
Abstract
Introduction
Influenza in hospitalized intensive care unit (ICU) patients with respiratory failure is associated with 25% mortality, despite timely oseltamivir treatment. A systematic review of randomized controlled trials (RCTs) was conducted to evaluate the efficacy and safety of alternative neuraminidase inhibitor (NAI) regimens compared to standard of care in patients hospitalized for H1N1, H3N2, or B influenza.
Methods
The Cochrane collaboration searching methods were followed in Cochrane Library, PubMed, and Web of Science databases (2009–2019). Eligibility criteria were RCTs comparing different regimens of NAIs in hospitalized patients (at least 1 year old) for clinically diagnosed influenza (H1N1, H3N2, or B). Pre-defined endpoints were time to clinical resolution (TTCR), overall mortality, hospital discharge, viral clearance, drug-related adverse events (AEs), and serious adverse events.
Results
Seven trials (1579 patients) were included. Two trials compared two regimens of oral oseltamivir therapy, and one trial compared two regimens of intravenous zanamivir therapy vs oral oseltamivir therapy. Four trials focused on intravenous peramivir therapy: two trials compared two different regimens and two trials compared two different regimens vs oral oseltamivir therapy. Overall, the different regimens were well tolerated, with no significant differences in AEs; nonetheless non-significant differences were reported among different regimens regarding TTCR, mortality, and viral clearance.
Conclusion
Higher compared to standard doses of NAIs or systemic peramivir therapy compared to oral oseltamivir therapy did not demonstrate benefit.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Influenza in hospitalized patients in the ICU with acute respiratory failure is associated with overall 25% mortality, despite timely oseltamivir treatment. |
What was learned from the study? |
Systemic administration of neuraminidase inhibitor regimens to treat hospitalized patients with influenza infections are equally safe but do not modify meaningful clinical outcomes when compared with orally administered oseltamivir 75 mg tid. |
For hospitalized patients with influenza H1N1, H3N2, or B, higher doses of neuraminidase inhibitors compared to standard of care (oral oseltamivir therapy or intravenous peramivir therapy) do not modify meaningful clinical outcomes when compared with the standard dose. |
Introduction
Influenza virus infection is a worldwide problem and it is the leading cause of respiratory viral disease in hospitalized patients [1,2,3]. Both the World Health Organization (WHO) and the European Centre for Disease Control (ECDC) recommend the use of neuraminidase inhibitors (NAI) for hospitalized adults with influenza [4]. The Infectious Diseases Society of America (IDSA) released guidelines on influenza management in 2018, identifying NAIs as first-line therapy in hospitalized patients regardless of illness duration prior to hospitalization, with no differences between oral oseltamivir therapy, intravenous peramivir therapy, or inhaled zanamivir therapy [5]. While there is a consensus on dosing and duration of treatment for outpatients and high-risk population, management of influenza treatment in hospitalized and severely ill patients is suboptimal. In spite of early initiation of NAIs, mortality rates exceed 25% in primary influenza pneumonia with acute respiratory failure (ARF). Critically ill patients are characterized by a variety of conditions that may alter drug absorption, like altered gastrointestinal motility, and pharmacokinetics, such as the need for renal replacement therapy or extracorporeal membrane oxygenation. Furthermore, in mechanically ventilated patients, administration of inhaled zanamivir is contraindicated because of reported fatal complications, and oseltamivir has to be administered via nasogastric tube [6]. The IDSA recommends against the routine use of higher doses of Food and Drug Administration (FDA)-approved NAI drugs for therapy of seasonal influenza [5]. Double oseltamivir dose has been used as salvage therapy in presence of ARF in some settings, but robust data are lacking [7]. Peramivir is the only FDA-approved intravenously administered drug for influenza, but optimal dosing regimen remains controversial [5]. To date, there are no unanimous data on NAI use for the treatment of hospitalized patients because treatment dosing, administration route, and duration are still debated in these patients, who require intensive care admission, and effect on outcomes and safety of different therapies is not clear.
