
Abstract
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and irreversible lung disease. Licensed treatment options for IPF are pirfenidone and nintedanib. The aim of this study was to assess the impact of antifibrotic therapy in patients with IPF with preserved lung function based upon a forced vital capacity (FVC) above 80%.
Method
This is a retrospective single-centre cohort study, performed as part of a service evaluation, between January 2007 and September 2018. Patient demographic, treatment and lung function profiles were collected using electronic patient records. A linear mixed model and Kaplan–Meier estimator were utilised to assess changes in FVC and survival over 36 months.
Results
A total of 161 patients were included in this study. Mean age was 72 ± 4. Twenty-four (14.9%) received pirfenidone, 86 (53.4%) received nintedanib and 18 (11.2%) received both antifibrotics provided by a compassionate use program (CUP), as the National Institute of Heath and Clinical excellence (NICE) criteria for antifibrotics in the UK is restricted to an FVC 50–80%. Thirty-three (20.5%) patients did not receive treatment. Patients without antifibrotic therapy had a statistically higher baseline FVC compared to other groups: 3.55 l (100%) vs 2.85 l (89.7%) pirfenidone (p = 0.012), vs 2.99 l (93.5%) nintedanib (p = 0.04) and 3.10 l (92.7%) (p = 0.07) for both antifibrotics. FVC decline over 1 year was similar in groups receiving pirfenidone, nintedanib or no treatment [3.72% (158.1 ml) untreated vs 2.77% (139 ml) pirfenidone vs 2.96% (131 ml) nintedanib]; however, it was significantly greater in patients who received both antifibrotics [6.36% (233 ml), p = 0.01]. Use of antifibrotics was associated with a higher median survival post diagnosis; 3.5, 3 and 3.75 years respectively in pirfenidone, nintedanib and both antifibrotic cohorts, compared to the untreated cohort (2.5 years).
Conclusion
One in five untreated patients with an average FVC of 100% die within a median of 2.5 years. Antifibrotic therapy was associated with a higher median survival of 3–3.75 years despite treatment groups having lower baseline lung function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Presently, there is limited unanimity on when antifibrotic treatment (pirfenidone and nintedanib) in patients with idiopathic pulmonary fibrosis (IPF) should be started. |
The UK restricts antifibrotic treatment to patients with a forced vital capacity (FVC) between 50% and 80%, and major clinical trials testing the efficacy of these drugs have used patients with moderate baseline lung function in their studies. |
The hypothesis of this study is that antifibrotic treatment (pirfenidone and nintedanib) in patients with IPF with preserved lung function (FVC above 80%) is associated with favourable outcomes with regards to disease progression and survival. |
What was learned from the study? |
Whilst FVC decline over 36 months was similar in pirfenidone, nintedanib and untreated groups; it was significantly greater in patients receiving both antifibrotics. Patients receiving antifibrotic treatment had favourable outcomes with regards to survival (3.5 years with pirfenidone, 3 years with nintedanib, 3.75 years with both antifibrotics compared to 2.5 years with no treatment), despite the treatment groups having a lower baseline FVC. |
This study found that patients with higher baseline FVC do deteriorate over time, and one in five patients in our no treatment group died, showing that this is not a ‘milder phenotype’ cohort. |
In our study of patients with preserved baseline FVC, antifibrotic treatment is associated with preferential outcomes with regards to survival, therefore highlighting the favourable effects that pirfenidone and nintedanib can have in this cohort. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13034141.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most prevalent of the interstitial lung diseases (ILD) and is associated with the worst mortality within this family of respiratory disorders [1]. It typically affects adults above the age of 70 and is a chronic, diffuse and progressive disease state, causing irreversible fibrosis of the lung parenchyma and respiratory failure [1]. Men are affected at a significantly greater proportion than women, and a smoking history of greater than 20 pack years constitutes an increased likelihood of acquiring IPF [2]. As a result of the decline in pulmonary function over time, it is an extremely debilitating condition and is associated with a poor prognosis; the median survival length after diagnosis is 3 years [3].
