an isolated defect in HCO3– reabsorption, but more typically occurs in association with Fanconi syndrome, characterized by a widespread proximal tubular dysfunction resulting in the loss of phosphate, glucose, uric acid, amino acids, and low molecular weight proteins, as well as HCO3– [33, 34]. Although proximal RTA is not associated with nephrolithiasis or nephrocalcinosis, patients with proximal RTA and Fanconi syndrome may develop skeletal abnormalities, such as osteomalacia [38,39,40,41]. Skeletal abnormalities result from impaired phosphate reabsorption, which causes chronic hypophosphatemia due to renal phosphate wasting [7, 38, 41]. Active vitamin D deficiency may be present as a result of impaired conversion of 25(OH) vitamin D3 to 1,25 (OH)2 vitamin D3 in the proximal tubule [40]. Osteopenia may also be evident as a result of acidosis-induced bone demineralization [42].
Proximal RTA in association with Fanconi syndrome can occur following exposure to some medications, including tenofovir [34], ifosfamide [43], sodium valproate [44], and topiramate [45]. Topiramate is a carbonic anhydrase inhibitor that can cause simultaneous defects in both proximal and distal acidification mechanisms, presenting as type 3 RTA [46]. Proximal RTA can occur secondary to metabolic diseases, such as hereditary fructose intolerance and glycogen storage disease [47, 48]. As illustrated by Clinical Case Presentation 2, the most common cause of acquired Fanconi syndrome in adults is multiple myeloma [49].
The clinical manifestations of anemia, hypercalcemia, and bone pain suggest a diagnosis of multiple myeloma. The patient’s condition is also complicated by proximal RTA, characterized by impaired proximal HCO3– reabsorption, hypokalemia, and variable urine pH. As the most common cause of proximal RTA in adults is multiple myeloma, this diagnosis should be excluded in all adults with proximal RTA unless another cause is found.
Hyperkalemic RTA (Type 4)
Clinical Case Presentation 3 A 55-year-old man with long-standing diabetes is referred for evaluation and treatment of diabetic nephropathy. His only medication is celecoxib 200 mg/day for treatment of mild degenerative joint disease. Physical examination is significant for a blood pressure of 146/92 mmHg and trace pedal edema. Laboratory test results show Na+ 142 mmol/L, K+ 5.7 mmol/L, Cl– 108 mmol/L, HCO3– 18 mmol/L, serum creatinine 2.0 mg/dL, protein 4.6 g/24 h, and an arterial blood gas test shows pH 7.5 and pCO2 33 mmHg. His primary care physician has been reluctant to start either an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin II receptor blocker (ARB) because of increased serum K+ concentrations. |
Hyperkalemic (type 4) RTA commonly develops in patients with diabetes or interstitial nephritis and is characterized by a disturbance in distal nephron function, leading to a reduction in the excretion of H+ and K+ in the cortical collecting duct that results in hyperkalemic, hyperchloremic, normal anion gap acidosis (Table 1) [50, 51]. Patients with hyperkalemic type 4 RTA are often asymptomatic and are typically diagnosed during routine laboratory analyses. When symptomatic, manifestations may include muscle weakness or palpitations due to cardiac arrhythmias [7, 52]. Hyperkalemic RTA may be diagnosed by the presence of hyperkalemia, normal anion gap metabolic acidosis, and abnormal serum aldosterone levels, although the GFR may be near-normal or only moderately reduced (45 to less than 60 mL/min/1.73 m2) [7, 8]. As illustrated by Clinical Case Presentation 3, patients usually have diabetes with mild-to-moderate decreases in GFR, a serum HCO3– concentration of 18–22 mmol/L, and a serum K+ concentration of 5.5–6.5 mmol/L [7]. Other causes of hyperkalemia and normal anion gap acidosis include selective aldosterone deficiency, or defects in K+ or H+ secretion resulting from aldosterone resistance in the kidney (sickle cell nephropathy) [7, 52]. Urinary obstruction can give rise to type 4 RTA [52, 53]. In patients with obstructive uropathy and hyperkalemic metabolic acidosis, those who are unable to acidify their urine pH to less than 5.5 are thought to have a voltage-dependent defect in Na+ transport in the distal nephron (i.e., voltage-dependent distal RTA) [52, 53].
