
Abstract
Anti-CD19 chimeric antigen receptor (CAR) T-cell therapies can be effective for diffuse large B-cell lymphoma (DLBCL), a cancer with limited treatment options and poor outcomes, particularly for patients with relapsed or refractory (r/r) disease. Axicabtagene ciloleucel (axi-cel) and tisagenlecleucel (tisa-cel) are CAR T-cell therapies approved by regulatory bodies for certain patients with r/r DLBCL on the basis of demonstrated treatment effects in their pivotal single-arm trials, ZUMA-1 and JULIET, respectively. In the absence of head-to-head trials, the question of whether a valid indirect treatment comparison (ITC) between axi-cel and tisa-cel could be performed using existing evidence is of interest to patients, physicians, payers, and other stakeholders. This article addresses that question by summarizing the current evidence from clinical trials and real-world studies and discussing the challenges and limitations of potential analytical approaches associated with an ITC. Two ITC approaches attempting to adjust for cross-trial heterogeneity between ZUMA-1 and JULIET, matching-adjusted indirect comparison and regression-prediction model analysis, were evaluated. After evaluating the current clinical trial data and real-world evidence, and present and prior ITC analyses of axi-cel and tisa-cel, the authors conclude that a valid comparative analysis is not currently feasible. The substantial differences (e.g., timing of leukapheresis and enrollment, use of bridging chemotherapy [90% in JULIET vs. 0% in ZUMA-1], lymphodepleting regimens) between the two trials’ designs and patient populations preclude a robust and reliable ITC. No other approaches are able to account for such differences. The current real-world data are still too immature to be used for ITCs. Thus, drawing conclusions from such ITCs should be avoided to prevent misinforming treatment choices or limiting patient access to effective treatment options. Additional data from ongoing or future real-world studies with appropriate statistical analyses are needed to provide insights into the comparative effectiveness and safety of these two treatments.
Avoid common mistakes on your manuscript.
Why carry out this study? |
Anti-CD19 chimeric antigen receptor (CAR) T-cell therapies can be effective for diffuse large B-cell lymphoma (DLBCL), a cancer with limited treatment options and poor outcomes, particularly for patients with relapsed or refractory (r/r) disease. |
Axicabtagene ciloleucel (axi-cel) and tisagenlecleucel (tisa-cel) are CAR T-cell therapies approved for r/r DLBCL on the basis of demonstrated efficacy and manageble safety in their pivotal clinical trials, ZUMA-1 and JULIET, respectively. |
As there are no head-to-head trials comparing axi-cel and tisa-cel, this article explored the current clinical trial data and real-world evidence (RWE) to assess whether a valid indirect treatment comparison (ITC) could be performed. |
What was learned from this study? |
The substantial differences between JULIET and ZUMA-1 trials in study designs and patient populations preclude a robust and reliable ITC; ITC approaches are unable to account for such differences without substantial and unrealistic assumptions. Current real-world data are also too immature to be used for ITCs. |
No comparative conclusions from ITC using existing data can be made, as there would be significant risk of misinforming decision-making or limiting patient access to these treatments. |
Additional data from ongoing or future real-word studies with appropriate statistical approaches are needed to provide insights into the comparative effectiveness and safety of these two CAR T-cell treatments. |
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma (NHL), constituting up to 40% of NHL cases globally [1]. In the US, DLBCL comprises 25% of NHL cases and affects 5.6 per 100,000 people [2, 3]; in Europe, it affects 3.8 per 100,000 people [4]. Around 50–70% of patients with DLBCL are cured after conventional first-line immunochemotherapy [5]. The remaining 30–40% of patients either relapse or exhibit refractory disease after the initial response. High-dose chemotherapy followed by autologous stem cell transplantation (ASCT) is the standard of care during second-line treatment. However, not all patients are eligible to receive ASCT because of age, medical history, organ dysfunction, disease stage, comorbidities, or other reasons [6]. Patients who are ineligible for or have failed ASCT can receive salvage immunochemotherapies, but these therapies are associated with worse outcomes compared with ASCT [5]. The CORAL extension studies retrospectively reviewed the outcomes of patients with DLBCL who either did or did not receive ASCT after third-line treatments and reported the median overall survival (OS) to be 10.0 months and 4.4 months, respectively [7, 8]. SCHOLAR-1, a multi-cohort retrospective study, also reviewed the outcomes of patients with relapsed or refractory (r/r) DLBCL and reported a median OS of 6.3 months [9]. These poor outcomes highlight the unmet need for innovative treatments in patients with r/r DLBCL.
Anti-CD19 chimeric antigen receptor (CAR) T-cell therapies can be effective treatments for patients with r/r DLBCL. CAR T-cell therapy involves the genetic modification of a patient’s autologous T-cells to express a chimeric antigen receptor specific for a tumour antigen [10]. When re-infused into the patient, the CAR T-cells bind to the antigen on the cancer cell, exerting a cytotoxic effect. As of 2020, two CAR T-cell therapies have been approved by regulatory bodies for the treatment of certain adult patients with r/r DLBCL. The US Food and Drug Administration (FDA) approved axicabtagene ciloleucel (axi-cel) in October 2017 and tisagenlecleucel (tisa-cel) in May 2018, while the European Medicines Agency (EMA) licensed both CAR T-cell therapies in 2018. Other CAR T-cell therapies have been under development. For example, lisocabtagene maraleucel is under review by regulatory agencies but not yet approved and thus is not discussed in this article.
Both axi-cel and tisa-cel have demonstrated efficacy and manageable toxicity profiles in separate single-arm clinical trials and in the real-world setting, providing patients with promising new treatment options for r/r DLBCL beyond conventional therapies [11–16]. Because these two CAR T-cell therapies became available at around the same time, understanding their comparative effectiveness is of interest to patients, clinicians, payers, and other stakeholders to help inform clinical decisions and maximize patient benefit. Randomized controlled trials (RCTs) are the gold standard for assessing comparative efficacy and safety; however, a head-to-head RCT has not been conducted for these two treatments. In the absence of direct evidence from RCTs, comparative efficacy can be assessed using data from separate clinical trials and/or real-world studies in an indirect treatment comparison (ITC). However, an ITC should be carried out with extreme caution if there are substantial differences across studies. For example, data from separate trials could introduce bias as a result of heterogeneity in study design and patient population, while real-word data could be limited by susceptibility to multiple sources of bias, such as lack of quality control surrounding data collection, variability in follow-up procedures, and selection bias (i.e., the choice of a CAR T-cell therapy depends on patient profiles based on assumed product attributes). While all non-randomized comparisons face limitations, it is important to assess whether the magnitude of the limitations overwhelms any value of interpreting comparative findings. Conclusions on comparative effectiveness based on immature or unreliable data could mislead treatment choice, limit patient access to effective treatment options, and impair resource allocation for health and medical care systems.