The hypothesis was that in patients admitted to hospital with influenza infection, the optimization of NAI administration may improve outcomes. Thus, the study’s aim was to perform a systematic review (SR) of randomized controlled trials (RCT) to evaluate the efficacy and safety of alternative NAI regimens compared with 75 mg orally administered oseltamivir twice/daily or 600 mg intravenously administered peramivir once/daily in patients hospitalized for H1N1, H3N2, or B influenza.
Methods
Protocol and Registration
This report describes the results of the SR following the guidance of the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) statement [8]. PICO (Population, Intervention, Comparator, Outcome) questions are detailed in the supplementary material 1. The protocol was published in the National Institute for Health Research International Prospective Register of Systematic Reviews (PROSPERO), registration number CRD42018110060.
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Sources
A global search strategy was systematically performed in three databases: MEDLINE database through the PubMed search engine, the Cochrane Library Database, and Web of Science database. Websites from ClinicalTrials.gov and clinicaltrialsregister.eu were consulted for other ongoing trials. Search terms were detailed in the supplementary material 2. Restrictions in the search were applied regarding the language: only studies published in English, Spanish, French, Italian, and Portuguese were considered. Abstracts presented at scientific conferences, unpublished studies, and other unpublished data deriving from industry sites were excluded. A restriction was also applied to the publication period of time, between 2009 and 2019, partially because before 2009 there were no diagnostic tests of influenza and also since the outbreak of A/H1N1 in 2009 [9], the use of NAIs has increased. The first search was performed in January 2019 and repeated in November 2019.
Data Extraction and Study Selection Process
Two authors (ST and LC) independently evaluated all the studies identified in the literature search by screening their titles, abstracts, and full text. In case of disagreement, a third author (CSL) independently determined eligibility. A predesigned spreadsheet was used to collect study data in a standardized way. Data extracted from each trial included were the study design, quality assessment, characteristics of the study populations, method used for confirmation of the influenza infection, characteristics of compared treatment arms, the intention to treat (ITT) population and the subgroup of patients with laboratory-confirmed influenza infection, as well as data regarding the effectiveness and safety outcomes.
Studies were considered eligible for inclusion in the SR if they were a RCT that enrolled patients older than 1 year of age, requiring hospitalization with clinically diagnosed influenza (with H1N1, H3N2, or B) or influenza-like syndrome, with or without laboratory confirmation. Pre-defined treatments for inclusion were oseltamivir (oral administration), zanamivir (oral, intravenous, or inhaled administration), peramivir (oral or intravenous administration), and laninamivir (inhaled administration). Studies involving children less than 1 year old, NAIs against other serotypes of influenza such as H5N1, pregnant women, immunocompromised patients (more than 30% of the overall population), or outpatients were excluded. Also, observational cohort studies or studies with different intervention of NAIs such as polymerase inhibitors (baloxavir marboxil) treatment were excluded.
Definitions and Outcomes
Clinically suspected influenza was defined by the presence of respiratory symptoms (sore throat, cough, nasal congestion) and fever (≥ 37.7 °C) within 48 h of study enrollment, regardless of prior symptoms duration. Influenza infection was defined by the presence of a positive polymerase chain reaction (PCR), immunofluorescence assay, or rapid antigen test (RAT) for influenza virus. The ITT population included all patients randomized to receive the respective study regimens. The influenza-positive population included only patients with confirmed influenza. Time to clinical resolution (TTCR), defined by the individual study protocol as the time from initiation of the study treatment until resolution of vital sign abnormalities (the supplementary material 3), and overall mortality were considered as the primary effectiveness outcomes of this SR. Secondary effectiveness outcome was viral clearance, defined as the proportion of influenza virus-negative patients detected by PCR on nasopharyngeal samples at 5 day. Samples analyzed with different methods (e.g., viral culture) or at different time frames were excluded from the comparison. Safety was evaluated in terms of occurrence of respiratory and/or systemic drug-related adverse events (AEs) and serious adverse events (SAEs).