Type 4 RTA is typically caused by selective aldosterone deficiency or intrinsic defects in the cortical collecting duct that lead to aldosterone resistance, which causes impaired distal H+ and K+ secretion [7, 52]. Hypoaldosteronism causes reduced principal cell Na+ reabsorption and a decrease in transepithelial voltage in the cortical collecting duct, which results in diminished excretion of H+ and K+. Increased serum K+ concentrations inhibit NH3 synthesis in the proximal tubule, further reducing the kidney’s capacity to excrete acid. As described above, NH3 availability is critical for normal distal H+ secretion. Lack of adequate NH3 buffer results in a drop in urine pH creating a steep pH gradient, which impedes distal H+ secretion. Aldosterone deficiency can cause Na+ wasting, leading to decreased plasma volume that stimulates proximal Na+ reabsorption. The reduction in distal Na+ delivery secondarily inhibits secretion of K+ and H+ in the distal nephron [7, 52]. Hyperkalemia reduces NH3 production in the proximal tubule and inhibits NH4+ transport in the thick ascending limb as the high luminal K+ concentrations compete with NH4+ for Na+/K+/2Cl– cotransporters and apical K+ channels [54]. Reductions in urine NH3 can be detected by a positive urine anion gap and a failure to increase the urine osmolal gap.
In Clinical Case Presentation 3, the patient’s long history of diabetes led to an increased risk for type 4 RTA due to development of hyporeninemic hypoaldosteronism. Renin is suppressed because of salt retention, which causes volume expansion as well as atrophy and suppression of the juxtaglomerular apparatus, which leads to reduced renin secretion [7, 51]. A reduction in renin release or development of aldosterone resistance can be caused by chronic interstitial fibrosis, which commonly develops in CKD, particularly in patients with diabetic kidney disease [52]. Impaired cortical collecting duct function may also occur as a result of kidney structural damage due to interstitial kidney disease or obstructive uropathy [52]. Lupus nephritis is another cause of type 4 RTA due to tubulointerstitial damage and hyporeninemic hypoaldosteronism [55].
Patients with CKD are also at risk of developing RTA because of the progressive loss of functional kidney mass [7]. Initially, there is an adaptive increase in NH4+ production and acid secretion [56]; however, as kidney impairment progresses (GFR 30–40 mL/min/1.73 m2), this adaptive increase is unable to maintain sufficient net H+ excretion to keep pace with endogenous acid production. Patients develop a hyperchloremic normal gap acidosis, often referred to as RTA of kidney insufficiency [7]. In patients with more advanced CKD (GFR < 15–20 mL/min/1.73 m2), the ability to excrete phosphate and other anions is reduced and a high anion gap metabolic acidosis develops; the acidosis at this stage is termed uremic acidosis. During this transition, patients frequently manifest features of both a normal and increased anion gap metabolic acidosis [7]. Furthermore, patients with stage 3–5 CKD and hyperkalemia commonly develop metabolic acidosis as progressive kidney impairment leads to compromised maintenance of electrolyte and acid–base balance [57].
Pseudohypoaldosteronism is a genetic condition associated with hyperkalemic, hyperchloremic metabolic acidosis with normal kidney function and either normal or high aldosterone levels [52]. Type 1 pseudohypoaldosteronism may be caused by mutations in genes encoding the mineralocorticoid receptor or ENaC [52], whereas type 2 pseudohypoaldosteronism is caused by mutations in genes encoding the with-no-lysine (WNK) family of kinases [58]. Mutations in ENaC cause Na+ wasting and are characterized by increased aldosterone levels, while WNK mutations give rise to Na+ retention and either normal or low circulating aldosterone levels [52, 58].
Hyperkalemic type 4 RTA may be caused by medications, including K+-sparing diuretics (e.g., spironolactone, eplerenone, and amiloride), antibiotics (e.g., trimethoprim and pentamidine), nonsteroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase-2 (COX-2) inhibitors, ACE inhibitors, and heparin or low molecular weight heparin [5, 59]. These agents cause hyperkalemic RTA by reducing aldosterone synthesis (ACE inhibitors), release (NSAIDs and heparin), or receptor binding (spironolactone, eplerenone), or through inhibition of ENaC (amiloride, trimethoprim, and pentamidine) [5]. Hyperkalemic RTA may also be caused by immunosuppressant therapy with calcineurin inhibitors (e.g., tacrolimus and ciclosporin) [60,61,62]. Calcineurin inhibitors block K+ and H+ secretion from the collecting duct through inhibition of basolateral Na+/K+-ATPase and Na+/K+/2Cl– cotransporter activity [62, 63]. Calcineurin inhibitors also suppress expression of mineralocorticoid receptors, resulting in aldosterone resistance [64].
Clinical Case Presentation 3 highlights the need to monitor patients with diabetes for development of hyperkalemic RTA. The patient’s COX-2 inhibitor therapy potentially contributed to the development of hyperkalemia and should be discontinued. Although treatment with an ACE inhibitor or ARB was avoided because of the patient’s refractory hyperkalemia, the patient has diabetic nephropathy and would benefit from use of a RAAS inhibitor to slow the subsequent progression of CKD and to provide cardiovascular protection. Strategies for correction and minimization of hyperkalemia are discussed in the next section.