This article addresses the question of whether a valid ITC of axi-cel and tisa-cel for r/r DLBCL is possible by summarizing the existing evidence from clinical trials and real-world studies and discussing the challenges and limitations of potential analytical approaches associated with an ITC. In addition, this article offers forward thinking on future avenues for comparative analysis based on additional sources of evidence that are not presently available. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Evidence from Clinical Trials
Both tisa-cel and axi-cel have demonstrated durable clinical benefit and manageable toxicities in their pivotal clinical trials JULIET (ClinicalTrials.gov identifier: NCT02445248) and ZUMA-1 (NCT02348216), respectively [11, 12]. JULIET is a single-arm, global, multi-center, phase II trial of tisa-cel conducted across centers in North America, Europe, Australia, and Japan [12]. As of July 2019, 167 patients had met the clinical eligibility criteria and underwent leukapheresis; of these, 115 patients were infused with tisa-cel. ZUMA-1 is an open-label, single-arm, multi-center, phase I-II trial conducted predominantly in the US (1 center in Israel) [11]. As of August 2018, 108 patients were infused with axi-cel across two phases of the trial (7 in phase I and 101 in phase II).
To assess the feasibility, strengths, and limitations of an ITC comparing JULIET and ZUMA-1, we assessed similarities and differences in trial designs, inclusion processes, outcome definitions, and patient populations (summarized in Table 1). When conducting an ITC, it is important to be able to adjust for cross-trial differences that have a known or suspected impact on patient outcomes. These may include prognostic factors, which impact outcomes regardless of treatment type, or effect modifiers, which have different effects on outcomes for each treatment [17, 18]. It is not usually possible to distinguish between prognostic factors and effect modifiers, especially for novel therapies. In addition, differences across trials may stem from the design, enrollment processes, and outcome definitions, which may or may not be amenable to statistical adjustment.
A best practice when conducting an ITC is to document all discernible similarities and differences across trials, to employ statistical analyses to adjust for factors that, based on clinical input, are known or suspected confounders, and to note the limitations arising from any cross-trial differences that are not amenable to statistical adjustment. There are some similarities between JULIET and ZUMA-1, principally that they both enrolled patients with refractory DLBCL, were open-label single-arm trials, required eligible patients to have received prior chemotherapy and have Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 at screening, and assessed similar end points (e.g., overall response rate [ORR] as the primary end point). However, there are multiple important differences as summarized below.
Patient Journey from Screening to CAR T-cell Infusion
A consideration of the patient journey from screening through eligibility, leukapheresis, enrollment, and treatment in JULIET and ZUMA-1 reveals some important differences in the trial designs (Fig. 1). These differences stem from the screening and manufacturing process that is necessary for CAR T-cell therapy. As CAR T-cells are designed to transduce a patient's autologous T-cells, the T-cells first need to be collected via leukapheresis and then genetically modified to be able to fight the cancer before infusing to patients. The manufacturing process of CAR T-cell therapy begins after leukapheresis and receipt of the cells by the processing laboratory. From this point, it may take 3–4 weeks before the modified T-cells are returned to the treatment center for infusion [19]. The two trials used different approaches to allocate manufacturing capacity to eligible patients. In ZUMA-1, enrollment occurred only once a manufacturing slot was confirmed and leukapheresis was then commenced. Conversely, in JULIET, patient enrollment was independent of manufacturing slot availability; leukapheresis could occur prior to a confirmed manufacturing slot, and the wait for a manufacturing slot could occur after enrollment for some patients.

Patient journey from screening to CAR T-cell infusion for JULIET and ZUMA-1. The figure was developed by the authors based on trial protocols of JULIET and ZUMA-1 and was validated by clinical experts. axi-cel axicabtagene ciloleucel, CAR T-cell chimeric antigen receptor T-cell, tisa-cel tisagenlecleucel
This difference creates several important sources of potential bias. First, patients and their physicians may have selected different trials based on patients’ clinical status and the expected times to receive therapy. Second, a trial with broader enrollment and longer times to infusion would be expected to have greater observed mortality between enrollment and infusion than a trial that enrolls more selectively to minimize the time to infusion. Finally, when measuring the effect of treatment, there are several time points of interest from which treatment can be considered to have started: enrollment, leukapheresis, and infusion. The aforementioned differences across trials would result in different biases for each of these time points. In particular, because r/r DLBCL is associated with a high mortality risk, longer waiting times can result in survival bias. To further complicate matters, the manufacturing timelines experienced in these trials prior to market authorization are not reflective of current real-world timelines. These differences in the process for patient inclusion and treatment result in complex and likely substantial sources of bias that cannot be addressed with statistical adjustments (i.e., the patients in JULIET and ZUMA-1 were subjected to different selection processes), and we cannot estimate how the ZUMA-1 patients would have fared under the enrollment process of JULIET or vice versa.
Bridging Therapy After Enrollment
A second substantial difference between trials concerns the use of bridging chemotherapy. The JULIET trial allowed the use of bridging treatment prior to the lymphodepleting chemotherapy (LDC) to maintain patients with a poor prognosis while they were waiting for infusion, and the majority (90%) of patients received bridging chemotherapy, whereas bridging treatment was not allowed in ZUMA-1 per the trial protocol. Patients in JULIET who received bridging chemotherapy generally experienced worse outcomes compared to those who did not (e.g., the 12 month progression-free survival [PFS] rates were 32% vs. 61%, respectively). Bridging chemotherapy is considered important for providing disease control while patients are awaiting CAR T-cell infusion, and most patients who are treated with either tisa-cel or axi-cel in the real-world setting have received bridging chemotherapy [15, 16, 20–25]. Similarly, a real-world study of patients receiving axi-cel reported that those who received bridging chemotherapy before infusion had significantly poorer OS and PFS compared to those who did not (OS: hazard ratio [HR] = 3.34, p < 0.01; PFS: HR = 1.43, p = 0.04). Given this negative impact on patient outcomes, any comparative analyses of JULIET and ZUMA-1 would be confounded by the drastically different use of bridging chemotherapy within them (i.e., 90% vs. 0% receiving it) [20, 21].
Even if one was to attempt to adjust for the difference in bridging therapy, the analysis would remain unreliable for two reasons. First and most importantly, the lack of bridging therapy does not have the same clinical meaning in JULIET and ZUMA-1. Specifically, all patients in JULIET could receive bridging therapy, but some did not because of clinical decisions made after enrollment (presumably because it was deemed unnecessary or they were not expected to tolerate it). In contrast, the protocol of ZUMA-1 did not permit bridging chemotherapy, and patients chose to enroll in a trial where it was not allowed or accessible, despite any changes in their clinical status after enrollment. Second, even if the lack of bridging therapy had the same meaning across trials, the proportion of patients in JULIET who did not receive it was so small that adjustment for this single factor would limit the JULIET population to 10% of its original size, precluding adjustment for other important confounding factors without extrapolation beyond the observed data.
