
Abstract
Introduction
Pegfilgrastim-cbqv was developed as a biosimilar of pegfilgrastim, a pegylated form of recombinant human granulocyte colony-stimulating factor approved for decreasing febrile neutropenia-associated infection in patients receiving myelosuppressive drugs. This multicenter, randomized, single-blind, partial-reference-replicated, three-sequence crossover study assessed pharmacokinetic and pharmacodynamic bioequivalence of pegfilgrastim-cbqv and pegfilgrastim in healthy subjects.
Methods
One hundred twenty-two subjects were randomized to one of three treatment sequences; each included one dose of pegfilgrastim-cbqv and two doses of pegfilgrastim separated by ≥ 28 days. The primary pharmacokinetic end points were area under the curve (AUC) from 0 to infinity (AUC0−∞) and maximum concentration (Cmax). The primary pharmacodynamic end points were maximum absolute neutrophil count (ANCmax) and ANC AUC from time 0 to the last measurable observation (ANC AUC0−last). Pharmacokinetic and pharmacodynamic bioequivalences were demonstrated if the 90% CI for the geometric mean ratio (GMR) of pegfilgrastim-cbqv to pegfilgrastim was within 80–125% for the primary end points.
Results
Pharmacokinetic bioequivalence criteria were met for Cmax (GMR 105.0; 90% CI 95.5–115.4) and AUC0−∞ (GMR 97.5; 90% CI 88.6–107.2). Pharmacodynamic bioequivalence criteria were met for ANCmax (GMR 99.6; 90% CI 96.2–103.2) and ANC AUC0−last (GMR 96.7; 90% CI 92.2–101.4). Adverse events occurred in 76.0%, 76.6%, and 73.1% of subjects for pegfilgrastim-cbqv, first pegfilgrastim, and second pegfilgrastim dosing periods across treatment sequences, respectively. Investigators found no drug-related serious adverse events.
Conclusion
This study established pharmacokinetic and pharmacodynamic bioequivalence of pegfilgrastim-cbqv to pegfilgrastim. The treatments displayed similar safety profiles, including immunogenicity, with no unexpected safety findings.
Clinical Trials Registration
ClinicalTrials.gov, NCT02650973, February 2016.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
In patients undergoing certain types of chemotherapy, prophylactic administration of human granulocyte colony-stimulating factor (e.g., filgrastim [Neupogen®], pegfilgrastim [Neulasta®]) is the standard of care to prevent febrile neutropenia, a serious decrease in immune cells that results in an increased risk of infection with potentially fatal outcomes. |
Biosimilars are being developed as lower cost, competitive alternatives to originator biologics, such as pegfilgrastim, and can help increase availability of life-saving therapies and potentially lower overall drug costs. |
This study evaluated whether the biosimilar pegfilgrastim-cbqv (Udenyca®) and the originator biologic pegfilgrastim (Neulasta®) were equivalent with respect to pharmacokinetics, pharmacodynamics, immunogenicity, and safety in healthy subjects. |
What was learned from the study? |
In this comparative clinical study, pegfilgrastim-cbqv (Udenyca®) and pegfilgrastim (Neulasta®) showed similar pharmacokinetics, pharmacodynamics, and safety profiles, including immunogenicity. |
These results demonstrate the bioequivalence of pegfilgrastim-cbqv (Udenyca®) and its reference product. |
Introduction
The availability of biosimilars has helped to increase access to important medications for patients, provide options for health care practitioners to more consistently meet patient needs, and decrease cost to payers [1]. Cost is a major factor that limits patient access to treatment. Biosimilars help to control drug prices by introducing competition into the market, offering a lower-cost alternative, and preventing uncontested price increases of the reference product. This allows for health care practitioners to ensure optimal patient treatment through adherence to consensus guidelines and increases the number of treatment options available to patients [1].
