
Abstract
Some patients with interstitial lung diseases (ILDs) other than idiopathic pulmonary fibrosis exhibit a progressive clinical phenotype. These chronic progressive fibrosing ILDs have a variety of underlying diseases, and their prevalence is currently unknown. Here we carry out the first systematic review of literature on the prevalence of fibrosing ILDs and progressive fibrosing ILDs using data from physician surveys to estimate frequency of progression among different ILDs. We searched MEDLINE and Embase for studies assessing prevalence of ILD, individual ILDs associated with fibrosis and progressive fibrosing ILDs. These were combined with data from previously published physician surveys to obtain prevalence estimates of each chronic fibrosing ILD with a progressive phenotype and of progressive fibrosing ILDs overall. We identified 16 publications, including five reporting overall ILD prevalence, estimated at 6.3–76.0 per 100,000 people in Europe (four studies) and 74.3 per 100,000 in the USA (one study). In total, 13–40% of ILDs were estimated to develop a progressive fibrosing phenotype, with overall prevalence estimates for progressive fibrosing ILDs of 2.2–20.0 per 100,000 in Europe and 28.0 per 100,000 in the USA. Prevalence estimates for individual progressive fibrosing ILDs varied up to 16.7 per 100,000 people. These conditions represent a sizeable fraction of chronic respiratory disorders and have a high unmet need.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
A proportion of interstitial lung diseases (ILDs) (other than idiopathic pulmonary fibrosis) that are associated with a variety of underlying conditions may have a common chronic progressive phenotype, but the prevalence of such progressive fibrosing ILDs is currently unknown |
We have reviewed the literature to estimate the prevalence of different fibrosing ILDs, but there are few data on prevalence of a progressive fibrosing phenotype |
The frequency of progression among different fibrosing ILDs has been estimated in a physician survey, and we have used these estimates in combination with published prevalence estimates for fibrosing ILDs to estimate the overall prevalence of progressive fibrosing ILD |
The overall prevalence estimates for progressive fibrosing ILDs were 2.2–20.0 per 100,000 in Europe and 28.0 per 100,000 in the USA, with 13–40% of ILDs estimated to develop a progressive fibrosing phenotype |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13247072.
Introduction
The term “interstitial lung disease” (ILD) encompasses a heterogeneous group of pulmonary disorders, characterised by diffuse parenchymal lung infiltration [1,2,3]. Pulmonary fibrosis is common to many ILDs with differing underlying diseases. Some ILDs can develop a progressive fibrosing phenotype, characterised by worsening fibrotic changes on high-resolution computed tomography (HRCT) of the chest, decline in lung function over time, worsening symptoms and quality of life, and early mortality [4,5,6]. Patients who develop this phenotype are collectively described as having progressive fibrosing ILD [5,6,7]. Idiopathic pulmonary fibrosis (IPF) is generally recognised as the archetypal progressive fibrosing ILD [8]. However, a progressive fibrosing phenotype can develop in other ILDs, including idiopathic non-specific interstitial pneumonia (iNSIP), hypersensitivity pneumonitis (HP) and ILDs associated with autoimmune disorders, in particular rheumatoid arthritis-associated ILD (RA-ILD) and systemic sclerosis-associated ILD (SSc-ILD) [5, 7]. Not all patients with these ILDs will develop a progressive fibrosing phenotype, but those that do demonstrate a similar disease course and prognosis to patients with IPF [6, 7]. Although there is a broad agreement among experts that these conditions are rare, of debilitating nature and associated with high mortality, there are few published data on overall prevalence of progressive fibrosing ILDs [9,10,11]. Estimation of prevalence of progressive fibrosing ILD has methodological challenges due to the rarity of the disease, the lack of an established definition, the broad spectrum of underlying diseases and difficulty in establishing specific diagnoses; the prior lack of approved treatment for most progressive fibrosing ILDs further compounds this challenge. ILDs with a chronic progressive phenotype have been the subject of recent review articles [4, 12, 13], but these have not attempted to estimate prevalence and rates of progression in a systematic manner.
A phase III clinical trial has investigated the efficacy and safety of nintedanib in patients with differing clinical ILD diagnoses [7] who presented with features of a diffuse fibrosing lung disease of greater than 10% extent on chest HRCT and had clinical signs of progression (Table S1 in the electronic supplementary material) [7]. Compared with placebo, nintedanib significantly reduced the annual rate of decline in forced vital capacity (FVC) in these patients [14]. The potential availability of treatment for progressive fibrosing ILD amplifies the importance of better understanding the epidemiology of these conditions, thereby enabling a more precise quantification of the unmet medical needs.