Clinical Management of RTA
Differential Diagnosis of RTA
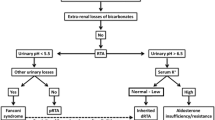
An approach to diagnosing RTA in a patient who presents with metabolic acidosis is shown in Fig. 4 [7]. In all cases of metabolic acidosis, plasma or serum anion gap should be the first laboratory assessment; hyperchloremic metabolic acidosis with a normal anion gap is present in all types of RTA (Table 1) [8, 65]. Analysis of urine biochemistry may also be useful in the evaluation of RTA; in patients with hyperchloremic metabolic acidosis and alkaline urine (i.e., pH > 5.5), RTA of some type should be strongly suspected [8].

A suggested algorithm for diagnosing renal tubular acidosis. Reprinted by permission of Edizioni Minerva Medica from Minerva Endocrinologica 2019 December; 44(4):363–77 [7]
Confirmatory testing of distal versus proximal RTA involves assessment of the markers of urinary acid and HCO3– secretion [8]. An NH4+ loading test is used to confirm distal RTA in patients with hypokalemic, hyperchloremic metabolic acidosis and urine pH > 5.5; at 6 h after oral ingestion of NH4Cl 100 mg/kg, patients with distal RTA will develop a positive urine anion gap (UAG; i.e., urine Na+ + urine K+ − urine Cl–) [8]. As the NH4Cl test is associated with gastrointestinal adverse effects (e.g., nausea and vomiting), the furosemide–fludrocortisone test may be used as an alternative method of diagnosing distal RTA [66, 67].
In patients with distal RTA, measurement of the urine-to-blood (U-B) pCO2 gradient during an NaHCO3 infusion can be used to diagnose H+-ATPase secretory defect [68]. Patients with an H+-ATPase defect have an abnormally low U-B pCO2 gradient (30 mmHg or lower), indicating impaired distal H+ secretion [68]. In patients with a gradient defect, as occurs with amphotericin B-induced distal RTA [13], the U-B pCO2 gradient is normal (greater than 30 mmHg), suggesting an intact H+ secretory mechanism [69]. In these patients, secreted H+ diffuses back into the cell as a result of a luminal permeability defect [13].
A diagnosis of proximal RTA may be confirmed using a NaHCO3 loading test; during an intravenous infusion of NaHCO3, the increase in serum HCO3– above the reabsorption threshold will lead to fractional excretion of HCO3– greater than 15% or urine pH > 7.5 in patients with proximal RTA [8]. Patients with proximal RTA should also be evaluated for Fanconi syndrome by assessment of serum and urine samples for glycosuria, hypophosphatemia, and hypouricemia.
The differential diagnosis of type 4 hyperkalemic RTA may be divided into conditions associated with hypoaldosteronism or those caused by cortical collecting duct defects. A urine pH of less than 5.5 indicates defects caused by impaired aldosterone activity and a more severe reduction in NH3 availability than impairment of H+ secretion [7]. In contrast, when the primary defect is caused by structural damage to the cortical collecting duct, the urine pH is more alkaline [7]. Selective aldosterone deficiency may be confirmed after other causes of hyperkalemia are excluded, including transcellular shifts in K+ or the use of KCl, K+-sparing diuretics, or RAAS inhibitors [52]. After correction of serum K+, persistently low aldosterone levels indicate aldosterone deficiency; in most cases plasma renin levels are low. In patients with hyperkalemia, urinary K+ below 40 mmol/L or fractional K+ excretion below 20% are indicative of a defect in kidney K+ secretion [52]. In patients with hypoaldosteronism, but normal renin levels, possible causes include adrenal gland damage, Addison disease, critical illness (i.e., direct renal suppression), RAAS inhibitor therapy, or heparin-induced suppression of aldosterone synthesis [50].
Extrarenal conditions may mimic RTA by causing normal gap metabolic acidosis through increased production of endogenous H+ and accelerated loss of extrarenal HCO3– [7]. Extrarenal metabolic acidosis is associated with elevated levels of H+ and urinary NH4+ excretion [7]. Severe or chronic diarrhea commonly causes hyperchloremic metabolic acidosis through the loss of large amounts of gastrointestinal HCO3–, particularly as HCO3– concentrations are usually higher in diarrheal fluid than in plasma [8, 9]. The reduction in plasma volume triggers an increase in kidney NaCl reabsorption which, in combination with HCO3– losses, leads to normal anion gap metabolic acidosis [7]. Low serum pH, as well as hypokalemia caused by gastrointestinal losses, promotes ammoniagenesis in the proximal tubule; the increased NH3 concentrations allow for increased H+ excretion by the distal nephron. This can result in an increased urine pH in patients with chronic diarrhea because of increases in kidney NH3 metabolism [7]. Chronic laxative abuse has been reported to mimic distal RTA through intestinal losses of HCO3– and K+ [70]. The normal increase in urinary NH3 excretion in these situations is reflected by a negative urinary anion gap and an increased urine osmolal gap.