Biosimilars undergo an extensive and robust assessment process before approval to ensure their safety and efficacy. To be considered a biosimilar two criteria must be met. The biosimilar must be highly similar in structure and function to the existing reference product approved by the US Food and Drug Administration (FDA), “notwithstanding minor differences in clinically inactive components,” and it must have no clinically meaningfully differences regarding safety, potency, and purity [2, 3]. The Biologics Price Competition and Innovation Act of 2009 created an alternative approval pathway in the US for biosimilar biologics that are shown to be highly similar to the reference biologic. A biosimilar is distinct from a generic drug in that it is produced in living systems with inherent variability. Thus, a biosimilar will not be identical to the reference biologic, unlike a small-molecule generic drug, which is generally produced by chemical synthesis with highly defined structures and is identical to the brand-name drug. There are no clinically meaningful differences between an approved biosimilar and the reference product in terms of safety, purity, and potency. Approval of a biosimilar requires a stepwise approach in which the totality of evidence from physicochemical, functional, preclinical, and clinical studies is reviewed by regulatory agencies before approval of the biosimilar [2, 3]. This approach ensures first that the proposed biosimilar product and the reference product are highly similar through rigorous analytical and nonclinical (i.e., functional) comparison. Consistent with the biosimilar pathway, the nonclinical pathway is abbreviated as appropriate for the proposed biosimilar product [2, 3]. The nonclinical program consists of in vitro functional assays to assess any potential difference in biologic activity [2, 3]. If deemed necessary to obtain additional information to support biosimilarity or assess toxicity, animal studies can be conducted, but in general the in vivo studies are abbreviated [2, 3]. Subsequently clinical assessment of pharmacokinetic (PK) bioequivalence and efficacy or pharmacodynamic (PD) bioequivalence (if a biomarker is available) is performed to compare the proposed biosimilar and reference products [2, 3]. A crossover study design is preferred, when feasible, for products with a short half-life [2]. Bioequivalence criteria are established by the FDA to be when the calculated 90% confidence interval is within 80–125% for the geometric mean ratio (GMR) of the test and reference product for predefined PK and/or PD end points [2]. Finally, clinical efficacy and safety (including immunogenicity) in an appropriately sensitive population are required, depending on the level of uncertainty about the similarity of the biosimilar product to the reference biologic [2]. A clinical study in a patient population is not always necessary if the mechanism of action of the proposed biosimilar and reference products is the same in healthy volunteers and the patient population, as is the case for pegfilgrastim [2]. The FDA has also recognized that healthy subjects may be the most sensitive study population in which to evaluate biosimilarity, if the biologic can be safely administered to healthy subjects, because they may have fewer confounding factors (e.g., comorbidities, concomitant medications) and be more sensitive to detect potential differences in PK, PD, and immunogenicity.
Filgrastim (Neupogen®, 1991) was the first myeloid growth factor approved by the FDA and the European Medicines Agency (EMA) for the prevention of infection as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive chemotherapy [4]. Pegfilgrastim (Neulasta®, 2002), a long-acting pegylated form of filgrastim that requires only a once-per-cycle administration, was first introduced by Amgen, Inc. (Thousand Oaks, CA) [5]. Pegylation of filgrastim prevents glomerular filtration in the kidney; thus, pegfilgrastim is self-regulated through elimination by binding to the granulocyte colony-stimulating factor (G-CSF) receptor and being internalized by neutrophils and neutrophil precursor cells [6, 7], resulting in maturation and release of neutrophils.
The biosimilar pegfilgrastim-cbqv (Udenyca®, Coherus BioSciences, Redwood City, CA) was approved by the EMA and FDA in September 2018 and November 2018, respectively, for the same indication as the reference product [8]. In compliance with EMA and FDA guidance [2, 3], a stepwise approach was used to demonstrate that pegfilgrastim-cbqv is highly similar to pegfilgrastim. Structural and functional similarity was established (data on file), and a targeted clinical development program was executed to confirm bioequivalence with respect to PK, PD (as measured by absolute neutrophil count [ANC], an established surrogate PD biomarker of clinical efficacy), immunogenicity, and safety. Herein, we present the evidence for the established PK and PD bioequivalence between pegfilgrastim-cbqv and pegfilgrastim in healthy subjects.
Methods
Study Design
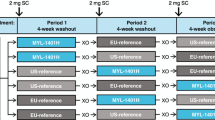
This was a randomized, single-blind, partial-reference-replicated, three-sequence, three-period crossover study in healthy subjects (Fig. 1) confined to a clinical pharmacology unit for dosing and immediate follow-up to assess PK, PD, and safety, including the immunogenicity of pegfilgrastim-cbqv and pegfilgrastim (ClinicalTrials.gov, NCT02650973). The study took place at four clinical sites across the US (Cincinnati, OH; Cypress, CA; West Bend, WI; San Antonio, TX). Each subject was randomly assigned in a 1:1:1 ratio (stratified by site and sex) to a treatment sequence group (A, B, or C) that included one dose of pegfilgrastim-cbqv and two doses of pegfilgrastim, each administered in a separate period with at least 28 days between doses of study drug (Fig. 1).