The objective of this systematic literature review was to assess the overall prevalence of ILDs, the overall prevalence of progressive fibrosing ILDs and the prevalence of individual ILDs that may develop a progressive fibrosing phenotype, in Europe and North America.
Methods
Systematic Literature Review
Search Strategy
Literature searches were conducted in August 2017 using MEDLINE and Embase electronic bibliographical databases to identify publications reporting the prevalence of ILD, the prevalence of individual ILDs and the prevalence of progressive fibrosing ILDs. Thesaurus terms (MeSH and Emtree for MEDLINE and Embase, respectively) and subject headings were combined with free-text keywords; full search strategies are outlined in Table S2.
To ensure that relevant publications were not missed as a result of indexing delays, manual searches of the American Journal of Respiratory and Critical Care Medicine, New England Journal of Medicine and European Respiratory Journal websites were performed for the final 2 years of the search period (2016 and 2017) using the terms “prevalence” and “interstitial lung disease”. In addition, manual reviews of reference lists of publications that met the inclusion criteria were performed to maximise the comprehensiveness of the search.
Eligibility Criteria
Inclusion criteria for the literature search included: non-interventional studies in humans; studies conducted in Europe or North America; studies performed in the general population or an ILD patient population; and studies assessing the prevalence of ILD, the prevalence of individual ILDs and/or the proportions of patients with individual ILDs. Results of the search were restricted to full-text publications in English or German, published between January 1, 1990 and August 7, 2017. Abstracts, case reports, case series, editorials, letters and opinion pieces were excluded. Studies reporting only on IPF or SSc-ILD were not included within our literature search, as these conditions have been the subject of previous systematic literature searches [15, 16].
Study Selection
Following the removal of duplicate publications, study selection was accomplished through a dual-level screening process. First, abstracts were reviewed to determine whether the study was observational in nature, performed in one of the target locations, included an eligible patient population and reported an outcome of interest. Full-text assessment of the qualifying publications was then performed by two reviewers according to the eligibility criteria outlined above.
Data Extraction and Quality Assessment
Data extraction was performed and checked for consistency by two reviewers. Extracted data included: location; data source; study design; study period; study population; study setting; study objectives; case definition; exclusion criteria; baseline demographics; number of cases; prevalence; and proportion of fibrosing ILDs and progression (if reported). Where possible (if not already reported in the publication), prevalence estimates were recalculated using the total population as the denominator.
Some publications did not report the prevalence of individual ILDs, but instead reported the overall prevalence of ILD and the proportion of patients within the ILD population with individual ILDs. In these cases, estimates of the prevalence of each individual ILD were obtained by multiplying the overall prevalence of ILD by the reported proportion of patients with each individual ILD.
Prevalence of Fibrosing ILD and Progressive Fibrosing ILDs
Prevalence estimates for individual ILDs with a progressive fibrosing phenotype were calculated (per 100,000 people). Estimates were based on the prevalence of individual ILDs reported in the literature and, on the basis of these, the proportion of patients with the given ILD expected to develop a progressive fibrosing phenotype was calculated. Proportions expected to progress were not, for the most part, available from published data so an exploratory approach based on a quantitative physician survey was taken in which expert clinicians, including both pulmonologists and rheumatologists who had a high publication record and had participated in guideline development and/or ILD clinical trials, provided estimates of the proportion of patients with individual ILDs that may develop a progressive fibrosing phenotype [17].
The individual estimated prevalence values for different ILDs with a progressive fibrosing phenotype were then summed to estimate the overall prevalence of progressive fibrosing ILDs in each publication, so that a prevalence range for progressive fibrosing ILDs in Europe and North America could be estimated. Only data from studies that reported the overall prevalence of ILD, as well as the prevalence or proportion of individual ILDs, were used for prevalence estimates of progressive fibrosing ILDs. Studies reporting only on individual ILDs and their rates of progression were not used for prevalence estimates of progressive fibrosing ILDs because of differences in study design, setting, methodology and patient population between studies.
A conceptual diagram of prevalence estimation for progressive fibrosing ILDs is presented in Fig. 1 [5].