Lymphodepleting Chemotherapy Regimen
Prior to the CAR T-cell infusion, LDC can prevent an anti-CAR immune response and help the infused CAR T-cells proliferate. Both JULIET and ZUMA-1 allowed LDC, but the regimens differed. All patients in ZUMA-1 received fludarabine-cyclophosphamide (Flu/Cy) chemotherapy (30 mg/m2 and 500 mg/m2, respectively, for 3 days), while JULIET used a more flexible dosing protocol. Specifically, 74% of JULIET patients were treated with Flu/Cy chemotherapy at a lower dose (25 mg/m2 and 250 mg/m2, respectively, for 3 days), and 19% received bendamustine (90 mg/m2 intravenously [IV] daily for 2 days). In addition, 7% of patients in JULIET did not receive any LDC if their white blood cell count was ≤ 1000 cells/µL within 1 week prior to tisa-cel infusion [21]. In a post-hoc analysis of the JULIET trial, patients who received Flu/Cy LDC achieved numerically better outcomes compared to those who received bendamustine or no LDC (ORR: 57.6% for Flu/Cy vs. 40.9% for bendamustine vs. 25% for no LDC) [21]. Such a discrepancy in the application of LDC regimens could potentially confound an ITC.
Patient Baseline Characteristics
Several differences exist in the baseline characteristics of patients in the JULIET and ZUMA-1 trials. Although both trials enrolled patients with DLBCL and transformed follicular lymphoma, ZUMA-1 also enrolled patients with primary mediastinal B-cell lymphoma (PMBCL). In a subgroup analysis of ZUMA-1, patients with PMBCL achieved better outcomes compared to patients with DLBCL [26]. In addition, the ECOG Performance Status scores, the proportions of patients who received prior ASCT, the numbers of prior lines of therapy, the proportions of patients who relapsed < 12 months post ASCT, and the proportions of patients with double/triple gene hits (c-MYC, BCL-2, and/or BCL-6) were different between the two trials.
Subsequent Therapies
There were also differences in subsequent treatments after the initial study drug infusion. Some eligible patients in ZUMA-1 received a second infusion of axi-cel, while no patients in JULIET were re-treated after the initial infusion of tisa-cel. Also, a higher proportion of patients received a stem cell transplant (SCT) after infusion in ZUMA-1 (11%) than in JULIET (6%).
Outcome Definitions
Outcomes were measured differently in the two trials. For example, the primary end point of the trials, ORR, was assessed in JULIET using the Lugano classification by a central independent review committee, while in ZUMA-1, ORR was assessed using the International Working Group Response Criteria as determined by the study investigators. Additionally, different systems were applied to grade cytokine release syndrome (CRS); the Penn grading system was used in JULIET while the Lee grading system was used in ZUMA-1. Compared to the Lee grading system, the Penn grading system is more likely to result in a higher evaluated grade of CRS [27, 28]. In addition, adverse events (AEs) were reported over different time periods in the trials. In JULIET, all AEs were reported through the 12-month visit and selected AEs were reported through the 60-month visit. In ZUMA-1, the reporting period was shorter. All AEs were reported for 3 months after infusion, and selected AEs were reported up to 24 months after infusion or disease progression.
Overall, these cross-trial differences are substantial in magnitude and involve factors that are already known to be predictive of outcomes in DLBCL. For these reasons, we believe it is evident that an ITC between JULIET and ZUMA-1 would be subject to major limitations precluding any reliable conclusions about comparative efficacy or safety.
Investigation of Potential Analytical Approaches to Adjust for Cross-trial Differences Between JULIET and ZUMA-1
To further explore and illustrate the limitations of conducting a valid ITC, we investigated well-established and commonly used analytical approaches in an attempt to adjust for cross-trial heterogeneity between the two pivotal trials. Given that JULIET and ZUMA-1 are single-arm trials, anchor-based ITC approaches (e.g., Bucher’s method [29] and network meta-analysis [NMA] [30]) are not feasible [29]. In this scenario, a population-adjusted ITC approach in the unanchored setting is usually considered [17, 18]. Without access to patient-level data in both of the trials, traditional statistical methods such as propensity score matching cannot be used to adjust for heterogeneity between the trials.
Two analytical approaches, matching-adjusted indirect comparison (MAIC) and simulated treatment comparison (STC) [31–34], are discussed in this article because both are commonly used ITC approaches considered by heath technology (HTA) assessment bodies (e.g., the National Institute for Health and Care Excellence [NICE]) [35–38]. Both approaches adjust for between-trial differences in the distribution of variables that influence outcomes, utilizing patient-level data for one treatment along with aggregate trial-level data for the other treatment [17, 35–38]. Specifically,
-
(1)
The MAIC method matches average patient characteristics between trials by applying propensity score-based weights (estimated via a propensity model) to each patient from the trial with patient-level data available [31]. Weighted outcomes are then compared across balanced trial populations.
-
(2)
The STC method fits a prediction model (i.e., a regression model) of relevant patient characteristics to each outcome using patient-level data from one trial [33, 34]. This model is then used to predict the outcomes for a population having the same baseline profile as in the other trial; the contrast between the predicted and observed outcomes in this population serves as the estimated treatment effect [17].
Critical assumptions for both approaches are that all confounding factors have been included in the adjustment and that the model is correctly specified (i.e., the propensity score model for a MAIC and the prediction model in an STC). This assumption is strong and may not be met in some situations [17]. Before interpreting a MAIC or STC, it is necessary to consider whether all important known or suspected confounding factors can be addressed through either of these statistical methods.
Two MAICs and an STC (referred to as a CAR-T prediction model analysis) were evaluated in this review. The OS outcome was selected for these analyses because it was consistently defined across trials (i.e., as the time from infusion to death due to any cause for infused patients), unlike other response and safety outcomes.
Matching-Adjusted Indirect Comparison
Two MAIC analyses have been conducted to indirectly compare tisa-cel and axi-cel using data from JULIET and ZUMA-1.
-
(1)
One was conducted by the authors, which reweighted JULIET patients to match the characteristics of ZUMA-1 patients using patient-level data from JULIET and aggregate data from ZUMA-1. After matching, the estimated HR between tisa-cel and axi-cel was 1.90 (95% confidence intervals [CI]: 1.28, 2.82). Figure 2 illustrates the OS curves from the adjusted JULIET data and the observed ZUMA-1 data.