Treatment of RTA
Dietary Considerations
In general, plant-based diets contain a lower net acid load and less bioavailable phosphate than animal-based diets [71]. In patients with metabolic acidosis, the dietary acid load can be decreased by limiting acid-producing foods (e.g., animal protein) and increasing alkali-producing foods (e.g., fruits and vegetables) (Table 1) [72,73,74]. Reduction in the consumption of dietary protein obtained from animal sources also results in increases in serum total CO2 concentrations [74].
Changes in dietary consumption of citrus fruit and juices, as well as restricted Na+, oxalate, fructose, and animal protein intake, in combination with normal calcium intake may also benefit patients with distal RTA and nephrolithiasis [75]. Increased fruit and vegetable intake is associated with increased urinary citrate excretion in patients with hypocitraturia and nephrolithiasis [75, 76]. In patients with hyperkalemic RTA, dietary restriction of K+ has previously been the standard of care [5]; however, new data suggest that increased intake of alkali-producing fruits and vegetables (which are often high in K+) and limiting intake of acid-producing foods may correct acidosis [72].
Pharmacotherapy
Table 1 summarizes the different pharmacotherapy options for patients with RTA.
Alkali therapy may be used to correct acidosis in patients with distal or proximal RTA [7]. In patients with distal RTA, alkali therapy also corrects for hypokalemia. Alkali therapy with 1–2 mmol/kg/day NaHCO3 or KHCO3 is normally sufficient to equal daily acid production [5]; however, in patients with nephrolithiasis or nephrocalcinosis, the elevated Na+ load with NaHCO3 therapy may cause increased urine calcium excretion, which can precipitate kidney stone formation. In these patients, K-citrate administration is preferable; this will also increase urine citrate excretion and prevent recurrence of kidney stones [75]. Patients with severe hypokalemia should also receive K+ replacement (i.e., with KCl or K-citrate) to prevent further lowering of serum K+ concentrations and symptomatic hypokalemia [7]. Long-term treatment of distal RTA generally requires a combination of NaHCO3 and KHCO3 [7, 14]. Children with distal RTA require sufficient NaHCO3 or KHCO3 (usually 4–8 mmol/kg/day) to maintain normal serum HCO3– concentrations and prevent growth retardation [5].
In contrast, treatment of proximal RTA is often challenging and patients require larger quantities of alkali therapy (10–15 mmol/kg/day), which are usually administered as a K+ salt (e.g., K-citrate) to avoid worsening hypokalemia [5, 7]. However, as exogenous alkali is rapidly excreted in the urine, correction of acidosis is often impossible despite administration of large amounts of alkali therapy [7]. In addition to alkali therapy, patients with Fanconi syndrome are treated with fluid and electrolyte replacement to prevent volume depletion, as well as supplementation with vitamin D and phosphate (1–3 g/day) to prevent bone disease [77]. Impaired proximal HCO3– reabsorption leads to most of the administered HCO3– being lost in the urine with minimal increases in serum concentrations [78], while increased distal Na+ delivery and elevated aldosterone levels cause increased renal K+ wasting [7]. Hydrochlorothiazide may be beneficial in increasing HCO3– reabsorption capacity, preventing volume expansion, and increasing the effectiveness of alkali therapy; however, supplemental K+ or K+-sparing diuretics are needed to prevent hypokalemia [5, 77]. After initiation of therapy, patients should be closely monitored for severe electrolyte abnormalities. Children with proximal RTA often require aggressive alkali therapy (5–15 mmol/kg/day) to mitigate bone disease and growth retardation [79], whereas alkali therapy is typically administered in adults with serum HCO3– concentrations of less than 18 mmol/L to prevent severe acidosis [7].
NaHCO3 therapy is often associated with gastrointestinal adverse effects, most commonly bloating and belching, and should be taken on an empty stomach [72, 74]. If these adverse effects limit patient adherence, Na-citrate or enteric-coated NaHCO3 (where available) may be considered [74].