Study design. Subjects were randomly assigned to treatment sequence groups a, b, or c. Each sequence group included 1 dose of pegfilgrastim-cbqv and 2 doses of pegfilgrastim, each administered in a separate period with at least 28 days between doses of study drug. SC subcutaneous.
Compliance with Ethics Guidelines
The protocol and informed consent form were submitted to and approved by the sites’ institutional review boards before initiation of the study. IRB approval was obtained from Schulman Institutional Review Board (Cincinnati, OH), Alpha Independent Review Board (San Clemente, CA), Chesapeake Institutional Review Board (Columbia, MD), and IntegReview Independent Review Board (Austin, TX) in the US. This study was conducted in accordance with all applicable laws and regulations, and it complied with the International Conference for Harmonisation E6 Guideline on Good Clinical Practice and the Declaration of Helsinki. The rationale of the study, procedural details, and investigational goals were explained to each subject, along with potential risks and benefits. Each subject was assured of his/her right to withdraw from the study at any time. Before the initiation of any study procedures, each subject signed and dated an approved informed consent form.
Treatment
A single dose of pegfilgrastim-cbqv (6 mg/0.6 ml sterile single-use prefilled syringe) or pegfilgrastim (6 mg/0.6 ml 6 ml sterile single-use prefilled syringe) was administered subcutaneously on day 1 of each period, according to the subject’s assigned treatment sequence. Treatment of male subjects was spaced by no fewer than 28 days, whereas female subjects were dosed only at 28-day intervals (± 2 days) to ensure that they were dosed at the same time in their menstrual cycles to decrease intrasubject variability. This study was single-blinded, as the pegfilgrastim-cbqv prefilled syringe is not identical to the pegfilgrastim prefilled syringe. Each syringe was labeled with a unique number, and an unblinded site pharmacist matched the appropriate unique syringe number to the appropriate subject. The subjects, investigational site staff, biostatisticians, and data managers remained blinded to the subject treatment for the duration of the study. The subjects and investigational site staff responsible for assessing adverse events (AEs) and injection site reactions (ISRs) were blinded to study treatments. To strengthen the blinding of the study, access to the randomization scheme was limited, unblinded plasma concentrations were not shared with the study site, and investigational site staff responsible for assessing safety were not the same as the staff performing the injections.
Study Population
Eligible subjects were healthy male and female participants aged 18–45 years (Table 1). Inclusion criteria were body weight 50–100 kg (110–220 lb), with a body mass index of 18–28 kg/m2. The subjects were required to be medically healthy, with normal hematologic, coagulation, hepatic, and renal function and a fasting lipid profile that did not require medications to control lipid levels. Women of childbearing potential who were not actively breastfeeding must have had a negative pregnancy test and agreed to use an approved method of birth control; alternatively, women could have been > 1 year postmenopausal, and men must have been willing to use barrier contraception and refrain from donating sperm.
Exclusion criteria included previous exposure to pegfilgrastim or filgrastim; presence of hematologic disorder, cancer, diabetes, or any clinically significant condition that might interfere with lymphatic flow or PK of the drug; history of chronic or acute respiratory illness within the past 3 months; use of drugs (prescription, nonprescription, or recreational) or alcohol at screening or during study period; participation in high-impact exercise or activities that could result in blunt trauma to the truncal area; consumption of > 400 mg of caffeine per day; participation in a clinical study within 30 days before screening; blood and/or plasma donation ≥ 500 ml in the previous month; HIV or hepatitis B or C infection; clinically significant food or drug allergies; any condition that would complicate or compromise the study or well-being of the subject. Subjects who did not meet the ANC criteria for dosing (1.7–7. 2 × 109/l) were precluded from subsequent dosing.
End Points
The primary PK end points for this study were the peak plasma concentration (Cmax) and the area under the concentration-time curve from time 0 to infinity (AUC0−∞). The primary PD end points were the maximum absolute neutrophil count (ANCmax) and the area under the curve of the ANC from time 0 to the last sample (ANC AUC0−last). Secondary end points included the characterization of the PK profile of pegfilgrastim-cbqv using standard parameters and the characterization of the safety profile (including immunogenicity) and tolerance of pegfilgrastim-cbqv, as assessed by AEs, AEs of special interest (AESIs), laboratory variables, vital signs, incidence of antidrug antibodies/neutralizing antibodies (ADAs/NAbs), and ISRs.