Fig. 2 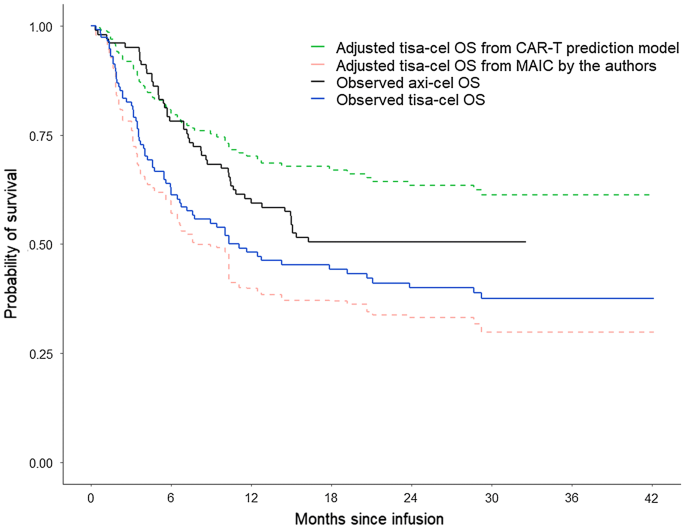
Observed tisa-cel OS, observed axi-cel OS, and adjusted tisa-cel OS based on the MAIC and CAR-T prediction model1−4. axi-cel axicabtagene ciloleucel, CAR-T chimeric antigen receptor T-cell therapy, ITC indirect treatment comparison, LDC lymphodepleting chemotherapy, MAIC matching-adjusted indirect comparison, OS overall survival, tisa-cel tisagenlecleucel. 1Adjusted tisa-cel OS from the CAR-T prediction model: expected OS for tisa-cel assuming that tisa-cel had treated patients with similar patient characteristics as those from ZUMA-1. The prediction model was built based on tisa-cel patient-level data from JULIET. Due to the small number of events for several key predictors (e.g., only 11 out of 115 patients did not receive bridging chemotherapy in JULIET), this method was not reliable. 2Adjusted tisa-cel OS from the MAIC: expected OS for tisa-cel among patients who had similar patient characteristics to those from ZUMA-1. Due to missingness of important effect modifiers (i.e., use of bridging chemotherapy and LDC regimens), this method was also not reliable. 3Substantial differences (i.e., enrollment, bridging chemotherapy usage, LDC regimens, etc.) between JULIET and ZUMA-1 preclude any reliable ITC being conducted; if conducted, two ITC methods (MAIC and CAR-T prediction model) provide contradictory conclusions as shown in the figure. 4The proportional hazards assumption was not rejected in any Cox models in the analyses
-
(2)
The other MAIC was conducted by Oluwole et al. and matched ZUMA-1 patient characteristics to those of JULIET patients using patient-level data from ZUMA-1 and aggregate data from JULIET [39]. The HR estimated between tisa-cel and axi-cel was 2.27 (1.47, 3.45) when matching to JULIET patients.

The two MAICs would not be expected to produce identical results, even if both were valid and interpretable, as the two analyses compared OS for different matched populations (i.e., adjusting JULIET patients to match the ZUMA-1 population and vice versa). Besides matching to different patient populations, different matching factors were considered in the two analyses. For example, the MAIC by the authors matched more baseline characteristics (including sex, prior ASCT, and bulky disease) than the MAIC by Oluwole et al. It is not clear how these factors could be ruled out as being potential confounding factors. Conversely, disease stage was matched in the MAIC by Oluwole et al. but not in the one by the authors, because no published ZUMA-1 data on disease stage were available at the time of our analyses. The detailed list of adjusted variables for both MAICs is summarized in Table 2.
Besides the differences in matched baseline characteristics, a larger issue is that both MAICs are subject to major limitations due to cross-trial differences that cannot be addressed via statistical adjustment. As described in the prior section, the timing of leukapheresis and enrollment, use of bridging chemotherapy, LDC regimen, and the treatment regimens received following CAR T-cell infusion systematically differed between trials and could not be included in the adjustments. Because these variables may be prognostic of patient outcomes, the inability to adjust for them would render the results from both analyses unreliable. In addition, given the multiple substantial and interrelated differences across trials, it is not possible to discern the likely direction or magnitude of any bias.
CAR-T Prediction Model Analysis
A CAR-T prediction model analysis was also performed by the authors to extrapolate OS for tisa-cel in a hypothetical population similar, in terms of patient characteristics, to those enrolled in the ZUMA-1 trial [17] (the list of adjusted variables is shown in Table 2). Compared to the MAICs, which cannot directly extrapolate beyond the observed data, the CAR-T prediction model approach allows extrapolation of outcomes to populations that are not well represented by the source population. In particular, this approach was used to extrapolate the JULIET outcomes to a population who did not receive bridging therapy and received different LDC regimens. While this extrapolation incurs multiple additional limitations (i.e., extrapolation in general is unreliable and, as described above, the meaning of lack of bridging therapy differs across trials), it is helpful here as an exploratory and illustrative tool.
In this CAR-T prediction model analysis, a multivariable Cox model was developed for OS using patient-level data from the JULIET trial. The model was used to predict the efficacy of tisa-cel, via extrapolation, in a hypothetical patient population with a similar set of patient characteristics as ZUMA-1, including 0% receiving bridging chemotherapy and 100% with fludarabine-based LDC. In this analysis, tisa-cel was associated with a numerically longer OS than axi-cel (tisa-cel vs. axi-cel, HR [95% CI]: 0.75 [0.48, 1.18]), contrary to both of the MAIC results described above. The adjusted tisa-cel OS curve assuming tisa-cel had been used to treat the ZUMA-1 patient population and the observed ZUMA-1 OS curve are shown in Fig. 2.
The CAR-T prediction model made extrapolations based on factors that could potentially impact OS, particularly bridging chemotherapy and LDC regimens, which could not be accounted for using MAIC. However, it may have induced additional bias by assuming that the predicted outcome for the average patient equalled the average of the predicted outcomes across patients. Such an assumption is not likely to be met in non-linear prediction models (e.g., a Cox model of OS) [33]. In addition, only 11 patients in JULIET did not receive bridging chemotherapy. The small number of events introduced additional bias as those patients may not be representative of those who require bridging chemotherapy.
The ITCs investigated here are commonly employed analytic approaches, but there is no guarantee that any methodology, no matter its operating characteristics and general acceptability, can produce a valid and reliable comparison across non-randomized treatment groups. In the case of comparison between JULIET and ZUMA-1, both approaches are subject to major limitations due to substantial differences in trial designs and patient populations that cannot be accounted for by statistical adjustments. It is notable that the different methods led to contradictory conclusions, further demonstrating the high risk of drawing incorrect conclusions by any comparison across trials that substantially differ.
Real-World Evidence
In addition to data from clinical trials, real-world evidence (RWE) is another potential data source for comparative analysis. Real-world clinical practice does not always follow clinical trial specifications, which potentially reflects the evolution of clinical practice and the broader and more heterogeneous patient population in the real world. Regulatory bodies such as the FDA have a long history of using RWE to inform trial design and to monitor safety and evaluate effectiveness after drug approval [40]. RWE has played an increasingly important role in treatment evaluations, particularly in the settings of oncology and rare diseases.
A number of real-world studies of patients receiving tisa-cel or axi-cel have been conducted in the US and in Europe since the approval of these therapies. Riedell et al. retrospectively analyzed data from patients who underwent apheresis for commercial use of tisa-cel or axi-cel from eight US academic centers [15]. Data collection in that study began following the FDA approval of tisa-cel when centers had the choice to prescribe either tisa-cel or axi-cel. Among 244 patients who received a CAR T-cell treatment, 158 were treated with axi-cel and 86 were treated with tisa-cel. More axi-cel than tisa-cel patients were treated in the inpatient setting (92% vs. 37%, respectively), and the majority of both cohorts received bridging chemotherapy (61% and 75%, respectively). The high rates of bridging chemotherapy could indicate that, in real-world practice, most patients cannot wait without bridging therapy during manufacture before receiving infusion. The two cohorts described in Riedell et al. had different demographic and clinical characteristics. For example, tisa-cel recipients were older than axi-cel recipients (median age: 67 vs. 59 years, respectively) and more heavily pre-treated (86% vs. 73% with ≥ 3 prior therapies). Riedell et al. concluded that the efficacy in the commercial setting appeared similar to outcomes observed in the pivotal clinical trials. At day 90 post-infusion, 64% of axi-cel patients achieved objective response (OR) with 53% achieving complete response (CR), while 51% of tisa-cel patients achieved OR including 42% with CR. Median OS was not reached for either cohort due to the short follow-up (median: 7.6 and 6.2 months for the axi-cel and tisa-cel cohorts, respectively).