In patients with hyperkalemic type 4 RTA, lowering of serum K+ concentrations often leads to correction of metabolic acidosis [7]. The reduction in serum K+ leads to increased NH3 production in the proximal tubule and medullary transfer, thereby increasing the availability of buffer supply for distal acidification [7]. Any non-essential medications affecting renal K+ excretion or aldosterone synthesis or activity should be discontinued [5, 7]. In Clinical Case Presentation 3 described above, discontinuation of the COX-2 inhibitor celecoxib would be beneficial in reducing serum K+ concentrations. ACE inhibitors and ARBs reduce urinary K+ excretion through inhibition of aldosterone secretion in the adrenal gland and may cause hyperkalemia in patients with pre-existing conditions that cause impaired K+ excretion [80]. For this reason, RAAS inhibitor therapy was avoided in Clinical Case Presentation 3; however, ACE inhibitors and ARBs are normally continued in patients with CKD because of their cardiovascular and renoprotective benefits [7]. Low-dose fludrocortisone therapy may also be effective in managing hyperkalemia and hyponatremia in patients with type 4 RTA who do not have heart failure or hypertension [52, 81]. Since thiazide diuretics are largely ineffective in patients with an estimated GFR of less than 30 mL/min/1.73 m2, loop diuretics and NaHCO3 therapy may be beneficial in patients with type 4 RTA, particularly when fludrocortisone is not tolerated. Loop diuretics may reduce serum K+ and help control volume overload by increasing Na+ delivery and flow rates to the cortical collecting duct while lowering blood pressure [52]. The Kidney Disease-Improving Global Outcomes guidelines suggest administration of oral HCO3– therapy to maintain serum HCO3– in the normal range in patients with CKD and serum HCO3– less than 22 mmol/L [82]. NaHCO3 administration will correct metabolic acidosis and further minimize the risk of hyperkalemia, and the addition of Na+ would not be problematic in the setting of effective diuretic therapy. However, patients on NaHCO3 therapy should be closely monitored for volume overload and hypertension [7].
Novel Pharmacotherapy
For patients with hyperkalemic type 4 RTA, there are newer K+-binding agents available (i.e., patiromer and sodium zirconium cyclosilicate [SZC]), which can be used to treat hyperkalemia and improve acidosis. Patiromer is a polymeric cation exchange resin that binds K+ ions in exchange for calcium ions in the colon [83], whereas SZC is non-polymeric, selective K+-binder, which entraps K+ and NH4+ ions in exchange for H+ and Na+ ions throughout the gastrointestinal tract [84]. Both patiromer and SZC are effective in patients receiving RAAS inhibitor therapy, which allows continuation of these therapeutic agents in the presence of hyperkalemia [85, 86]. SZC has also been associated with significant increases in serum HCO3– concentrations by approximately 2 mmol/L versus placebo during the initial 48-h treatment period and 2–3 mmol/L during maintenance therapy for 1 month, regardless of CKD stage [85]. This increase in serum HCO3– with SZC is most likely as a result of gastrointestinal NH4+ binding ions and associated decreases in serum urea concentrations [87]. Reducing serum K+ concentrations with secondary increases in NH3 synthesis may be an additional mechanism by which acidosis is improved with SZC. Whether patiromer produces similar effects on serum HCO3– remains to be determined; however, treatment with SZC may provide therapeutic benefits in patients with hyperkalemic RTA. Studies are needed to confirm the efficacy and safety of SZC or patiromer in hyperkalemic RTA.
Veverimer (TRC101) is a treatment for metabolic acidosis currently in clinical development. Veverimer selectively binds H+ in the gastrointestinal tract, thereby increasing serum HCO3– concentrations and correcting acidosis [88]. In patients with CKD and metabolic acidosis, veverimer significantly increased serum HCO3– concentrations within 24–72 h of administration and continued to be effective over 2 weeks [89] or 12 weeks [90]. In a 1-year extension study, veverimer therapy corrected metabolic acidosis, was well tolerated, and was associated with improved patient-reported physical function outcomes [88]. Although studies in patients with RTA are needed to confirm the efficacy and safety of veverimer in RTA, this drug represents an emerging alternative to alkali therapy.
Conclusions
In clinical practice, patients with RTA can present with a wide range of signs and symptoms. Accurate diagnosis of RTA is important in selecting the appropriate treatment strategy. Diet modifications and alkali therapy are treatment options, and the availability of newer K+ binders may assist in the management of hyperkalemic RTA. The H+-binding agent veverimer may be an additional emerging treatment option.
References
Hamm LL, Nakhoul N, Hering-Smith KS. Acid-base homeostasis. Clin J Am Soc Nephrol. 2015;10:2232–42.
Scialla JJ, Asplin J, Dobre M, et al. Higher net acid excretion is associated with a lower risk of kidney disease progression in patients with diabetes. Kidney Int. 2017;91:204–15.
Halperin ML, Jungas RL. Metabolic production and renal disposal of hydrogen ions. Kidney Int. 1983;24:709–13.
Hamilton PK, Morgan NA, Connolly GM, Maxwell AP. Understanding acid-base disorders. Ulster Med J. 2017;86:161–6.
Soleimani M, Rastegar A. Pathophysiology of renal tubular acidosis: core curriculum 2016. Am J Kidney Dis. 2016;68:488–98.
Palmer BF. Normal acid-base balance. In: Johnson RJ, Feehally J, Floege J, editors. Comprehensive clinical nephrology. 5th ed. Philadelphia: Elsevier; 2014. p. 142–8.