The UK has the highest incidence rate of IPF in Europe [4]. Currently, there are two licensed medications for IPF which are effective in reducing the progression of lung fibrosis over time. These are pirfenidone and nintedanib; both of these drugs decelerate lung fibrosis in patients with mild to moderate disease [5]. Whilst existing landmark clinical trials have demonstrated the efficacy of these two agents in cohorts with moderate forced vital capacity (FVC) impairment [6,7,8], post hoc studies have in fact demonstrated favourable outcomes of the drugs even in groups with a higher baseline lung function (i.e. FVC above 80%) [9, 10]. Pirfenidone has been shown to decelerate lung function decline in patients with IPF in a number of clinical trials [7, 8]. In a phase III study, the ASCEND trial, the cohort receiving pirfenidone showed a linear decline in FVC of 164 ml compared to 280 ml in the placebo group (p < 0.01, mean difference 42%) [8]. Nintedanib also effectively preserves lung function in patients with IPF [11], which is shown by two concurrent large phase III trials, INPULSIS-1 and INPULSIS-2 [6]. In INPULSIS-1, there was a rate of decline in FVC in the control group of − 114.7 ml and − 239.9 ml in the placebo group (p < 0.01). In INPULSIS-2, the control group saw a yearly decline of − 113.6 ml compared to − 207.3 ml in the placebo group (p < 0.01) [6].
There is currently limited unanimity on when treatment with antifibrotics should be started in patients with IPF. For instance, in the UK, both pirfenidone and nintedanib are restricted to patients with an FVC between 50% and 80%, based upon a health economic rationale [12,13,14]. However post hoc studies have demonstrated favourable effects of antifibrotic irrespective of baseline lung function. Albera et al. showed that pirfenidone had a similarly favourable effect in a group with moderate baseline lung function (FVC below 80%) versus a group with preserved FVC (at least 80%) over 12 months [9]. Moreover, Kolb et al. also showed comparable results with respect to nintedanib, as patients with preserved lung function (FVC above 90%) benefited similarly from nintedanib compared to patients with a lower FVC [10]. Nintedanib was in fact better tolerated in patients with a pre-treatment FVC greater than 80% compared to patients who had an FVC between 50% and 80% in another study [15]. Consequently, in our real-world study we aim to assess a cohort of patients with preserved lung function and assess their response to antifibrotic treatment with respect to lung function and survival.
Methods
This is a retrospective single-centre observational cohort study of patients with a multidisciplinary diagnosis of IPF with an FVC greater than 80% between January 2007 and September 2018 (n = 161). In the UK, these patients were ineligible to receive antifibrotic therapy as the National Institute of Health and Care Excellence (NICE) criteria for prescribing antifibrotics in the UK is restricted to patients with an FVC 50–80%. A cohort received antifibrotics via a compassionate use program (CUP) provided by the drug manufacturers. Lung function data was analysed across a 36-month period.
The patients were split into four groups: those who were receiving pirfenidone, nintedanib and both antifibrotics, and patients not receiving any antifibrotic treatment. A retrospective review of electronic patient records was conducted using results from pulmonary function tests (PFTs), outpatient clinical letters and radiological imaging reports. Various parameters were collected including patient demographic data, treatment records, adverse effects and lung function data.
Results were analysed using IBM SPSS statistics (version 25). Continuous data was represented as mean ± standard deviation (SD), whilst nominal variables were represented as frequencies (percentage). The normality of the distribution was tested using the Shapiro–Wilk test, whilst an independent-samples t test was used to compare the mean demographics in each treatment group. A p value less than 0.05 denoted statistical significance. A linear mixed model was performed to assess the changes in lung function over time. Moreover, a Kaplan–Meier analysis was used to assess survival across the three treatment groups and the untreated patients over 36 months.
We consulted the NHS Health Research Authority decision aid to ascertain whether ethical approval was required. It was deemed that approval was not required, as this work represents a service evaluation/surveillance, which utilises anonymised data collected as part of the routine delivery of a clinical service. In addition, we confirmed approval from our Caldicott Guardian.