Quality Assessment
Risk of bias was assessed for each included study independently by ST on the basis of the Cochrane Handbook for Systematic Reviews of Interventions [10] and using the Cochrane Review Manager 5.3 risk of bias tool which takes account of allocation sequence generation, concealment of allocation, masking of participants and investigators, incomplete outcome reporting, selective outcome reporting, and other sources of bias. Each potential source of bias was graded to determine whether studies were considered at high, low, or moderate risk of bias. In case of disagreement, a second author (CSL) independently determined the quality assessments.
Data Analysis
For categorical outcomes, the numbers of patients who had each outcome and denominator were extracted, and for continuous outcomes, sample size, mean [standard deviation (SD)], or median [interquartile range (IQR)] were extracted on the basis of the information provided within studies. Where results were not reported in the same format for analysis, we used recommended methods from the Cochrane Collaboration to extract or estimate effects including contacting study authors and using formulae for conversion of medians (IQR) to estimated mean (SD) as previously described [11].
Results
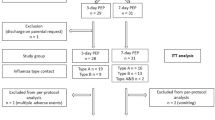
A total of 6692 studies were identified: 5732 studies in the MEDLINE (PubMed), 563 in Web of Science, and 397 in the Cochrane Library databases. Seven trials and 1579 ITT patients were included. The PRISMA flow diagram of the studies’ selection is presented in Fig. 1. A summary of the risk of bias of the included RCT is detailed in Fig. 2.

Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) flow diagram of the study selection

a “Risk of bias” graph: authors’ judgments about each risk of bias item presented as percentages across all included studies. b “Risk of bias” summary: authors’ judgments about each risk of bias item presented as percentages for each of the included study
Interventions
A total of seven trials were included, analyzing different NAI regimens. Main characteristics of the included studies are described in Table 1. Two trials focused on oral oseltamivir therapy [12, 13], comparing high dose (150 mg twice/daily) vs standard dose (75 mg twice/daily). One trial compared two regimens of intravenous zanamivir therapy [14] (300 mg vs 600 mg twice/daily) vs standard dose of orally administered oseltamivir (75 mg twice/daily). Two trials compared two different regimens of intravenous peramivir therapy [15, 16] (300 mg vs 600 mg once daily; or 200 mg vs 400 mg once daily) vs standard dose of orally administered oseltamivir (75 mg twice/daily), and two trials of intravenous peramivir therapy [17, 18] compared high dose (600 mg once/daily) vs standard dose (300 mg twice/daily or once/daily). No study analyzed inhaled zanamivir, given the contraindication of its use in severely ill patients on mechanical ventilation [5]. No laninamivir trial respecting all the inclusion criteria was found; hence laninamivir was not included in the SR.
Patient Characteristics
A total of 1579 patients were included in the seven analyzed trials. Of these, 1312 (83.0%) had confirmed influenza infection, and 205 (12.9%) were vaccinated against influenza. Baseline characteristics of the population included are described in Table 2. Five trials involved only adult patients (≥ 16 years of age), whereas the remaining two trials involved children and adults (≥ 6 years or ≥ 1 year old). A total of 545 (34.5%) patients received other antiviral treatment prior to study drug initiation and 342 (21.6%) patients needed admission to the ICU. The most common underlying diseases were chronic obstructive pulmonary disease (COPD) (15.0%), diabetes (11.0%), and asthma (7.2%).
Outcomes
All data on outcomes extracted from each trial included are presented in Table 3.