Palmer BF, Clegg DJ. Hyperchloremic normal gap metabolic acidosis. Minerva Endocrinol. 2019;44:363–77.
Yaxley J, Pirrone C. Review of the diagnostic evaluation of renal tubular acidosis. Ochsner J. 2016;16:525–30.
Berend K. Review of the diagnostic evaluation of normal anion gap metabolic acidosis. Kidney Dis (Basel). 2017;3:149–59.
Trepiccione F, Prosperi F, de la Motte LR, et al. New findings on the pathogenesis of distal renal tubular acidosis. Kidney Dis (Basel). 2017;3:98–105.
Watanabe T. Improving outcomes for patients with distal renal tubular acidosis: recent advances and challenges ahead. Pediatric Health Med Ther. 2018;9:181–90.
Rodriguez SJ. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13:2160–70.
Emmett M, Kelepouris E. Overview and pathophysiology of renal tubular acidosis and the effect on potassium balance. https://www.uptodate.com/contents/overview-and-pathophysiology-of-renal-tubular-acidosis-and-the-effect-on-potassium-balance. Accessed Feb 17, 2020.
Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018;31:511–22.
Vasquez-Rios G, Westrich DJ Jr, Philip I, Edwards JC, Shieh S. Distal renal tubular acidosis and severe hypokalemia: a case report and review of the literature. J Med Case Rep. 2019;13:103.
Gueutin V, Vallet M, Jayat M, et al. Renal beta-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest. 2013;123:4219–31.
Oh JY, Shin JI. Syndrome of inappropriate antidiuretic hormone secretion and cerebral/renal salt wasting syndrome: similarities and differences. Front Pediatr. 2014;2:146.
Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90.
Smith AN, Skaug J, Choate KA, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–5.
Stover EH, Borthwick KJ, Bavalia C, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803.
Karet FE, Gainza FJ, Gyory AZ, et al. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci U S A. 1998;95:6337–42.
Sawaya BP, Briggs JP, Schnermann J. Amphotericin B nephrotoxicity: the adverse consequences of altered membrane properties. J Am Soc Nephrol. 1995;6:154–64.
Weiner ID, Leader JP, Bedford JJ, et al. Effects of chronic lithium administration on renal acid excretion in humans and rats. Physiol Rep. 2014;2:e12242.
Bichard L, Toh D. Ibuprofen-induced distal (type 1) renal tubular acidosis and hypokalaemia: the dangers of ibuprofen-codeine combination over-the-counter preparations. Intern Med J. 2017;47:707–9.
Ho K, Dokouhaki P, McIsaac M, Prasad B. Renal tubular acidosis as the initial presentation of Sjogren's syndrome. BMJ Case Rep. 2019;12:e230402.
Francois H, Mariette X. Renal involvement in primary Sjogren syndrome. Nat Rev Nephrol. 2016;12:82–93.
Agrwal S, Mantan M, Dabas A. An unusual case of familial systemic lupus erythematosus with distal renal tubular acidosis and hemolytic anemia. Iran J Kidney Dis. 2019;13:337–9.
Goutaudier V, Szwarc I, Serre JE, Pageaux GP, Argiles A, Ribstein J. Primary sclerosing cholangitis: a new cause of distal renal tubular acidosis. Clin Kidney J. 2016;9:811–3.
Bharani A, Manchanda R, Singh RK, Prashant S. Distal renal tubular acidosis in sickle cell anemia. Saudi J Kidney Dis Transpl. 2018;29:1000–4.
Subrahmanyam DK, Vadivelan M, Giridharan S, Balamurugan N. Wilson’s disease: a rare cause of renal tubular acidosis with metabolic bone disease. Indian J Nephrol. 2014;24:171–4.
Clericetti CM, Milani GP, Lava SAG, Bianchetti MG, Simonetti GD, Giannini O. Hyperammonemia associated with distal renal tubular acidosis or urinary tract infection: a systematic review. Pediatr Nephrol. 2018;33:485–91.
Fuster DG, Moe OW. Incomplete distal renal tubular acidosis and kidney stones. Adv Chronic Kidney Dis. 2018;25:366–74.
Kashoor I, Batlle D. Proximal renal tubular acidosis with and without Fanconi syndrome. Kidney Res Clin Pract. 2019;38:267–81.
Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant. 2012;27:4273–87.
Unwin RJ, Capasso G. The renal tubular acidoses. J R Soc Med. 2001;94:221–5.
Igarashi T, Inatomi J, Sekine T, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999;23:264–6.
Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2171–7.
Gutierrez JO, Zurita MF, Zurita LA. Sjogren’s syndrome associated with Fanconi’s syndrome and osteomalacia. Am J Case Rep. 2018;19:392–6.
Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int. 2000;58:1267–77.