Adapted with permission of the © ERS 2020. European Respiratory Journal 51 (5) 1800692; https://doi.org/10.1183/13993003.00692-2018 Published 17 May 2018
Conceptual diagram of prevalence estimation for progressive fibrosing ILDs [5]. Figure is not to scale. “Autoimmune ILDs” includes RA-ILD, SSc-ILD, PM-ILD, DM-ILD, Sjögren’s-ILD, SLE-ILD, CTD-ILD and MCTD-ILD; “Non-IPF IIPs” includes iNSIP and unclassifiable IIP; “Environmental ILDs” includes asbestosis, silicosis and coal worker’s pneumonitis. CTD-ILD connective tissue disease-associated interstitial lung disease, DM-ILD dermatomyositis-associated interstitial lung disease, HP hypersensitivity pneumonitis, IIP idiopathic interstitial pneumonia, ILD interstitial lung disease, iNSIP idiopathic non-specific interstitial pneumonia, IPF idiopathic pulmonary fibrosis, MCTD-ILD mixed connective tissue disease-associated interstitial lung disease, PM-ILD polymyositis-associated interstitial lung disease, RA-ILD rheumatoid arthritis-associated interstitial lung disease, SLE-ILD systemic lupus erythematosus-associated interstitial lung disease, SSc-ILD systemic sclerosis-associated interstitial lung disease.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Results
Systematic Literature Review
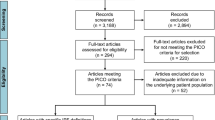
A flow diagram of the publications identified for inclusion is presented in Fig. S1 in the electronic supplementary material. In total, 1633 publications were identified through literature searches conducted in MEDLINE and Embase. After the removal of 340 duplicates, 1251 publications were excluded on the basis of their abstracts, as they either did not report on the outcomes of interest or were not conducted in North America or Europe. Of the remaining 42 publications, 33 did not meet the eligibility criteria. Web-based searches and manual review of reference lists yielded an additional seven references, providing a total of 16 publications from which data were extracted [1,2,3, 18,19,20,21,22,23,24,25,26,27,28,29,30]. Six of the studies were conducted in the USA and 10 were conducted in Europe; 10 were cohort studies and six had a cross-sectional design; studies were conducted between 1988 and 2013 and published between 1990 and 2013. Of the 16 identified publications, five reported data on overall ILD prevalence and 11 reported data on the prevalence of individual ILDs or the proportion of individual ILDs in the prevalent ILD population (Table S3).
ILD Prevalence
Overall ILD prevalence, as reported in five identified publications from the literature review, ranged from 6.3 to 76.0 per 100,000 people in four studies in Europe [1, 24, 26, 30] and was 74.3 per 100,000 people in one study in the USA [21] (Fig. 2 [1, 21, 24, 26, 30]). Two of these studies also reported the prevalence of progressive fibrosing ILD specifically [21, 24]. The first study, conducted in France, estimated fibrotic idiopathic interstitial pneumonia (including IPF, iNSIP and cases with the International Classification of Diseases [ICD] 10th Revision code for pulmonary fibrosis) prevalence at 12.6 per 100,000 people [24]. The second study, conducted in New Mexico, USA, estimated the prevalence of ILD with pulmonary fibrosis (based on the presence of ICD code 515) as 29.0 per 100,000 for men and 27.0 per 100,000 for women [21].

The prevalence of other individual ILDs was reported (or calculated on the basis of the proportion of patients with individual ILDs within the ILD population) in 11 identified publications from the literature review and varied up to 21.5 per 100,000 people. Some studies reported zero prevalence for particular ILDs. The estimated prevalence or prevalence range for each ILD, including data from studies that reported prevalence estimates for individual ILDs, is presented in Fig. 3 [2, 16, 19,20,21,22,23,24,25,26, 30].