When AEs were examined in Riedell et al., tisa-cel was found to be associated with fewer CRS and neurologic events compared to axi-cel, which impacted healthcare resource use. For instance, patients treated with axi-cel had longer hospital stays than tisa-cel patients (median: 16 vs. 2 days, respectively) as well as increased incidence of intensive care unit transfer (39% vs. 7%) and greater use of tocilizumab (61% vs. 15%) and steroids (53% vs. 8%) [15]. Additionally, it has been observed that the management and grading of CRS in the real world (using the American Society of Transplantation and Cellular Therapy system) differ from that of clinical trials [15]. Early use of corticosteroids and prophylactic use of tocilizumab in the real world may have reduced the recorded incidence of severe CRS in patients with DLBCL receiving CAR T-cell therapies. While it might be expected that deviations from strict clinical trial specifications might lead to poorer outcomes, Riedell et al.’s reported outcomes in patients treated with tisa-cel and axi-cel in the real-world are similar to those reported in the pivotal clinical trials [15]. However, the values should not be compared directly without controlling for differences in patient populations, such as the difference in bridging chemotherapy rates, which may have an impact on the outcomes [20].
The Center for International Blood and Marrow Transplant Research (CIBMTR) registry is another source providing RWE for both tisa-cel and axi-cel [13, 14], although the follow-up times are still very short at the time of reporting (4.5 months for tisa-cel and 6.2 months for axi-cel). The tisa-cel cohort included 116 patients (median age of 65 years); 41% had double/triple hit lymphoma and 27% had transformed lymphoma. The ORR for tisa-cel was 58% with 40% of patients achieving CR. The OS and PFS rates at 3 months were 79.6% and 61.6%, respectively. While the efficacy results for tisa-cel in the real world were similar to the efficacy data reported in the JULIET trial, the rates of CRS (4% for grade 3+) and neurotoxicity (5% for grade 3+) were lower [13]. The axi-cel cohort included 533 patients (median age of 61 years); 36% had double/triple hit lymphoma and 30% of patients had transformed lymphoma. The ORR rate was 74%, with 14% of patients with reported CRS (grade 3+) and 61% with reported neurotoxicity (any grade). In the real-world setting, similar efficacy and safety for axi-cel were observed as in the ZUMA-1 trial, with additional data focusing on older patients (aged ≥ 65 years) whose results were similar to the younger patients (aged < 65 years) [14]. However, it is noted that there were differences in baseline characteristics among patients treated with tisa-cel or axi-cel. For example, the patients who received tisa-cel were older than those who received axi-cel (median age: 65 vs. 61 years, respectively). In addition, more patients who received tisa-cel had double/triple gene hits than those who received axi-cel (41% vs. 36%, respectively). The CIBMTR registry plans to follow 1500 treated patients for each therapy for 15 years and will provide more evidence for comparative analyses between tisa-cel and axi-cel when longer follow-up data become available. With the availability of long-term CIBMTR data, analytical approaches such as propensity score matching or weighting using patient-level data can be potentially implemented to adjust for the differences in patient characteristics.
In addition to the above-mentioned studies, a number of other real-world studies evaluating the efficacy and safety of tisa-cel and axi-cel have been conducted all over the world [16, 20, 22–25, 41–46]. These studies will help provide further information on the treatment outcomes and clinical practice in the real-world setting. At the time of writing this article, those studies are limited by short follow-up periods (the reported median follow-up times ranged from 4–7 months) and small sample sizes (e.g., < 100 in most studies). Thus, the currently existing evidence from real-world studies of tisa-cel and axi-cel are still too immature to allow for comparative analysis.
Discussion
The clinical trials and RWE have demonstrated that tisa-cel and axi-cel are both effective treatments for patients with r/r DLBCL. While robust comparative data between these two therapies are needed for clinical and economic decision-making, a valid ITC using the existing evidence from clinical trials and RWE is not currently feasible. The substantial differences in trial design and patient characteristics between JULIET and ZUMA-1 preclude a reliable ITC between these two treatments. Due to these cross-trial differences, different ITC methods (i.e., MAIC and a CAR-T prediction model) can lead to different conclusions about comparative OS. Such differences have also been noted by other researchers, who suggested that further studies are needed in the absence of a head-to-head trial between the therapies [19, 47–51].
Existing real-world studies provide valuable information on the safety and short-term effectiveness of tisa-cel and axi-cel, especially in the studies where both CAR T-cell therapies were available and prescribed to patients in a similar setting (e.g., allowance of bridging chemotherapy) [15, 42, 45, 52, 53]. However, there are noticeable differences in the patient populations using tisa-cel and axi-cel in the real-world, and the data are still too immature to allow for comparative analysis because of the short follow-up times and small sample sizes. Additional data collected from ongoing real-world studies, such as Riedell et al., Jaglowski et al., and Pasquini et al. [13–15], as well as similar future real-world studies, can provide further insights and valuable data to assess these two therapies. Besides ongoing and planned studies in the US, several real-world studies are being conducted in Europe which can provide more RWE. For instance, the European Society for Blood and Marrow Transplantation (EBMT) is collecting information on patients treated with tisa-cel or axi-cel for the EBMT Registry [54]. In addition, access to patient-level data of both therapies by the investigators of these real-world studies can allow them to control for imbalances in the patient population and thus could potentially overcome the issue of selection bias and ensure a fair comparison. However, there may still be unobserved effect modifiers, such as physicians’ preferences in assigning different treatments based on patients’ disease status and safety concerns as well as reimbursement restrictions that could limit patients’ access to both therapies.
As demonstrated by the contradictory conclusions from the MAICs and the CAR-T prediction model analysis, the risk of drawing incorrect conclusions from unreliable analyses is high. It is critical to note the limitations associated with such ITC analyses and interpret any ITC results with extreme caution, especially where there are substantial differences between the trials. Clinical and treatment decisions should not be informed based on the current ITC results. Incorrect interpretation of such results could have a substantial impact on patients and on healthcare systems. Patients could be denied access to therapies that appeared to demonstrate poor efficacy and/or safety in comparative effectiveness studies that were subject to severe limitations. Furthermore, any restriction of patient access to medically necessary care and/or medications could prolong suffering and reduce the potential for patients to make a full recovery [55]. A 2019 study reported that the social value of CAR T-cell therapy for r/r DLBCL in the US was greatly limited by treatment delays, with 4.2%, 11.5%, and 46.0% of social value lost after 1, 2, and 6 months of treatment delays, respectively [56]. In addition, such restrictions fail to acknowledge that practitioners and patients should make individualized treatment decisions and to recognize the unique and non-interchangeable nature of medical care [57, 58]. Patient access to comprehensive, quality health care services is essential for promoting and maintaining physical, social, and mental health, preventing and managing disease, reducing unnecessary disability and premature death, improving quality of life, and achieving health equity [56, 59–61].