Yamaguchi S, Maruyama T, Wakino S, et al. A case of severe osteomalacia caused by tubulointerstitial nephritis with Fanconi syndrome in asymptomotic primary biliary cirrhosis. BMC Nephrol. 2015;16:187.
Brenner RJ, Spring DB, Sebastian A, et al. Incidence of radiographically evident bone disease, nephrocalcinosis, and nephrolithiasis in various types of renal tubular acidosis. N Engl J Med. 1982;307:217–21.
Lee YS, Kim BK, Lee HJ, Dan J. Pathologic femoral neck fracture due to Fanconi syndrome induced by adefovir dipivoxil therapy for hepatitis B. Clin Orthop Surg. 2016;8:232–6.
Negro A, Regolisti G, Perazzoli F, Davoli S, Sani C, Rossi E. Ifosfamide-induced renal Fanconi syndrome with associated nephrogenic diabetes insipidus in an adult patient. Nephrol Dial Transplant. 1998;13:1547–9.
Knights M, Thekkekkara T, Morris A, Finlay E. Sodium valproate-induced Fanconi type proximal renal tubular acidosis. BMJ Case Rep. 2016;2016:bcr2015213418.
Izzedine H, Launay-Vacher V, Deray G. Topiramate-induced renal tubular acidosis. Am J Med. 2004;116:281–2.
Sacré A, Jouret F, Manicourt D, Devuyst O. Topiramate induces type 3 renal tubular acidosis by inhibiting renal carbonic anhydrase. Nephrol Dial Transplant. 2006;21:2995–6.
Nagai T, Matsuo N, Tsuchiya Y, Cho H, Hasegawa Y, Igarashi Y. Proximal renal tubular acidosis associated with glycogen storage disease, type 9. Acta Paediatr Scand. 1988;77:460–3.
Richardson RM, Little JA, Patten RL, Goldstein MB, Halperin ML. Pathogenesis of acidosis in hereditary fructose intolerance. Metabolism. 1979;28:1133–8.
Mathur M, Chacko B, Vankalakunti M, Patil C. Fanconi syndrome due to light chain proximal tubulopathy in a patient with multiple myeloma. Saudi J Kidney Dis Transpl. 2016;27:805–7.
Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol. 2009;20:251–4.
Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med. 2015;373:548–59.
Batlle D, Arruda J. Hyperkalemic forms of renal tubular acidosis: clinical and pathophysiological aspects. Adv Chronic Kidney Dis. 2018;25:321–33.
Batlle DC, Arruda JA, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructive uropathy. N Engl J Med. 1981;304:373–80.
Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol. 2014;9:1974–86.
Sánchez-Marcos C, Hoffman V, Prieto-González S, Hernández-Rodríguez J, Espinosa G. Renal tubular acidosis type IV as a complication of lupus nephritis. Lupus. 2016;25:307–9.
Garibotto G, Sofia A, Robaudo C, et al. Kidney protein dynamics and ammoniagenesis in humans with chronic metabolic acidosis. J Am Soc Nephrol. 2004;15:1606–15.
Cook E, Davis J, Israni R, et al. Prevalence of metabolic acidosis among patients with CKD and hyperkalemia [abstract 89]. Am J Kidney Dis. 2020;75:561–2.
Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–12.
Harris RC, Breyer MD. Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol. 2006;1:236–45.
Lin W, Mou L, Tu H, et al. Clinical analysis of hyperkalemic renal tubular acidosis caused by calcineurin inhibitors in solid organ transplant recipients. J Clin Pharm Ther. 2017;42:122–4.
Riveiro-Barciela M, Campos-Varela I, Tovar JL, et al. Hyperkalemic distal renal tubular acidosis caused by immunosuppressant treatment with tacrolimus in a liver transplant patient: case report. Transplant Proc. 2011;43:4016–8.
Schmoyer C, Mishra S, Fulco F. Tacrolimus-induced type IV renal tubular acidosis following liver transplantation. Case Rep Hepatol. 2017;2017:9312481.
Tumlin JA, Sands JM. Nephron segment-specific inhibition of Na+/K+-ATPase activity by cyclosporin A. Kidney Int. 1993;43:246–51.
Heering PJ, Kurschat C, Vo DT, Klein-Vehne N, Fehsel K, Ivens K. Aldosterone resistance in kidney transplantation is in part induced by a down-regulation of mineralocorticoid receptor expression. Clin Transplant. 2004;18:186–92.
Santos F, Ordonez FA, Claramunt-Taberner D, Gil-Pena H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. 2015;30:2099–107.
Kyono Y, Nozu K, Nakagawa T, et al. Combination of furosemide and fludrocortisone as a loading test for diagnosis of distal renal tubular acidosis in a pediatric case. CEN Case Rep. 2020;9:81–6.
Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007;71:1310–6.