Assessments
For each period, subjects were admitted to a clinical pharmacology unit for the first 4 nights of the treatment period and discharged the morning of day 5. Subjects returned for daily blood samples on days 6–13 and day 21 of each period as well as day 28 of period 3. The on-treatment blood sampling schedule for the plasma drug levels and ANC took place before dose administration and at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 60, 72, 84, and 96 h after dose and on days 6, 7, 8, 9, 10, 11, and 13. On days 21 and 28 (period 3 only), ANC was assessed, including white blood cell and differential counts. Plasma samples were analyzed for pegfilgrastim concentrations using a validated enzyme-linked immunosorbent assay. For the immunogenicity assessment, plasma samples for ADAs and NAbs were collected before dose administration on days 1 and 11 in each period and on day 28 following the last dose of the study drug. The presence of ADAs was assessed using a validated electrochemiluminescent bridging assay. Confirmed ADA-positive samples were characterized for binding specificity to polyethylene glycol (PEG) or G-CSF and semiquantitation (titer) and tested in a validated cell-based assay to determine if they were neutralizing. NAb-positive samples were further tested to assess the neutralization of endogenous G-CSF.
Safety assessments took place at the intervals specified in the protocol and included serum chemistry measurements, hematologic measurements, urinalysis, vital signs, physical examination (including an abdominal examination), clinical AE reports, ISRs, immunogenicity assessment, electrocardiograph, and collection of concomitant medications. AESIs included those related to the spleen, acute respiratory distress syndrome, anaphylaxis, sickle cell crisis, leukocytosis, glomerulonephritis, potential for tumor growth stimulatory effects on malignant cells, and cytokine release/capillary leak syndromes.
Statistical Analysis
A sample size of 120 subjects was determined based on statistical methods provided by Golkowski et al. and through simulations to assess intrasubject variation and power [9]. This study was designed to demonstrate PK bioequivalence (PK-BE) with approximately 90% power with 78 evaluable subjects based on a two-sided 90% CI to evaluate the GMR of pegfilgrastim-cbqv/pegfilgrastim for Cmax and AUC0−∞. The planned sample size assumed an intrasubject coefficient of variation (CV) ≤ 44% for Cmax and AUC0−∞ and an expected true GMR of pegfilgrastim-cbqv/pegfilgrastim of 1.05 for Cmax and AUC0−∞. Additionally, 78 evaluable subjects would provide > 95% power to demonstrate PD bioequivalence based on a two-sided 90% CI to evaluate GMR of pegfilgrastim-cbqv/pegfilgrastim for ANCmax and ANC AUC0−last. This planned sample size assumed an intrasubject CV ≤ 25% for ANCmax and ANC AUC0−last and an expected true GMR of pegfilgrastim-cbqv/pegfilgrastim of 1.0 for ANCmax and ANC AUC0−last.
A planned interim analysis of intrasubject CV for pegfilgrastim 1/pegfilgrastim 2 for AUC0-∞ was conducted when the period 3 PK samples from the initial cohort were available to allow for readjustment of the sample size, if necessary. A predefined cutoff of ≤ 43% was prospectively determined to maintain adequate study power. When PK samples from period 3 were available, the intrasubject CV of AUC0−∞ for pegfilgrastim 1/pegfilgrastim 2 was calculated by an unblinded independent statistician in subjects who had received at least two doses of pegfilgrastim and had sufficient plasma concentration–time data to calculate at least one of the primary PK end points [10]. Because the intrasubject CV was ≤ 43%, it was determined that no additional subjects were required.
Pegfilgrastim plasma concentration data were summarized for each time point using descriptive statistics from subjects who received at least two doses of study drug—one of pegfilgrastim-cbqv and at least one of pegfilgrastim—and had sufficient plasma-concentration time data to calculate at least one of the primary PK end points (PK-BE evaluable population). Standard PK variables were calculated using standard noncompartmental methods using plasma concentration data from the collected blood samples and summarized by treatment group and study period using the PK bioequivalence evaluable population. For AUC and Cmax, the pegfilgrastim average was the geometric mean of the two reference pegfilgrastim doses of each subject if both were available; otherwise, the single available measurement was used. ANC data were summarized for each time point using descriptive statistics for subjects who received at least two doses of study drug (one of pegfilgrastim-cbqv and at least one of pegfilgrastim) and had sufficient data to calculate at least one of the key PD end points (PD evaluable population). The safety population consisted of all randomly assigned subjects who received ≥ 1 dose of either study drug. Descriptive analysis was conducted in PK, PD, or safety profile based on sex or ethnicity. Summary statistics of clinical laboratory data and vital signs were generated by treatment group and time point. The presence of ADAs and NAbs was listed by subject and summarized descriptively by treatment and time point. The ADA/NAb impacts on PK, PD, and safety were evaluated.