Time to Clinical Resolution
The median days of clinical resolution was assessed in five studies. The study by Lee et al. [12], focused on oral oseltamivir therapy, reported a non-significant TTCR decrease in the group of patients treated with standard dose twice/daily (1 day [75 mg twice/daily] vs 2 days [150 mg twice/daily], p = 0.48). The study by Marty et al. [14], focused on intravenous zanamivir therapy, reported a non-significant TTCR decrease in the group of patients treated with high dose twice/daily (5.58 days [300 mg twice/daily] vs 5.15 days [600 mg twice/daily], p = 0.25). Three trials focused on intravenous peramivir therapy: Kohno et al. [17] reported a significant decrease of TTCR in the group of patients treated with high dose once/daily (4.7 day [300 mg once/daily] vs 1.7 days [600 mg once/daily], p < 0.001), whereas Ison et al. [18] reported a significant decrease in the group of patients treated with standard dose twice/daily (1.9 day [300 mg twice/daily] vs 6.9 days [600 mg once/daily], p < 0.001). Ison et al. [15] reported a non-significant decrease of TTCR in the group of patients treated with high dose once/daily (1.3 days [300 mg once/daily] vs 1 day [600 mg once/daily] vs 1.5 days [75 mg orally administered oseltamivir twice/daily], p = 0.306).
Mortality
Overall mortality was assessed in six studies. The two studies that focused on oral oseltamivir therapy [12, 13] reported a non-significant mortality decrease in the group of patients treated with standard dose twice/daily (0.9% [75 mg twice/daily] vs 2.4% [150 mg twice/daily], p > 0.99 [12] and 5.8% [75 mg twice/daily] vs 7.6% [150 mg twice/daily], p = 0.54 [13]). Marty et al. [14] reported a non-significant difference of mortality between zanamivir groups (7% [300 mg twice/daily] vs 7% [600 mg twice/daily], p = 0.91). Ison et al. [15] and Ison et al. [18], focused on intravenous peramivir therapy, reported a non-significant mortality decrease in the group of patients treated with low dose in both studies (7% [300 mg twice/daily] vs 12% [600 mg once/daily], p = 0.19 [18] and 0% [200 mg once/daily] vs 2% [400 mg once/daily] vs 0% [75 mg orally administered oseltamivir twice/daily], p = 0.32 [15]). The study by Lee et al. [16] reported only one death out of 70 patients.
Viral Clearance
Viral clearance, defined as the proportion of influenza virus-negative patients detected by PCR on nasopharyngeal samples at 5 days, was analyzed in four studies. The two studies that focused on oral oseltamivir therapy [12, 13] reported a non-significant increase of viral clearance in the group treated with high dose twice/daily in both studies (40.2% [75 mg twice/daily] vs 44.7% [150 mg twice/daily], p = 0.634 [12] and 68.2% [75 mg twice/daily] vs 72.3% [150 mg twice/daily], p = 0.42 [13]). Two trials focused on intravenous peramivir therapy: Ison et al. [18] reported a non-significant clearance increase in the group of patients treated with standard dose twice/daily (62.2% [300 mg twice/daily] vs 51% [600 mg once/daily], p = 0.303). The second trial by Lee et al. [16] reported a significant increase of viral clearance in the group of patients treated with overall peramivir once/daily (43.8% [overall peramivir once/daily] vs 39.0% [75 mg orally administered oseltamivir twice/daily], p = 0.744).
Drug-Related Adverse Events
The drug-related AEs were assessed in six studies. Two studies focused on oral oseltamivir therapy [12, 13] reported a significant decrease of AEs incidence in the group of patients treated with low dose twice/daily in one study [12] (5.3% [75 mg twice/daily] vs 22% [150 mg twice/daily], p < 0.01), and a non-significant decrease of AEs incidence in the group of patients treated with high dose twice/daily [13] (5.6% [75 mg twice/daily] vs 3% [150 mg twice/daily], p = 0.25). Marty et al. [14] reported a non-significant difference of AEs (12% [300 mg twice/daily] vs 11% [600 mg twice/daily], p = 0.54). Three trials focused on intravenous peramivir therapy: Kohno et al. [17] reported a significant decrease of AEs in the group of patients treated with standard dose once/daily (28.6% [300 mg once/daily] vs 38.1% [600 mg once/daily], p = 0.51), whereas Ison et al. [18] reported a non-significant decrease of AEs rate in the group of patients treated with high dose once/daily (19% [300 mg twice/daily] vs 16% [600 mg once/daily], p = 0.56). The third trial by Lee et al. [16] reported a total of 20% of patients with drug-related AEs.