Prevalence of fibrosing ILDs in Europe and the USA. aAs reported by or calculated from [2, 24, 26]. bAs reported by or calculated from [24, 26, 30]. cAs reported by or calculated from [2, 21, 24, 26, 30]. dAs reported by or calculated from [24, 26]. eAs reported by or calculated from [2, 21, 24, 26, 30]. fAs reported by [21, 26]. gAs reported by [16, 21, 26]. hAs reported by or calculated from [24, 30]. iAs reported by or calculated from [2, 19,20,21,22,23,24,25,26, 30]. CTD-ILD connective tissue disease-associated interstitial lung disease, DM-ILD dermatomyositis-associated interstitial lung disease, HP hypersensitivity pneumonitis, IIP idiopathic interstitial pneumonia, ILD interstitial lung disease, iNSIP idiopathic non-specific interstitial pneumonia, IPF idiopathic pulmonary fibrosis, MCTD-ILD mixed connective tissue disease-associated interstitial lung disease, PM-ILD polymyositis-associated interstitial lung disease, RA-ILD rheumatoid arthritis-associated interstitial lung disease, SLE-ILD systemic lupus erythematosus-associated interstitial lung disease, SSc-ILD systemic sclerosis-associated interstitial lung disease
Five additional publications were identified that did not report prevalence data, but did assess the proportion of patients diagnosed with individual ILDs within the prevalent ILD population [3, 18, 27,28,29], with IPF (27.0–32.5%) and sarcoidosis (33.7–44.7%) being the most commonly reported (Table S1).
Estimates of Progression
Excluding IPF (which is by definition progressive), 13–40% of patients with ILD were estimated to develop a progressive fibrosing phenotype, depending on the underlying disease (Table 1) [17, 31,32,33,34]. Four studies describing the proportion of patients with RA-ILD [31], SSc-ILD [32], polymyositis/dermatomyositis-associated ILD [33] and sarcoidosis [34] who had a progressive fibrosing phenotype were identified in the literature (Table 1). Two of these studies used pre-specified criteria (diffusing capacity of the lung for carbon monoxide [DLCO] falling below 40% predicted [31]; relative decline in FVC of at least 10% or in DLCO of at least 15% [33]) to define a progressive fibrosing phenotype. While progression was not defined in the other two studies, patients whose disease progressed were identified retrospectively on the basis of worsening lung function [32] or worsening respiratory symptoms [34].
Prevalence of Progressive Fibrosing ILDs
Of the five studies that reported overall ILD prevalence, two also reported the prevalence of various individual ILDs, such as RA-ILD [26, 30], and two reported the proportion of patients with individual ILDs within the ILD population [21, 24], thus enabling prevalence estimates to be calculated. For these four studies, for which the prevalence of individual ILDs could be determined [21, 24, 26, 30], calculated prevalence estimates for individual ILDs with a progressive fibrosing phenotype varied up to 16.7 per 100,000 people (Table 2) [21, 24, 26, 30]. The collective estimated prevalence of progressive fibrosing ILDs ranged from 2.2 to 20 per 100,000 people in Europe [24, 26, 30] and was 28 per 100,000 people in the USA [21].
Discussion
There have been few epidemiological studies investigating individual ILDs or ILDs overall. In view of the potential availability of treatment for progressive fibrosing ILDs, based on clinical trials that have investigated whether therapies that slow progression in IPF can serve a similar function in progressive fibrosing ILDs, quantification of the prevalence of this group of diseases has become increasingly important. We present the first systematic review to estimate the overall prevalence of ILD and progressive fibrosing ILDs in Europe and the USA. We found that both prevalence estimates were generally low. Even without applying the criteria for a progressive fibrosing phenotype, the estimated prevalence for each specific ILD was low. Besides IPF (16.7 per 100,000 people), connective tissue disease-associated interstitial lung disease (0.5–10.2 per 100,000 people) and sarcoidosis (1.9–66.1 per 100,000 people) were most commonly reported. On the basis of estimates from the literature review and the results from the physician interviews and survey [35], 13–40% of patients with individual ILDs (100% for IPF) were estimated to develop a progressive fibrosing phenotype, resulting in prevalence estimates for the progressive fibrosing phenotype in individual ILDs of up to 16.7 per 100,000 people for the four studies analysed. The overall prevalence of progressive fibrosing ILDs was subsequently estimated at 2.2–28.0 per 100,000 people (2.2 in Belgium, 6.0 in Greece, 20.0 in France and 28.0 in the USA). Progressive fibrosing ILDs can, therefore, be considered rare, affecting fewer than 200,000 people in the USA (a prevalence of approximately 80 per 100,000 people, based on the 2016 adult population) and having an estimated prevalence below the 50 per 100,000 people threshold for orphan status in Europe [37]. It should also be noted that variations between countries may reflect the methodologies used to calculate the estimates rather than true differences in prevalence [37].