PK and PD bioequivalence analysis used a two one-sided tests procedure for an unscaled average bioequivalence approach for the partial reference-replicated three-treatment sequence, three-period design [10]. Bioequivalence was demonstrated if the 90% CIs for the GMR of pegfilgrastim-cbqv:pegfilgrastim for the primary PK and PD end points were entirely within the range of 80–125%.
Results
A total of 122 subjects were randomly assigned to the study (Fig. 1, Table 1). All subjects received their initial treatment, 94 subjects received their second treatment, and 69 subjects received their third treatment (Fig. 2). A total of 58 subjects discontinued the study early; the most common reason for early discontinuation across all three sequences was that subjects did not meet the ANC criteria for dosing (A: 8 subjects; B: 5 subjects; C: 4 subjects) for 5 and 12 subjects during the pegfilgrastim-cbqv or pegfilgrastim treatment, respectively (Table 2). The number of subjects who discontinued the study early was anticipated, and a post hoc analysis of the impact of dropouts on end points was conducted. The post hoc analysis consisted of a missing not at random tipping point analysis conducted to explore the robustness of the biosimilarity conclusions through an assessment of how much missing data could introduce bias on the PK data because of issues with sequential sampling and subject withdrawal. The tipping point analysis demonstrated that the subjects who discontinued the study early did not introduce bias. The mean age of subjects was 29.9 years, and approximately 71% were male. Average body weight was 72 kg (159 lb) and average body mass index was 24.03 kg/m2 (normal range 18.5–24.9 kg/m2). Patient demographics and baseline characteristics were similar between treatment sequences (Table 1).

CONSORT diagram for the study evaluating the pharmacokinetic and pharmacodynamic equivalence of pegfilgrastim-cbqv and pegfilgrastim. Sequence A: pegfilgrastim-cbqv/pegfilgrastim/pegfilgrastim. Sequence B: pegfilgrastim/pegfilgrastim-cbqv/pegfilgrastim. Sequence C: pegfilgrastim/pegfilgrastim/pegfilgrastim-cbqv
Pharmacokinetics
Plasma levels in the PK-BE evaluable population (n = 84) are shown in Fig. 3. In all treatment groups, mean pegfilgrastim blood levels peaked at hour 16 after dose administration and returned to near baseline levels by hour 96. Mean pegfilgrastim concentrations for pegfilgrastim-cbqv were similar to those of pegfilgrastim at and after the time to maximum plasma concentration (Tmax) in all three treatment sequences. In addition, there did not appear to be a treatment by period interaction (i.e., order of treatments administered). Therefore, it was concluded that the observed period effect did not bias the results. The summary statistics of PK parameters were also similar between treatment groups (Table 3).

Mean serum pegfilgrastim concentrations for pegfilgrastim-cbqv and pegfilgrastim in the PK-BE evaluable population. Concentrations for pegfilgrastim were the average of the two periods. N indicates the number of subjects in the PK-BE evaluable population. The 90% CIs for the GMR of pegfilgrastim-cbqv relative to pegfilgrastim in the PK-BE evaluable population AUC0−∞ and Cmax were calculated. AUC0−∞ area under the plasma concentration-time curve extrapolated from 0 to infinity; BE bioequivalence; Cmax maximum plasma concentration; CI confidence interval; GMR geometric mean ratio; PK pharmacokinetic; SD standard deviation
PK bioequivalence assessment between products was based on the GMR of AUC and Cmax for pegfilgrastim-cbqv relative to pegfilgrastim (Table 3). As the 90% CIs for the GMRs of pegfilgrastim-cbqv:pegfilgrastim were entirely within the range of 80–125% for AUC0−∞ and Cmax, bioequivalence was demonstrated between pegfilgrastim-cbqv and pegfilgrastim in terms of PK response. Furthermore, the GMRs were close to 1 for all AUCs and Cmax.
Pharmacodynamics
PD bioequivalence was assessed by ANC levels, given that the primary pharmacologic effect of pegfilgrastim is an increase in ANC. Blood levels of ANC by time point for the PD evaluable population (n = 85) are shown in Fig. 4. ANCmax occurred at hour 60 after dose administration and returned to baseline levels by hours 480–648 (days 21–28). The 90% CI of the GMRs for pegfilgrastim-cbqv:pegfilgrastim were entirely within the boundary of 80–125% for ANC AUC0–last and ANCmax, and PD bioequivalence was established between pegfilgrastim-cbqv and pegfilgrastim (Table 1).