Serious Adverse Events
The SAEs were assessed in four studies. One study, focused on oral oseltamivir therapy [13], reported a non-significant decrease of SAEs in the group of patients treated with high dose twice/daily (0.6% [75 mg twice/daily] vs 0% [150 mg twice/daily], p = 0.31). Marty et al. [14], focused on intravenous zanamivir therapy, reported a non-significant decrease of SAEs in the group of patients treated with high dose of zanamivir twice/daily (19% [300 mg twice/daily] vs 16% [600 mg twice/daily], p = 0.41). Two trials focused on intravenous peramivir therapy: Ison et al. [18] reported a non-significant SAEs decrease in the group of patients treated with standard dose twice/daily (18% [300 mg twice/daily] vs 22% [600 mg once/daily], p = 0.45), and Ison et al. [15] reported a significantly decreased rate of SAEs in the group of patients treated with low dose once/daily (4% [200 mg once/daily] vs 17% [400 mg once/daily], p = 0.48 vs 9% [75 mg orally administered oseltamivir twice/daily], p = 0.306).
Discussion
This is the first SR of RCTs that have evaluated the efficacy and safety of different dosage and/or regimens of systemic NAIs in an important clinical and public health challenge, such as hospitalized patients with seasonal or pandemic influenza. Our data suggest that alternative regimens are safe to use in a hospitalized population but do not significantly change mortality in the efficacy analyses. Also, the evidence is inconclusive for other meaningful outcomes, such as TTCR or viral clearance. Our findings confirm the variability of efficacy of antiviral treatment regimen for severe hospitalized patients with influenza infection.
Several SR, including both RCT and observational studies, conducted in the past years have addressed the efficacy and safety of NAI treatment, demonstrating the effectiveness of NAI treatment to reduce severity of influenza in outpatients, and mortality in hospitalized patients, compared without treatment [19,20,21,22,23]. Furthermore, it is widely accepted that the efficacy of NAI treatment is higher if administered within 48 h from symptoms onset [5]. Nonetheless, given the variety of the population enrolled in the published studies, involving both in- and outpatients, treated with different NAI regimens, no consensus exists on which NAI represents the best option in hospitalized patients with influenza. In a meta-analysis [23] of individual participant data in 29,234 hospitalized patients from 78 studies with influenza A (H1N1)pdm09 with infection, NAI therapy was associated with a reduction of mortality in the subgroup of ICU patients, compared with no treatment. Moreover, treatment within 2 days of symptoms onset was associated with a reduction in mortality compared with the late administration. In our SR, time from symptoms onset to NAI treatment was heterogeneous among the included studies, from 48 h to 10 days, with median duration of illness from 2 to 5 days, adding a confounding factor. Standardized RCT protocols might help in reducing controllable variables, to equalize studies conducted in different settings, and further investigate NAI time-efficacy.