When interpreting these results, the following factors should be considered. Studies were conducted across several distinct geographical locations in Europe and the USA, with population differences potentially resulting in varying prevalence estimates. In addition, studies were conducted in different settings, leading to potential over- or underestimation of prevalence. Some studies were based on insurance claims and codes used for these, without clinical verification [20, 22], while others were based on cross-referencing several sources of data to validate each individual diagnosis [3, 18, 23, 25]. In the analysis of patient registry data for residents of Bernalillo County, New Mexico, USA, prevalence estimates were likely to be high owing to the presence of mining as a major industry and, consequently, higher occupational exposure to coal and silica relative to the general population [21]. Conversely, prevalence estimates from Flanders, Belgium were potentially low because of the exclusion of occupational ILDs [30]. Studies were based on data from differing sources, including patient registries, claims databases, medical records and surveys. For instance, only cases observed at respiratory centres where physicians volunteered to participate were included in the analysis of Greek survey data [26]. When medical records data from residents of the Seine-Saint-Denis department in France were analysed, approximately one-quarter of cases identified from hospital and community records could not be reviewed because charts were not available [24]. Studies were also conducted during different time frames, ranging from 1988 to 2013; classification of ILDs has changed markedly over this time [38–40], which may have further contributed to the variation in estimates between studies and could impact the results. Differences in the terminology and diagnostic criteria used to define cases between studies further complicate the ability to directly compare estimates within a given condition; patients were identified using clinical criteria or diagnostic guidelines in some studies, whereas others used ICD coding to identify cases, which may result in differences in estimation. Progressive fibrosing ILD is a relatively new disease construct that is not consistently defined in either the clinical practice or research context. Nevertheless, these are the first estimates of prevalence of progressive fibrosing ILD based on published literature and inputs from practising physicians, which meet a current unmet need in this disease therapy area.
Potential limitations of this systematic literature review methodology should also be considered. The literature review was restricted to prevalence data from studies conducted in Europe or North America and published in English or German, identified using our search strategy. Although all journals indexed in MEDLINE and Embase were searched, only American Journal of Respiratory and Critical Care Medicine, New England Journal of Medicine and European Respiratory Journal were manually reviewed in case of indexing delays, limited to the years 2016 and 2017. Furthermore, studies reporting only on IPF and SSc-ILD were not included within our search because these diseases have been analysed in literature reviews previously [14, 15], meaning that more recent publications (not within the time frames of the existing reviews) were not considered. Regarding our approach to determine the prevalence of progressive fibrosing ILDs, estimates were based on four studies that reported overall ILD prevalence and the prevalence of individual ILDs or specific ILD proportions [20, 23, 25, 29]. All four studies did not report data for progressive fibrosing ILDs specifically, and estimates of progression were applied on all studies retrospectively based on findings from the physician survey. The use of expert opinion to predict prevalence of progression in this way has not been validated and is open to question. Studies reporting only prevalence estimates for individual ILDs, and not for ILD overall [28, 31–33], were not included in the calculation of the overall prevalence of progressive fibrosing ILDs, as it was not considered methodologically sound because of significant differences in study design, setting, methodology and patient population. Consideration of the estimates from these studies may have resulted in differences in the estimated overall prevalence of progressive fibrosing ILDs, especially given the small number of studies upon which the prevalence calculations were based. In addition, proxies were used to determine a progressive fibrosing phenotype for data from the literature; thus, definitions of progression vary compared with the physician survey, as well as between publications [28,29,30,31]. Another limitation is related to the progressive phenotype of the disease being defined by lung function decline and/or worsening symptoms or imaging, independent of prior treatment received. In the INBUILD trial of nintedanib in this setting [7], patients were selected on the basis of a progressive phenotype despite conventional therapy. As prior therapy could not be assessed from the literature review of these 16 studies, it is unclear what proportion of the cases here were identified as having a progressive phenotype that may in fact be responsive to immunomodulation therapy, possibly overestimating the prevalence of the progressive phenotype.
Although the prevalence estimates calculated in this review should be considered preliminary (i.e. as the first estimate based on a literature review), this represents an important step in understanding the epidemiology of progressive fibrosing ILDs.