Results
Demographical data, duration of treatment, adverse effects and reasons for discontinuation of medication are summarised in Table 1. A total of 161 patients with a baseline FVC above 80% were included in this study; 74.5% (n = 120) were male. The mean age was 72 ± 4 years. A total of 128 patients received treatment with antifibrotic medication through the CUP: 24 (14.9%) patients received pirfenidone and 86 (53.4%) received nintedanib whilst 18 (11.2%) patients received both pirfenidone and nintedanib. Thirty-three (20.5%) patients had no treatment. There was no significant difference between the three groups with respect to comorbidities and smoking history. The most common comorbidities were hypertension (14.7%), ischaemic heart disease (13.7%) and gastro-oesophageal reflux disease (11.3%). Among the patients, 70.8% were former smokers, 5.6% were current smokers and 23.6% had never smoked. Patients receiving pirfenidone were on treatment for a mean 21.8 ± 15.9 months whilst patients who received nintedanib received treatment for a mean of 24.3 ± 16.1 (p = 0.420). Appetite loss (19.6% vs 9%; p < 0.01), constipation (0% vs 3%; p = 0.03), fatigue (17.9% vs 9.8%; p = 0.02) and skin rash (8.9% vs 0%; p < 0.01) were statistically higher in those treated with pirfenidone compared to nintedanib therapy, whereas diarrhoea (33.8% vs 5.4%; p < 0.01) was most frequent in patients treated with nintedanib compared to pirfenidone. The discontinuation rate was 21% in both pirfenidone and nintedanib groups; the main reason for this was intolerable adverse effects.
Baseline lung function parameters were statistically higher in the untreated cohort compared to those treated with antifibrotics. The baseline FVC was 3.55 (100.5%) in the untreated cohort compared to 2.85 (90%) and 2.99 (93%) in those treated with pirfenidone and nintedanib (p = 0.012 and p = 0.04 respectively). Patients who had received both treatments also had a lower baseline FVC of 3.10 l (92.7%), compared to the no treatment group (p = 0.07). Similarly baseline diffusing capacity of the lungs for carbon monoxide (DLCO) was statistically higher in the untreated cohort (4.13; 54.3%) compared to those treated with pirfenidone (3.14; 39.7%), p < 0.001 or nintedanib (3.54; 46.6%), p = 0.02. Over a period of 12 months the FVC declined by − 3.72% (158.1 ml) in the untreated cohort, − 2.77% (139 ml) in the cohort receiving pirfenidone, − 2.96% (131 ml) in the nintedanib-treated group and a significant decline of − 6.36 (232 ml) in the cohort who had received both antifibrotics (p = 0.01) (Table 1).
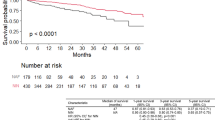
A Kaplan–Meier analysis of survival showed no major difference in survival despite the no treatment group having statistically greater lung function (p = 0.33) (Fig. 2). The median survival post diagnosis was 2.5 years in the untreated cohort compared to 3.5 years in those treated with pirfenidone, 3 years in the nintedanib-treated group and 3.75 years in patients who had received both antifibrotics.
Discussion
In the UK, NICE has restricted access of antifibrotics to patients with IPF who have an FVC between 50% and 80% [16]. This decision is based on a complex health economic rationale with the assumption that patients with an FVC above 80% are considered to have a “milder” phenotype of IPF [17]. The aim of this real-world study was to assess baseline demographics and natural progression with respect to lung function and survival in patients with IPF who have an FVC value above 80%. A total of 128 (79.5%) patients in this study were able to access pirfenidone or nintedanib through a CUP. The remaining patients (33, 20.5%) were untreated either because the CUP treatment window had closed or patient choice. The important findings of this study highlight that patients with untreated IPF characterised as “mild” in phenotype with a baseline FVC of 100% and DLCO 54% have a significantly reduced median survival post diagnosis of 2.5 years. This is compared to a survival of 3.5, 3 and 3.75 years in a cohort of patients with IPF treated with pirfenidone, nintedanib and both treatments respectively. This reduced survival in the untreated cohort is despite the fact that the antifibrotic treated cohort of patients with IPF have lower baseline lung function compared to the untreated group.
In this study, there were no significant imbalances observed between the baseline demographics of the untreated and antifibrotic-treated cohorts. The majority of patients were male with an average age of 73 years. This was similar to the INPULSIS trials [6]; however, the average age in the CAPACITY studies [7] was lower at 68 and 66.8 years (study arm 004 and 006 respectively). Additionally, the patients were mostly former smokers with comorbidities such as hypertension, ischaemic heart disease and gastro-oesophageal reflux disease.