A well-designed head-to-head RCT is the most reliable approach to assess comparative effectiveness. However, such an RCT is not always feasible or practical for innovative therapies (e.g., advanced therapy medicinal products) in rare diseases. To support timely patient access to those novel treatments in populations with high and urgent unmet needs, comparative evidence based on ITCs across suitably similar trials can be valuable for regulators and HTA bodies to inform decision making. Pathways to accelerate drug evaluation by leveraging additional sources of evidence have been developed [32]. Regulators, such as the the US FDA, have accepted data from single-arm trials as substantial evidence supporting accelerated approval of novel treatments [62–65]. Support from regulatory agencies and medical societies to harmonize the designs of single-arm trials would be desirable. While such harmonization is challenging in rapidly evolving therapeutic areas, it can increase the value of the patient data collected in a single-arm trial and enable decision-makers to receive higher quality evidence from external controls or indirect comparisons, which may be the best comparative evidence possible when randomized trials are not feasible or ethical. Best practices for indirect comparative analyses and externally controlled studies using data from single-arm trials have been put forward by multiple groups, and additional guidance is under development [40, 66–70]. Because indirect comparisons are retrospective and do not have the benefit of randomization, there is a heightened importance of the study process and governance to mitigate risk of bias. These practices include justifying the choice of included trials (regarding relevance of study design, comparator treatment, patient population, and outcome criteria), identifying and accounting for confounding factors (e.g., via a systematic review and input from medical experts), processes for selecting appropriate ITC methodologies and sensitivity analyses, defining a priori a statistical analysis plan, and discussing potential biases (in terms of measurement, selection, and attrition) and limitations [71–73].
In addition to indirect comparisons across trials, well-designed real-world studies that evaluate multiple treatments in real-world settings can provide valuable comparative evidence. For CAR T-cell therapies in DLBCL, it is essential to account for the multiple complexities of care patterns, patient characteristics, and outcome definitions that could impact comparative effectiveness research when designing or selected real-world data sources. These factors include patient inclusion/exclusion criteria, use of bridging chemotherapy (and reasons for use or non-use), the intervals between screening and infusion, LDC regimens, and retreatment with a CAR T-cell therapy. The same study processes as described above, including gaining clinical input on potential confounding factors and the pre-specification of appropriate statistical and sensitivity analyses, will be important for developing reliable comparative evidence for CAR T-cell therapies based on RWE.
Given the lack of head-to-head RCTs for CAR T-cell therapies in DLBCL and the challenges involved in conducting such trials, comparative evidence derived from ITCs and real-world data can play an especially important role in evaluating treatments for DLBCL, provided that the data sources are suitability similar and that comparative studies are well designed and conducted.
Conclusions
When considering all the available evidence from clinical trial and real-world settings, it is concluded that due to substantial differences in study designs and patient populations, an ITC to assess comparative effectiveness and safety between tisa-cel and axi-cel cannot currently provide reliable results. The pivotal trials of tisa-cel (JULIET) and axi-cel (ZUMA-1) have fundamental and irreconcilable differences in trial designs and patient populations. Real-world data have demonstrated that both CAR T-cell therapies produced durable clinical benefit. However, due to the short follow-up times, the RWE is still too immature to allow for valid comparative analyses between the two CAR T-cell therapies.
References
World Health Organization. Diffuse large B-cell lymphoma. https://www.who.int/selection_medicines/committees/expert/20/applications/DiffuseLargeBCellLymphoma.pdf. Accessed 4 Apr 2020
Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA-Cancer J Clin. 2016;66(6):443–59.
National Cancer Institute. Cancer stat facts: NHL- diffuse large B-call lymphoma (DLBCL). Available at: https://seer.cancer.gov/statfacts/html/dlbcl.html. Accessed 4 Apr 2020.
Tilly H, Gomes da Silva M, Vitolo U, Jack A, Meignan M, Lopez-Guillermo A, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v116–v12525.
Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematol-Am Soc Hematol. 2016;2016(1):366–78.
Holmberg LA, Deeg HJ, Sandmaier BM. Determining eligibility for autologous hematopoietic cell transplantation. https://www.uptodate.com/contents/determining-eligibility-for-autologous-hematopoietic-cell-transplantation. Accessed 19 May 2020
Van Den Neste E, Schmitz N, Mounier N, Gill D, Linch D, Trneny M, et al. Outcomes of diffuse large B-cell lymphoma patients relapsing after autologous stem cell transplantation: an analysis of patients included in the CORAL study. Bone Marrow Transplant. 2017;52(2):216–21.
Van Den Neste E, Schmitz N, Mounier N, Gill D, Linch D, Trneny M, et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the International CORAL study. Bone Marrow Transplant. 2016;51(1):51–7.
Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–8.
Miliotou AN, Papadopoulou LC. CAR T-cell therapy: a new era in cancer immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5–18.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Jaglowski S, Hu Z-H, Zhang Y, Kamdar M, Ghosh M, Lulla P, et al. Tisagenlecleucel chimeric antigen receptor (CAR) T-cell therapy for adults with diffuse large B-cell Lymphoma (DLBCL): Real world experience from the Center for International Blood & Marrow Transplant Research (CIBMTR) Cellular Therapy (CT) Registry. Blood. 2019;134(Supplement_1):766.
Pasquini MC, Locke FL, Herrera AF, Siddiqi T, Ghobadi A, Komanduri KV, et al. Post-marketing use outcomes of an anti-CD10 chimeric antigen receptor (CAR) T cell therapy, axicabtagene ciloleucel (axi-cel), for the treatment of large B cell lymphoma (LBCL) in the United States (US). Blood. 2019;134(Supplement_1):764.
Riedell PA, Brower J, Nastoupil L, Pennisi M, Maziarz RT, McGuirk JP, et al. A multicenter retrospective analysis of outcomes and toxicities with commercial axicabtagene ciloleucel and tisagenlecleucel for relapsed/refractory aggressive B-cell lymphomas. Presented at the Meeting of the American Society for Transplantation and Cellular Therapy on February 19–23, 2020, Orlando, FL.
Kuhnl A, Roddie C, Martinez-Cibrian N, Menne TF, Linton K, Lugthart S, et al. Real-world data of high-grade lymphoma patients treated with CD19 CAR-T in England. Blood. 2019;134(Supplement1):767.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200–11.
National Institute for Health Care and Excellence (NICE). NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. https://nicedsu.org.uk/wp-content/uploads/2018/08/Population-adjustment-TSD-FINAL-ref-rerun.pdf. Accessed 28 Apr 2020.