Kim S, Lee JW, Park J, et al. The urine-blood PCO2 gradient as a diagnostic index of H+-ATPase defect distal renal tubular acidosis. Kidney Int. 2004;66:761–7.
Stinebaugh BJ, Schloeder FX, Tam SC, Goldstein MB, Halperin ML. Pathogenesis of distal renal tubular acidosis. Kidney Int. 1981;19:1–7.
Sidler M, Mohebbi N, Hoorn EJ, Wagner CA. Gut it out: laxative abuse mimicking distal renal tubular acidosis. Kidney Blood Press Res. 2019;44:1294–9.
Carrero JJ, Gonzalez-Ortiz A, Avesani CM, et al. Plant-based diets to manage the risks and complications of chronic kidney disease. Nat Rev Nephrol. 2020;16:525–42.
Goraya N, Wesson DE. Management of the metabolic acidosis of chronic kidney disease. Adv Chronic Kidney Dis. 2017;24:298–304.
Navaneethan SD, Shao J, Buysse J, Bushinsky DA. Effects of treatment of metabolic acidosis in CKD: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2019;14:1011–20.
Raphael KL. Metabolic acidosis in CKD: core curriculum 2019. Am J Kidney Dis. 2019;74:263–75.
Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol. 2009;11:134–44.
Meschi T, Maggiore U, Fiaccadori E, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66:2402–10.
Karatzas A, Paridis D, Kozyrakis D, et al. Fanconi syndrome in the adulthood. The role of early diagnosis and treatment. J Musculoskelet Neuronal Interact. 2017;17:303–6.
Sebastian A, McSherry E, Morris RC Jr. On the mechanism of renal potassium wasting in renal tubular acidosis associated with the Fanconi syndrome (type 2 RTA). J Clin Invest. 1971;50:231–43.
Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann CM Jr. Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr. 1972;80:738–48.
Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004;351:585–92.
Dobbin SJH, Petrie JR, Lean MEJ, McKay GA. Fludrocortisone therapy for persistent hyperkalaemia. Diabet Med. 2017;34:1005–8.
Kidney Disease Improving Global Outcomes. KDIGO clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150.
Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J. 2011;32:820–8.
Stavros F, Yang A, Leon A, Nuttall M, Rasmussen HS. Characterization of structure and function of ZS-9, a K+ selective ion trap. PLoS One. 2014;9:e114686.
Roger SD, Lavin PT, Lerma EV, et al. Long-term safety and efficacy of sodium zirconium cyclosilicate for hyperkalaemia in patients with mild/moderate versus severe/end-stage chronic kidney disease: comparative results from an open-label, phase 3 study. Nephrol Dial Transplant. 2020. https://doi.org/10.1093/ndt/gfz285.
Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med. 2015;372:211–21.
Roger SD, Spinowitz BS, Lerma EV, et al. Sodium zirconium cyclosilicate increases serum bicarbonate concentrations among patients with hyperkalaemia: exploratory analyses from three randomized, multi-dose, placebo-controlled trials. Nephrol Dial Transplant. 2020. https://doi.org/10.1093/ndt/gfaa158.
Wesson DE, Mathur V, Tangri N, et al. Long-term safety and efficacy of veverimer in patients with metabolic acidosis in chronic kidney disease: a multicentre, randomised, blinded, placebo-controlled, 40-week extension. Lancet. 2019;394:396–406.
Bushinsky DA, Hostetter T, Klaerner G, et al. Randomized, controlled trial of TRC101 to increase serum bicarbonate in patients with CKD. Clin J Am Soc Nephrol. 2018;13:26–35.
Wesson DE, Mathur V, Tangri N, et al. Veverimer versus placebo in patients with metabolic acidosis associated with chronic kidney disease: a multicentre, randomised, double-blind, controlled, phase 3 trial. Lancet. 2019;393:1417–27.
Acknowledgements
Funding
The development of this manuscript and the journal’s rapid service and open access fees were supported by AstraZeneca.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript and take responsibility for the integrity of the work as a whole.
Medical Writing, Editorial, and Other Assistance
Sarah Greig, PhD (Auckland, New Zealand), and Meri Pozo, PhD, CMPP (New York, NY, USA) of inScience Communications provided medical writing support funded by AstraZeneca.
Disclosures
Biff F. Palmer and Deborah J. Clegg have nothing to disclose. Ellie Kelepouris discloses receipt of a grant from Mallinckrodt; personal fees for advisory board and/or consulting from Akebia, AstraZeneca, Mallinckrodt, and Relypsa/Vifor; personal fees for manuscript preparation from Up-to-Date. The authors have not received any funding for the current work.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Palmer, B.F., Kelepouris, E. & Clegg, D.J. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Adv Ther 38, 949–968 (2021). https://doi.org/10.1007/s12325-020-01587-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01587-5