Mean ANC for pegfilgrastim-cbqv and pegfilgrastim in the PD evaluable population. ANC for pegfilgrastim was the average of the two periods. The 90% CI for the GMR of pegfilgrastim-cbqv relative to pegfilgrastim in the PD evaluable population for ANC AUC0−last and ANCmax was calculated. ANC AUC0−last area under the absolute neutrophil count-time curve calculated from time 0 to the last measurable observation; ANCmax maximum observed neutrophil count; BE bioequivalence; CI confidence interval; GMR geometric mean ratio; PD pharmacodynamic
Safety
Overall, pegfilgrastim-cbqv demonstrated a comparable safety profile to pegfilgrastim (Table 4). The majority of AEs were mild or moderate in severity and were generally similar across treatment groups (Table 5). Severe AEs were reported in three subjects who received pegfilgrastim-cbqv and three subjects who received pegfilgrastim. Only one serious AE (stab wound) was observed across all treatment periods; this event was considered life-threatening and was not considered study drug related per investigator. Regarding AESIs (detailed in the Methods), three subjects experienced splenic pain or other symptoms suggestive of splenic involvement; one subject reported left shoulder and left upper quadrant abdominal pain, one subject reported left upper quadrant abdominal pain, and one subject had splenomegaly with splenic pain that was mild in severity. No other AESIs or clinically significant effects in vital signs, serum chemistry, or hematology were observed.
The immunogenicity assessment showed that similar percentages of subjects in each treatment group developed treatment-emergent ADA after the first dose (period 1): 28.6% of subjects treated with pegfilgrastim-cbqv (sequence A) and 33.3% of subjects treated with pegfilgrastim (sequences B or C). No new subject developed ADA upon treatment with pegfilgrastim-cbqv in periods 2 and 3, after switching from pegfilgrastim treatment. The ADA responses were transient and generally low titer and specific to the PEG portion of the molecule. Anti-PEG antibodies are commonly present in healthy subjects. No treatment-emergent NAbs were detected in any subject. The presence of ADAs did not impact the PK or PD response to pegfilgrastim or pegfilgrastim-cbqv and did not affect their safety profile.
Discussion
To demonstrate biosimilarity between two products, the FDA recommends a stepwise approach to evaluate any clinically meaningful differences between products by conducting structural analyses, functional assays, and animal testing; these should be followed by human PK and clinical efficacy or PD studies and clinical immunogenicity assessment [11]. In line with these biosimilar regulatory guidelines, this study was designed to demonstrate the bioequivalence of pegfilgrastim-cbqv to pegfilgrastim for PK and PD parameters, ensuring similar exposure of the reference product. Safety and immunogenicity were also assessed.
This study used a partial reference-replicate, three-sequence, three-period, crossover design in healthy subjects to allow for estimation of intrasubject CV for the two pegfilgrastim treatment groups and an adjustment of sample size at the interim analysis, if necessary, without compromising the primary end points. The design of this study was further informed by data from previous trials [10, 12] in which high intrasubject PK variability was observed (data on file). Intrasubject PK variability was reduced by modifying inclusion/exclusion criteria, standardization of drug administration procedures (e.g., a single-study drug administrator at each site, standardized injection sites), and addition of PK time points. The limitations of this study include nonidentical syringes of each treatment, which was overcome by additional measures to ensure subjects and investigational staff were blinded to the subject treatment. Also, data interpretation was complicated by subject withdrawal; however, post hoc tipping point analysis of the PK data demonstrated that the evidence of PK biosimilarity in healthy volunteers was sufficient.
The PK bioequivalence of pegfilgrastim-cbqv and pegfilgrastim was shown by similar pegfilgrastim plasma concentration profiles when comparing between treatment groups. In addition, the PD bioequivalence of pegfilgrastim-cbqv and pegfilgrastim was demonstrated using ANC as a surrogate end point of clinical efficacy [7]. ANC is an appropriate measure of PD because (1) ANC is a direct outcome and reflection of the mechanism of action of pegfilgrastim; (2) duration of severe neutropenia as measured by ANC is correlated with and hence relevant to the clinical outcome of febrile neutropenia; (3) ANC is measurable for a sufficient period of time after dosing with appropriate precision; (4) ANC has the sensitivity to detect clinically meaningful differences between the proposed product and the reference product. The ANC response observed was also consistent with that reported for pegfilgrastim [7, 12], further supporting the data establishing PD bioequivalence.