Abramson JS. Anti-CD19 CAR T-cell therapy for B-cell non-Hodgkin lymphoma. Transfus Med Rev. 2020;34(1):29–33.
Jain M, Jacobs M, Nastoupil L, Spiegel J, Feng G, Lin Y, et al. Characteristics and outcomes of patients receiving bridging therapy while awaiting manufacture of standard of care axicabtagene ciloleucel CD19 chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory large B-cell lymphoma: results from the US lymphoma CAR-T consortium. Blood. 2019;134:245.
Andreadis C, Tam C, Borchmann P, Jaeger U, McGuirk J, Holte H, et al. Correlation of bridging and lymphodepleting chemotherapy with clinical outcomes in patients with relapsed/refractory diffuse large B-cell lymphoma treated with tisagenlecleucel. Blood. 2019;134(Supplement1):2883.
Nasta SD, Hughes M, Namaglu E, Landsburg D, Chong E, Barta S, et al. A characterization of bridging therapies leading up to commerical CAR T-cell therapy. Blood. 2019;134(Supplement1):4108.
Paillassa J, Vercellino L, Di Blasi R, Bernard S, Moatti H, Bommier C, et al. Impact of bridging chemotherapy on clinical outcomes of CD19 CAR T therapy in relapse/refractory diffuse Large B-cell lymphoma in real world experience. Blood. 2019;134(Supplement1):2886.
Pinnix C, Gunther J, Dabaja B, Adkins S, Hawkins M, Westin J, et al. Radiation therapy can be an effective bridging strategy prior to axicabtagene ciloleucel therapy for relapsed/refractory large B-cell lymphoma. Blood. 2019;134(Supplement1):1609.
Thieblemont C, Le Gouill S, Di Blasi R, Cartron G, Morschhauser F, Bachy E, et al. Real-world results of an anti-CD19 CAR T-cell axicabtagene ciloleucel for patients with relapsed/refractory large B-cell lymphoma included in a French early access program. Presented at the 2nd European CAR T Cell Meeting on January 30-February 1, 2020, Sitges, Spain.
Kite Pharma Inc. Dossier zur Nutzenbewertung gemäß § 35a SGB V: Axicabtagen-Ciloleucel (YESCARTA) Modul 4A. https://www.g-ba.de/downloads/92-975-2746/2018-10-31_Modul4A_Axicabtagen-Ciloleucel.pdf. Accessed 13 April 2020.
Schuster SJ, Maziarz RT, Ericson SG, Rusch ES, Signorovitch J, Romanov VV, et al. Consensus grading of cytokine release syndrome (CRS) in adult patients with relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL) treated with tisagenlecleucel on the JULIET study. Blood. 2018;132(Supplement1):4190.
Schuster SJ, Maziarz RT, Rusch ES, Li J, Signorovitch JE, Romanov VV, et al. Grading and management of cytokine release syndrome in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020;4(7):1432–9.
Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683–91.
Dias S, Welton NJ, Sutton AJ, Ades A. National Institute for Health and Care Excellence (NICE); NICE DSU Technical Support Document 2: A generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. https://www.ncbi.nlm.nih.gov/books/NBK310366/. Accessed 7 Apr 2020.
Signorovitch JE, Wu EQ, Yu AP, Gerrits CM, Kantor E, Bao Y, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935–45.
Alliance for Regenerative Medicine. Getting ready for advanced therapy medicinal products (ATMPs) in Europe. https://alliancerm.org/indication-data/market-access-report/. Accessed 10 Apr 2020.
Caro JJ, Ishak KJ. No head-to-head trial? Simulate the missing arms. Pharmacoeconomics. 2010;28(10):957–67.
Ishak KJ, Proskorovsky I, Benedict A. Simulation and matching-based approaches for indirect comparison of treatments. Pharmacoeconomics. 2015;33(6):537–49.
National Institute for Health Care and Excellence (NICE). NICE technology appraisal guidance 319: Ipilimumab for previously untreated advanced (unresectable or metastatic) melanoma. https://www.nice.org.uk/guidance/ta319/resources/guidance-ipilimumab-for-previously-untreated-advanced-unresectable-or-metastatic-melanoma-pdf. Accessed 10 Apr 2020.
National Institute for Health Care and Excellence (NICE). Talimogene laherparepvec for treating unresectable metastatic melanoma: Technology appraisal guidance [TA410]. https://www.nice.org.uk/guidance/ta410. Accessed 10 Apr 2020.
Quinn C, Ma Q, Kudlac A, Palmer S, Barber B, Zhao Z. Indirect treatment comparison of talimogene laherparepvec compared with ipilimumab and vemurafenib for the treatment of patients with metastatic melanoma. Adv Ther. 2016;33(4):643–57.
Quinn C, Ma Q, Kudlac A, Palmer S, Barber B, Zhao Z. Relative efficacy of granulocyte-macrophage colony-stimulating factor, dacarbazine, and glycoprotein 100 in metastatic melanoma: an indirect treatment comparison. Adv Ther. 2017;34(2):495–512.
Oluwole OO, Jansen J, Lin V, Chan K, Navale L, Locke FL. PCN445 Indirect treatment comparison of axicabtagene ciloleucel (axi-cel) versus tisagenlecleucel (tisa-cel) in relapsed/refractory large B cell lymphoma (RR-LBCL). Value Health. 2019;22(Supplement3):S522.
United States Food and Drug Administration (FDA). Framework for FDA’s real-world evidence program. https://www.fda.gov/media/120060/download. Accessed 5 May 2020
Pennisi M, Jain T, Santomasso BD, Mead E, Wudhikarn K, Silverberg ML, et al. Comparing CAR T-cell toxicity grading systems: application of the ASTCT grading system and implications for management. Blood Adv. 2020;4(4):676–86.
Gajra A, Zettler ME, Phillips EG Jr, Klink AJ, Kish JK, Mehta S, et al. Neurological adverse events following CAR-T cell therapy: A real-world analysis of adult patients treated with axicabtagene ciloleucel or tisagenlecleucel. Blood. 2019;134(Supplement1):1952.
Strati P, Tummala S, Nastoupil LJ, Westin JR, Fayad L, Ahmed S, et al. Clinical and radiological correlates of neurotoxicity after standard of care axicabtagene ciloleucel in patients with relapsed/refractory large B-cell lymphoma. Blood. 2019;134(Supplement1):765.
Iacoboni G, Catala E, Sanchez-Pina J, Bailen R, Mussetti A, Sancho J, et al. Real-world evidence of the use of tisagenlecleucel for patients with relapsed/refractory aggressive B-cell lymphomas. The Spanish experience. Presented at the 2nd European CAR T Cell Meeting on January 30-February 1, 2020, Sitges, Spain.
Denlinger N, Braunstein Z, Agyeman A, Purdin Z, Neal A, Yucebay F, et al. CAR T-cell therapy: Clinical outcomes, patient selection and financial metrics with tisagenlecleucel and axicabtagene ciloleucel, a single center experience. Bio Blood Marrow Transplant. 2020;26(Supplement3):S266.