We reported similar profiles with respect to adverse effects and discontinuation rates compared to existing clinical trials. The most common adverse effect experienced by the pirfenidone cohort was loss of appetite (19.6%), fatigue (17.9%), indigestion (17.9%) and nausea/vomiting (12.5%). These are of lower frequency compared to the adverse effects noted in the CAPACITY trials: nausea (36%) and skin rash (32%) [8]. Similarly the most common adverse event experienced by the nintedanib cohort in our study was diarrhoea (33.8%); this again was less than those reported in the INPULSIS trials (61.5% and 63.2% in INPULSIS-1 and INPULSIS-2 respectively) [6]. Our adverse events frequencies were lower than those of clinical trials potentially because of reporter bias as clinical assessment and documentation perhaps would not be as robust as clinical trials. Among our patients, 21% permanently discontinued pirfenidone treatment; this rate was greater than that in the CAPACITY trial 006 (19.9%) [7]. Moreover, the discontinuation rate was greater in the patients who received both antifibrotics, as compared to patients receiving pirfenidone and nintedanib (27.8%, 21% and 21% respectively). The most common cause of termination in the case of both drugs was intolerable adverse effects. An average of about two separate adverse effects were reported per patient in the pirfenidone and nintedanib group, whilst about five adverse events were reported on average per patient in the category of patients who received both treatments.
The average baseline FVC in our untreated group was significantly higher than in both pirfenidone and nintedanib groups: 3.55 l (100%) vs 2.85 l (89.7%) pirfenidone (p = 0.012) vs 2.99 l (93.5%) nintedanib (p = 0.04) vs 3.10 (92.7%) both treatments (p = 0.07). One reasonable explanation for this could be the lack of randomisation regarding treatment options. Patients with higher FVC values may have had milder symptoms, and therefore may have declined treatment for this reason. The FVC decline was similar in the untreated group and in the groups receiving pirfenidone and nintedanib (Fig. 1); however, it is difficult to directly compare the groups as the untreated group in our study had a statistically higher baseline FVC, when compared to the treated groups. Patients treated with both antifibrotics had a statistically greater decline compared to those treated with single antifibrotic alone. Again this is because this group has a selection bias as the clinical decision to switch from one antifibrotic to another is based on the presence of an FVC decline of greater than 10%. This cohort of patients treated with both antifibrotics is therefore deemed to be more progressive in nature.

Decline in forced vital capacity, FVC (%), in untreated patients (blue line) compared to those treated with pirfenidone (red line), nintedanib (green line) or both antifibrotics (orange line), over a period of 36 months
When comparing to existing trials, the 004 arm of the CAPACITY trial showed an 8% FVC decline in the pirfenidone group and a 12.4% decline in the untreated placebo group over 72 weeks [7]. In our study both our pirfenidone and untreated cohorts declined at a much slower rate, by 4.16% and 5.1% in 72 weeks respectively (2.77% and 3.72% per year). The baseline FVC and DLCO in the pirfenidone and placebo arms of the CAPACITY 004 trial were an FVC of 73% and 73.6% respectively and DLCO of 45.4% and 43.7% respectively [7]. This was in comparison to an FVC of 89.7% and DLCO 39.7% for the pirfenidone-treated group and FVC of 100.5% and DLCO 54.3% in the untreated group in our study. The higher baseline FVC in our cohort may explain the slower observed rate of decline in FVC compared to published clinical trials. When comparing to the INPULSIS-1 trial [6] our study had a similar rate of decline in the nintedanib-treated group (− 130.9 ml vs − 104.3 ml) but a slower decline in the untreated group (− 158.1 ml vs 239.9 ml). Again, this is likely to be because of higher baseline lung function in our cohort compared to those in the clinical trials.
A Kaplan–Meier analysis of survival showed similarities between all groups (p = 0.33), despite the treatment groups having a significantly lower baseline lung function (Fig. 2). We found that patients in the treatment groups had a longer median survival length post diagnosis when compared to our untreated group; (3.5 years pirfenidone, 3 years nintedanib, 3.75 years both treatment and 2.5 years untreated). This was despite all of the treatment groups having lower overall baseline FVC and DLCO values. Furthermore, in the untreated cohort, FVC continued to decline at a similar rate to treatment arms in clinical trials and one in five patients died within the time period, despite having a higher FVC and DLCO compared to treated groups in this study or patients with IPF in clinical trials. This leads to the important finding that a higher FVC at diagnosis does not correlate with a milder disease phenotype and that these patients would benefit from treatment. Furthermore, our study showed that receiving antifibrotic treatment was associated with an increased median survival post diagnosis regardless of having a lower FVC, thereby showing the beneficial effects these drugs have with respect to survival in patients with an FVC above 80%. Comparably, in an analysis from the Australian IPF registry, Jo et al. showed that whilst patients with preserved baseline lung function (FVC at least 80%) have a greater survival rate, their disease progression over time is similar to groups with lower FVC values [18]. This shows that the natural history of the disease is analogous, irrespective of baseline FVC, and these groups would benefit considerably from antifibrotic therapy.