Conclusion
This systematic review provides an overview of the available literature on the prevalence of ILD, and the prevalence and proportion of patients with individual ILDs, in addition to the first estimate of prevalence of progressive fibrosing ILDs combining published literature and physician surveys. Progressive fibrosing ILDs have an estimated prevalence of between 2.2 and 20.0 per 100,000 people in Europe and 28.0 per 100,000 people in the USA, and can therefore be considered rare, based on current criteria. The data in this literature review lay the groundwork for other future epidemiological studies to be conducted on the prevalence of progressive fibrosing ILDs, and hopefully will give rise to a broadened awareness of these rare diseases and the unmet needs associated with them.
References
Gouder C, Fenech M, Montefort S. Interstitial lung disease in Malta. Malta Med J. 2012;24(2):11–5.
Roelandt M, Demedts M, Callebaut W, et al. Epidemiology of interstitial lung disease (ILD) in Flanders: registration by pneumologists in 1992–1994. Acta Clin Belg. 1995;50(5):260–8.
Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: a distinct entity. Chest. 2011;140(5):1292–9.
Kolb M, Vašáková M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. 2019;20(1):57.
Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J. 2018;51(5):1800692.
Cottin V, Hirani NA, Hotchkin DL, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381:1718–27.
Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–68.
Harari S. Beyond idiopathic pulmonary fibrosis: the world of progressive-fibrosing interstitial lung disease. Eur Respir Rev. 2018;27(150):180110.
Olson AL, Gifford AH, Inase N, Fernández Pérez ER, Suda T. The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. Eur Respir Rev. 2018;27(150):180077.
Holtze C, Flaherty K, Kreuter M, et al. Healthcare utilisation and costs in the diagnosis and treatment of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180078.
Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev. 2019;28(151):180100.
De Sadeleer LJ, Goos T, Yserbyt J, Wuyts WA. Towards the essence of progressiveness: bringing progressive fibrosing interstitial lung disease (PF-ILD) to the next stage. J Clin Med. 2020;9(6):1722.
Distler O, Gahlemann M, Maher TM. Nintedanib for systemic sclerosis-associated interstitial lung disease. Reply. N Engl J Med. 2019;381(16):1596–7.
Caminati A, Madotto F, Cesana G, Conti S, Harari S. Epidemiological studies in idiopathic pulmonary fibrosis: pitfalls in methodologies and data interpretation. Eur Respir Rev. 2015;24(137):436–44.
Bergamasco A, Hartmann N, Wallace L, Verpillat P. Epidemiology of systemic sclerosis and systemic sclerosis-associated interstitial lung disease. Clin Epidemiol. 2019;11:257–73.
Wijsenbeek M, Kreuter M, Olson A, et al. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35(11):2015–24.
Adegunsoye A, Oldham JM, Demchuk C, Montner S, Vij R, Strek ME. Predictors of survival in coexistent hypersensitivity pneumonitis with autoimmune features. Respir Med. 2016;114:53–60.
Arkema EV, Grunewald J, Kullberg S, Eklund A, Askling J. Sarcoidosis incidence and prevalence: a nationwide register-based assessment in Sweden. Eur Respir J. 2016;48(6):1690–9.
Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc. 2016;13(8):1244–52.
Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967–72.
Cragin LA, Laney AS, Lohff CJ, Martin B, Pandiani JA, Blevins LZ. Use of insurance claims data to determine prevalence and confirm a cluster of sarcoidosis cases in Vermont. Public Health Rep. 2009;124(3):442–6.
Deubelbeiss U, Gemperli A, Schindler C, Baty F, Brutsche MH. Prevalence of sarcoidosis in Switzerland is associated with environmental factors. Eur Respir J. 2010;35(5):1088–97.
Duchemann B, Annesi-Maesano I, de Naurois JC, et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J. 2017;50(2):1602419.
Erdal BS, Clymer BD, Yildiz VO, Julian MW, Crouser ED. Unexpectedly high prevalence of sarcoidosis in a representative U.S. metropolitan population. Respir Med. 2012;106(6):893–9.
Karakatsani A, Papakosta D, Rapti A, et al. Epidemiology of interstitial lung diseases in Greece. Respir Med. 2009;103(8):1122–9.
Schweisfurth H. Report by the Scientific Working Group for Therapy of Lung Diseases: German Fibrosis Register with initial results. Pneumologie. 1996;50(12):899–901.
Tinelli C, De Silvestri A, Richeldi L, Oggionni T. The Italian register for diffuse infiltrative lung disorders (RIPID): a four-year report. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22:S4–8.