The identification of subtle but potentially impactful differences in PK, PD, and immunogenicity is challenging in the cancer patient population owing to potentially confounding factors, such as underlying and/or concomitant disease, immunocompetence, antidrug antibody response, and concomitant medications, which complicate assessment of treatment-related AEs [2]. Healthy, immune-competent subjects are highly sensitive to potential differences in PK, PD, and immunogenicity, making them an appropriate study population in which to evaluate similarity. Similar PK and PD profiles have been reported for healthy subjects and patients with cancer before receiving chemotherapy [12]. While there are differences in absorption and half-life (elimination) of pegylated G-CSF between patients receiving chemotherapy (or radiation therapy) and healthy subjects because of reduced ANC in the former population compared with the latter, the mechanism of action of pegfilgrastim is the same in patients and healthy subjects, as is the neutrophil-mediated mechanism of clearance. Because the mechanism of action of pegfilgrastim is the same in healthy subjects as in patients with cancer, the PK and PD responses observed in this study are directly applicable to clinical efficacy in patients with cancer receiving myelosuppressive chemotherapy. Thus, based on the established PK and PD bioequivalence, pegfilgrastim-cbqv is expected to have the same efficacy as the reference product and to decrease the incidence of infection in patients receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia [13].
Consistent with the PK and PD responses indicating biosimilarity, pegfilgrastim-cbqv and pegfilgrastim had overall comparable safety and immunogenicity profiles. No unexpected AEs associated with pegfilgrastim-cbqv treatment were observed. A characteristic side effect of myeloid growth factors, such as filgrastim and pegfilgrastim, is skeletal pain [14]. Common AEs reported in this study that are reflective of that effect included back pain, extremity pain, neck pain, and arthralgia, and the incidence of these AEs was generally comparable between treatment groups. In contrast to studies in cancer patients undergoing chemotherapy, the safety profile observed in healthy subjects is also more likely to be more specific to pegfilgrastim because healthy subjects do not have confounding factors, such as disease state and chemotherapy treatment.
The immunogenicity profile was similar after treatment with pegfilgrastim-cbqv or pegfilgrastim, and there was no correlation between ADA positivity and incidence of AEs or AESIs. The ADA incidence after treatment with pegfilgrastim was higher in this study than originally reported for pegfilgrastim [5], likely owing to assay sensitivity, but is in line with recent published studies [15]. Importantly, there was no evidence of sensitization after treatment switching, and no treatment-emergent NAbs were detected in any subject after dosing with either pegfilgrastim-cbqv or pegfilgrastim.
Although biosimilars present an opportunity to increase patient access to important medications, concerns remain regarding the safety of switching from treatment with pegfilgrastim to a biosimilar in clinical practice [13]. This study included a partial reference-replicate design where subjects were switched between treatment groups (Fig. 1). No increased immunogenicity or loss of efficacy (as assessed by ANC) was observed following switching to and/or from pegfilgrastim-cbqv (Fig. 1). In this study, pegfilgrastim-cbqv and pegfilgrastim were shown to be bioequivalent regardless of treatment period. A sensitivity analysis showed that the sequence of administration of pegfilgrastim-cbqv and pegfilgrastim did not significantly change the results, suggesting that health care professionals should not observe any clinically significant differences in therapeutic outcome when switching in clinical practice.
Conclusions
In summary, this study demonstrated PK and PD bioequivalence between pegfilgrastim-cbqv and pegfilgrastim, no new safety signals, and similar immunogenicity in a healthy population. Pegfilgrastim-cbqv represents an opportunity to increase patient access to life-saving therapy and decrease overall costs of treatment.
References
Gascon P, Tesch H, Verpoort K, Rosati MS, Salesi N, Agrawal S, et al. Clinical experience with Zarzio(R) in Europe: what have we learned? Support Care Cancer. 2013;21(10):2925–32.
US Department of Health and Human Services. FDA guidance for industry,clinical pharmacology data to support a demonstration of biosimilarity to a reference product. December 2016. US Department of Health and Human Services, Silver Springs, MD. 2016. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-data-support-demonstration-biosimilarity-reference-product. Accessed December 5, 2019.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. European Medicines Agency, London, UK. 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf. Accessed June 26, 2019.
Amgen Inc. NEUPOGEN® (filgrastim) injection, for subcutaneous or intravenous use. Amgen Inc.: Thousand Oaks, CA; 2018. p. 45.
Amgen Inc. NEULASTA® (pegfilgrastim) injection, for subcutaneous use. Amgen, Inc.: thousand Oaks, Inc.; 2019. p. 60.