Kaplan–Meier survival analysis of untreated patients (blue line), those treated with pirfenidone (red line), nintedanib (green line) or both antifibrotics (orange line). (p = 0.33)
Our study has a number of limitations. Firstly, this study is a single-centre retrospective non-randomised real-world study. As a result of its retrospective nature, patients did not have standardised records in their consultations regarding occurrence of adverse events. Furthermore, in the real-world setting patients have the autonomy to decline treatment, which would introduce bias in this study. There was no randomisation with regards to treatment choice. Lastly, we would endeavour for a larger sample size in the future which would increase the reliability of our results.
Conclusion
This is a real-world assessment of patients with an FVC above 80% who either received compassionate use antifibrotic therapy or no treatment. Whilst the rates of decline in FVC were similar between groups, our study showed that antifibrotic use was associated with an increased median survival length after diagnosis in patients with a statistically lower baseline FVC compared to the untreated group. The important findings of this study highlight that patients with untreated IPF characterised as “mild” in phenotype have a significantly reduced median survival post diagnosis of 2.5 years. We would advocate that patients with IPF with an FVC above 80% would benefit from treatment with antifibrotic therapy.
References
Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155(1):242–8.
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46(3):795–806.
Ipatova AY, Koerner PH, Miller RT, Staskon F, Radi M. Retrospective analysis of medication utilization and clinical outcomes in patients with idiopathic pulmonary fibrosis treated with nintedanib or pirfenidone. Clin Med Insights Circ Respir Pulm Med. 2019;13:1179548419834922.
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Albera C, Costabel U, Fagan EA, et al. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur Respir J. 2016;48(3):843.
Kolb M, Richeldi L, Behr J, et al. nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72(4):340.
McCormack PL. Nintedanib: first global approval. Drugs. 2015;75(1):129–39.
NICE. How NICE measures value for money in relation to public health interventions United Kingdom. National Institute of Health and Care Excellence. 2013. https://www.nice.org.uk/Media/Default/guidance/LGB10-Briefing-20150126.pdf. Accessed July 2020.
NICE. Nintedanib for treating idiopathic pulmonary fibrosis United Kingdom. National Institute of Health and Care Excellence. 2016. https://www.nice.org.uk/guidance/TA379/chapter/1-Recommendations. Accessed July 2020.
NICE. Pirfenidone for treating idiopathic pulmonary fibrosis United Kingdom. National Institute for Health and Care Excellence. 2018. https://www.nice.org.uk/guidance/ta504. Accessed July 2020.
Fletcher SV, Jones MG, Renzoni EA, et al. Safety and tolerability of nintedanib for the treatment of idiopathic pulmonary fibrosis in routine UK clinical practice. ERJ Open Res. 2018;4(4):00049–2018.
NICE. Idiopathic pulmonary fibrosis in adults: diagnosis and management United Kingdom. National Institute of Health and Care Excellence. 2017. https://www.nice.org.uk/guidance/cg163/chapter/1-Recommendations.
NICE. NICE technology appraisal guidance United Kingdom. National Institute for Health and Care Excellence. 2020. https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-technology-appraisal-guidance.
Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med. 2018;18(1):19.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Saba Noor and Saira Nawaz declare that they have no conflict of interest. Nazia Chaudhuri is a member of the journal’s Editorial Board.
Compliance with Ethics Guidelines
Ethics committee approval was not required. We consulted the NHS Health Research Authority decision aid to ascertain whether ethical approval was required. It was deemed that approval was not required, as this work represents a service evaluation/surveillance, which utilises anonymised data collected as part of the routine delivery of a clinical service. In addition, we confirmed approval from our Caldicott Guardian.
Data Availability
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Noor, S., Nawaz, S. & Chaudhuri, N. Real-World Study Analysing Progression and Survival of Patients with Idiopathic Pulmonary Fibrosis with Preserved Lung Function on Antifibrotic Treatment. Adv Ther 38, 268–277 (2021). https://doi.org/10.1007/s12325-020-01523-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01523-7