Schweisfurth H, Kieslich C, Satake N, et al. How are interstitial lung diseases diagnosed in Germany? Results of the scientific registry for the exploration of interstitial lung diseases (“Fibrosis registry”) of the WATL. Pneumologie. 2003;57(7):373–82.
Thomeer M, Demedts M, Vandeurzen K, VRGT Working group on Interstitial Lung Diseases. Registration of interstitial lung diseases by 20 centres of respiratory medicine in Flanders. Acta Clin Belg. 2001;56(3):163–72.
Zamora-Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Progressive decline of lung function in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. 2017;69(3):542–9.
Hoffmann-Vold A-M, Aaløkken TM, Lund MB, et al. Predictive value of serial high-resolution computed tomography analyses and concurrent lung function tests in systemic sclerosis. Arthritis Rheumatol. 2015;67(8):2205–12.
Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard J-F. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis. A series of 107 patients. Arthritis Rheumatol. 2011;63(11):3439–47.
Baughman RP, Lower EE. Frequency of acute worsening events in fibrotic pulmonary sarcoidosis patients. Respir Med. 2013;107(12):2009–13.
Wijsenbeek M, Kreuter M, Fischer A, et al. Non-IPF progressive fibrosing interstitial lung disease (PF-ILD): the patient journey. Am J Respir Crit Care Med. 2018;197:A1678.
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431–40.
Spagnolo P, du Bois RM, Cottin V. Rare lung disease and orphan drug development. Lancet Respir Med. 2013;1(6):479–87.
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304.
Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–87.
Hoffmann-Vold A-M, Aalokken TM, Lund MB, et al. Predictive value of serial high-resolution computed tomography analyses and concurrent lung function tests in systemic sclerosis. Arthritis Rheumatol. 2015;67(8):2205–12.
Acknowledgements
Funding
The analysis reported in this manuscript was funded by Boehringer Ingelheim International GmbH, Germany, which was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. The journal’s Rapid Service and Open Access Fees were also funded by Boehringer Ingelheim International GmbH.
Medical Writing, Editorial, and Other Assistance
Medical writing assistance, in the form of the preparation and revision of the manuscript, was supported financially by Boehringer Ingelheim International GmbH and provided by Lianne Young, BSc (Hons), ISMPP CMPP™, of Complete HealthVizion, Ben Daniels, BSc (Hons), BA, of AMICULUM Limited, and John Carron, PhD, of Nucleus Global, under the authors’ conceptual direction and based on feedback from the authors. Zhou Lin aided with the conduct of the systematic literature review.
Authorship
The authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors. The authors take full responsibility for the scope, direction, content of, and editorial decisions relating to the manuscript, were involved at all stages of development and have approved the submitted manuscript. All authors contributed to the development of this manuscript in terms of critical review and amendment of initial drafted content, and approval of the final, submitted version.
Disclosures
Amy Olson has nothing to disclose. Mouhamad Nasser reports other fees from Boehringer Ingelheim, Roche and Actelion, outside the submitted work. Luca Richeldi reports grants and personal fees from InterMune; and personal fees from Sanofi-Aventis, Roche, ImmuneWorks, Shionogi, Boehringer Ingelheim, Celgene, Nitto, Fibrogen, Promedior, Bristol Myers Squibb, and DynaMed, outside the submitted work. Anna-Maria Hoffmann-Vold reports grants, non-financial support and other from Boehringer Ingelheim, non-financial support and other from Actelion, and other from Roche, outside the submitted work. Vincent Cottin reports personal fees and non-financial support from Actelion, grants, personal fees and non-financial support from Boehringer Ingelheim and Roche/Promedior, and personal fees from Bayer/MSD, Novartis, Sanofi, Celgene, Galapagos, Galecto Shionogi and AstraZeneca, outside the submitted work. Nadine Hartmann, Padmaja Patnaik, Laura Wallace and Rozsa Schlenker-Herceg are employees of Boehringer Ingelheim. The authors received no compensation related to the development of the manuscript.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Olson, A., Hartmann, N., Patnaik, P. et al. Estimation of the Prevalence of Progressive Fibrosing Interstitial Lung Diseases: Systematic Literature Review and Data from a Physician Survey. Adv Ther 38, 854–867 (2021). https://doi.org/10.1007/s12325-020-01578-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01578-6