Arvedson T, O'Kelly J, Yang BB. Design rationale and development approach for pegfilgrastim as a long-acting granulocyte colony-stimulating factor. BioDrugs. 2015;29(3):185–98.
Roskos LK, Lum P, Lockbaum P, Schwab G, Yang BB. Pharmacokinetic/pharmacodynamic modeling of pegfilgrastim in healthy subjects. J Clin Pharmacol. 2006;46(7):747–57.
Coherus BioSciences I. UDENYCA® (pegfilgrastim-cbqv) injection, for subcutaneous use. Coherus BioSciences, Inc: Redwood City, CA; 2018. p. 21.
Golkowski D, Friede T, Kieser M. Blinded sample size re-estimation in crossover bioequivalence trials. Pharm Stat. 2014;13(3):157–62.
US Food and Drug Administration. Draft guidance on progesterone. In: 2011. US food and drug administration, Silver Spring, MD. 2011. Accessed June 26, 2019.
US Department of Health and Human Services. FDA guidance for industry, scientific considerations in demonstrating biosimilarity to a reference product. US Department of Health and Human Services,, Silver Springs, MD. 2015. https://www.fda.gov/media/82647/download. Accessed December 5, 2019.
Yang BB, Morrow PK, Wu X, Moxness M, Padhi D. Comparison of pharmacokinetics and safety of pegfilgrastim administered by two delivery methods: on-body injector and manual injection with a prefilled syringe. Cancer Chemother Pharmacol. 2015;75(6):1199–206.
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–6.
Gregory S, Schwartzberg L, Mo M, Sierra J, Vogel C. Evaluation of reported bone pain in cancer patients receiving chemotherapy in pegfilgrastim clinical trials: a retrospective analysis. Commun Oncol. 2010;7:297–308.
Waller CF, Tiessen RG, Lawrence TE, Shaw A, Liu MS, Sharma R, et al. A pharmacokinetics and pharmacodynamics equivalence trial of the proposed pegfilgrastim biosimilar, MYL-1401H, versus reference pegfilgrastim. J Cancer Res Clin Oncol. 2018;144(6):1087–95.
Acknowledgements
We thank John Glaspy, Radhika Turnstall, and Paula O’Connor for their contributions to this manuscript. Medical writing assistance was provided by Andrew Occiano, PharmD, and Monique N. O’Leary, PhD (ApotheCom, San Francisco, CA), and was funded by Coherus BioSciences (Redwood City, CA).
Funding
This study and medical writing support, along with the Rapid Service and Open Access Fees, were funded by Coherus BioSciences (Redwood City, CA).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript and take responsibility for the integrity of the work as a whole. The first draft of the manuscript was written by Barbara Finck. All authors substantially contributed to revising previous versions of the manuscript for important intellectual content. All authors read and approved the final manuscript.
Disclosures
At the time of the study, Barbara Finck, Helen Tang, Francesca Civoli, Jennifer Hodge, Hillary O’Kelly, and Vladimir Vexler were employees of and owned stock in Coherus BioSciences (Redwood City, CA). Jennifer Hodge is now an employee of Myovant Sciences Inc. (Brisbane, CA).
Compliance With Ethics Guidelines
The protocol and informed consent form were submitted to and approved by the sites’ institutional review boards (IRB) before initiation of the study. IRB approval was obtained from Schulman Institutional Review Board (Cincinnati, OH), Alpha Independent Review Board (San Clemente, CA), Chesapeake Institutional Review Board (Columbia, MD), and IntegReview Independent Review Board (Austin, TX) in the United States. This study was conducted in accordance with all applicable laws and regulations, and it complied with the International Conference for Harmonisation E6 Guideline on Good Clinical Practice and the Declaration of Helsinki. The rationale of the study, procedural details, and investigational goals were explained to each subject, along with potential risks and benefits. Each subject was assured of his/her right to withdraw from the study at any time. Before the initiation of any study procedures, each subject signed and dated an approved informed consent form.
Data Availability
Coherus will review and consider providing data on a case-by-case basis. Since this study is one of two studies for support of the biosimilarity assessment for an FDA- and EMA-approved biosimilar, Udenyca, the data have undergone extensive review by FDA and EMA. Coherus does not generally provide raw or deidentified datasets to investigators or journals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12696491.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Finck, B., Tang, H., Civoli, F. et al. Pharmacokinetic and Pharmacodynamic Equivalence of Pegfilgrastim-cbqv and Pegfilgrastim in Healthy Subjects. Adv Ther 37, 4291–4307 (2020). https://doi.org/10.1007/s12325-020-01459-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01459-y