Thieblemont C, Legouill S, Di Blasi R, Cartron G, Morschhauser F, Bachy E, et al. Real-world results on CD19 CAR T-cell for 60 French patients with relapsed/refractory diffuse large B-cell lymphoma included in a temporary authorization for use program. Hematol Oncol. 2019;37:301.
Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol. 2019;10:2040620719841581.
Chow VA, Shadman M, Gopal AK. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood. 2018;132(8):777–81.
Quintás-Cardama A. CD19 directed CAR T cell therapy in diffuse large B-cell lymphoma. Oncotarget. 2018;9(52):29843–4.
Viardot A, Wais V, Sala E, Koerper S. Chimeric antigen receptor (CAR) T-cell therapy as a treatment option for patients with B-cell lymphomas: perspectives on the therapeutic potential of axicabtagene ciloleucel. Cancer Manag Res. 2019;11:2393–404.
Schuster SJ. CD19-directed CAR T cells gain traction. Lancet Oncol. 2019;20(1):2–3.
Riedell PA, Walling C, Nastoupil L, Pennisi M, Maziarz RT, McGuirk JP, et al. A multicenter retrospective analysis of clinical outcomes, toxicities, and patterns of use in institutions utilizing commercial axicabtagene ciloleucel and tisagenlecleucel for relapsed/refractory aggressive B-cell lymphomas. Blood. 2019;134(Supplement1):1599.
Dwivedy Nasta S, Hughes ME, Namoglu EC, Landsburg DJ, Chong EA, Barta SK, et al. A characterization of bridging therapies leading up to commercial CAR T-cell therapy. Blood. 2019;134(Supplement1):4108.
European Society for Blood and Marrow Transplantation. How do I report on patients treated with CAR-T cells to the EBMT Registry? https://www.ebmt.org/ebmt/news/how-do-i-report-patients-treated-car-t-cells-ebmt-registry. Accessed 28 April 2020.
Gillick MR. Choosing appropriate medical care for the elderly. J Am Med Dir Assoc. 2001;2(6):305–9.
Thornton Snider J, Brauer M, Kee R, Batt K, Karaca-Mandic P, Zhang J, et al. The potential impact of CAR T-cell treatment delays on society. Am J Manag Care. 2019;25(8):379–86.
American Association of Clinical Endocrinologists (AACE) and the American College of Endocrinology (ACE). Patient access to necessary and appropriate medical care. https://www.empoweryourhealth.org/magazine/vol9_issue2/patient_access_to_necessary_and_appropriate_medical_care. Accessed 7 Apr 2020.
National Pharmaceutical Council. Patient access to medicines: A growing challenge. https://www.npcnow.org/newsletter/content/patient-access-medicines-growing-challenge. Accessed 7 Apr 2020.
Office of Disease Prevention and Health Promotion (ODPHP). Access to health services. https://www.healthypeople.gov/2020/topics-objectives/topic/Access-to-Health-Services. Accessed 7 Apr 2020.
Institute of Medicine (US) Committee on Monitoring Access to Personal Health Care Services. Access to Health Care in America. Millman M, editor. Washington (DC): National Academies Press (US); 1993.
Franco E. Navigating patient access in the new age of health care. https://www.nthrive.com/blog/navigating-patient-access-in-the-new-age-of-health-care. Accessed 7 Apr 2020.
Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100(6):1965–71.
Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–40.
DeAngelo DJ, Yu D, Johnson JL, Coutre SE, Stone RM, Stopeck AT, et al. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: cancer and Leukemia Group B study 19801. Blood. 2007;109(12):5136–42.
Jeha S, Gaynon PS, Razzouk BI, Franklin J, Kadota R, Shen V, et al. Phase II study of clofarabine in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. J Clin Oncol. 2006;24(12):1917–23.
Langham S, Pooley N, Madrigal AM. The challenges of carrying out an indirect comparison with single-arm studies. Value Health. 2015;18(7):A558.
Berger ML, Sox H, Willke RJ, Brixner DL, Eichler HG, Goettsch W, et al. Good practices for real-world data studies of treatment and/or comparative effectiveness: recommendations from the joint ISPOR-ISPE Special Task Force on real-world evidence in health care decision making. Pharmacoepidemiol Drug Saf. 2017;26(9):1033–9.
National Institute for Health Care and Excellence (NICE), Bell H, Wailoo AJ, Hernandez M, Grieve R, Faria R, et al. The use of real world data for the estimation of treatment effects in NICE decision making. Report by the decision support unit. https://nicedsu.org.uk/wp-content/uploads/2018/05/RWD-DSU-REPORT-Updated-DECEMBER-2016.pdf. Accessed 5 May 2020
National Institute for Health Care and Excellence (NICE), Faria R, Hernadez Alava M, Manca A, Wailoo AJ. The use of observational data to inform estimates of treatment effectiveness in technology appraisal: methods for comparative individual patient data. https://nicedsu.org.uk/wp-content/uploads/2016/03/TSD17-DSU-Observational-data-FINAL.pdf. Accessed 5 May 2020
European Agency for the Evaluation of Medicinal Products. ICH topic E10—Choice of control group in clinical trials. Note for guidance on choice of control group in clinical trials. (CPMP/ICH/364/96). Presented at International Conference on Harmonization on July 27, 2000 at London, United Kingdom.
Cucherat M, Laporte S, Delaitre O, Behier J-M, d’Andon A, Binlich F, et al. From single-arm studies to externally controlled studies Methodological considerations and guidelines. Therapies. 2020;75(1):21–7.
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Auf den Punkt gerbracht. https://www.iqwig.de/download/2016_IQWiG_Auf_den_Punkt_gebracht.pdf. Accessed 28 Apr 28 2020
Kiefer C, Sturtz S, Bender R. Indirect comparisons and network meta-analyses. Dtsch Arztebl Int. 2015;112(47):803–8.
Acknowledgements
Funding
This study was funded by Novartis Pharmaceutical Corp. The sponsor funded the journal’s Rapid Service and Open Access fees.
Medical Writing Assistance
Medical writing assistance was provided by Wenxi Tang, Shelley Batts, and Emma Gray, who are employees of Analysis Group, Inc. Support for this assistance was funded by Novartis Pharmaceutical Corporation.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Disclosures
Jie Zhang and Qiufei Ma are employees of Novartis Pharmaceutical Corp. and hold stock/options. Junlong Li, Hongbo Yang, James Signorovitch, and Eric Wu are employees of Analysis Group, Inc., which has received consulting fees from Novartis Pharmaceutical Corp.
Data Availability
The datasets generated and/or analysed during the current study are not publicly available and are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12370136.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zhang, J., Li, J., Ma, Q. et al. A Review of Two Regulatory Approved Anti-CD19 CAR T-Cell Therapies in Diffuse Large B-Cell Lymphoma: Why Are Indirect Treatment Comparisons Not Feasible?. Adv Ther 37, 3040–3058 (2020). https://doi.org/10.1007/s12325-020-01397-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01397-9