Oseltamivir, zanamivir, peramivir, and laninamivir are the NAIs currently available, approved for a variety of indications and formulations by the different regulatory agencies (Table 4). Oral oseltamivir therapy is approved to treat patients with uncomplicated influenza by both the FDA and European Medicines Agency (EMA); no information is available on safety and efficacy in hospitalized patients [24]. For severely ill patients, double dose oseltamivir has been used in some settings but robust data on its efficacy are lacking, and guidelines recommend against its use [7]. A recent study [25] among adult patients with pandemic influenza requiring ventilator support concluded that oseltamivir had a good enteric absorption, and the dosage of 75 mg twice daily achieved adequate plasma concentrations, far in excess of those required to inhibit viral neuraminidase activity. Accumulation of oseltamivir in patients with extracorporeal membrane oxygenation and continuous venovenous hemodiafiltration lead to 4- to 5-fold increase in plasma levels [26]. If oral or enteral administration of oseltamivir is impossible or its absorption is altered, intravenously administered NAIs might be used. Zanamivir is typically used as inhaled drug in outpatients, but lack of safety in subjects with airway diseases limits its use in hospitalized and mechanically ventilated patients [5]. On the basis of the trial from Marty et al. [14], the EMA approved the use of intravenously administered zanamivir 600 mg twice/daily in complicated influenza [5]; this formulation is not FDA approved and not included in the latest IDSA guidelines. Given the recent introduction, to date there are only a few anecdotical case reports and a small case series of four ICU patients treated with intravenous zanamivir therapy, but they showed a high efficacy and tolerability [27, 28]. Intravenously administered zanamivir could represent a good therapeutic option in severely ill patients with influenza infection, not only when oral or aerosolized antiviral medication cannot be administered but also in the unlikely event of oseltamivir resistance. In these cases, intravenous peramivir therapy might also be considered, but it is only approved for uncomplicated influenza and no consensus has been reached on the appropriate dosing and duration of treatment [5]. A recent SR [19] confronted intravenous peramivir therapy vs oral oseltamivir therapy demonstrating peramivir efficacy in reducing TTCR only in outpatients, with no differences in mortality and length of hospital stay for both in- and outpatients. To date, European guidelines do not include indication for intravenously administered peramivir at dosages different from 600 mg single administration in outpatient settings, while IDSA guidelines suggest to consider administering a multiday dosing regimen, although the optimal regimen is unknown. Finally, laninamivir is approved only in Japan (2010), used as a single dose aerosol in outpatients, with no data available in inpatients [29].
Among the different NAIs available for treating patients with influenza, no consensus has been reached about which regimen should be recommended to treat hospitalized patients. Comorbidities, clinical conditions, and clinical setting might play an important role in guiding NAI choice. New drugs are being developed, and studied in severe hospitalized patients: baloxavir marboxil is a novel polymerase inhibitor approved in Japan, the USA, and other countries. Two phase III trials [30, 31] in non-hospitalized patients with placebo found that single dose was superior to placebo in alleviating influenza symptoms, and was superior to both oseltamivir and placebo in reducing viral replication. A double-blind RCT (NCT03684044) comparing the combination of oseltamivir and baloxavir marboxil to oseltamivir alone is currently in progress in hospitalized patients.
Limitations should be considered when interpreting the results of this systematic review. We judged that the included studies were generally of low quality based upon the selection bias. The main limitation is the heterogeneity in dosage and comparators that precluded a meta-analysis, as well as and the size of the study population (large RCTs are needed) and the inclusion of clinically diagnosed influenza in two studies. Despite identifying many studies (e.g., trials with outpatients or observational studies), there were few RCTs about hospitalized patients with influenza treated with NAIs. None of the included studies assessed the penetration of antivirals into the lung tissue or analyzed the effect of antiviral concentrations on alveolar viral load. No study involving laninamivir met the inclusion criteria. Finally, only a small percentage of mechanically ventilated (MV) patients with acute respiratory distress syndrome (ARDS) or pneumonia were enrolled, and the impact of viral susceptibility on treatment could not be analyzed because of the scarcity of data. Even if rare, NAI resistance might influence the outcomes of different treatment regimens. Only four out of seven studies analyzed viral strain susceptibility pre-treatment, and six studies conducted a post-treatment analysis, with overall only four new resistances identified. The small numbers did not allow a correlation with clinical outcomes; furthermore, different analysis methods were used, not allowing a standardized comparison. Despite these limitations, our study provides information that is not available in the published literature, being an important strength and having implications for further research. Furthermore, the results were based on RCTs, rather than observational cohort studies, so that it illustrates the need for research in the form of RCTs in the subset of patients with respiratory failure requiring hospitalization or ICU admission, focusing on meaningful pre-defined outcome criteria.
Conclusion
The evidence evaluated in this SR indicates that the alternative NAI regimens to orally administered oseltamivir 75 mg twice/daily or intravenously administered peramivir 600 mg once/daily to treat hospitalized patients with influenza infection are equally safe but do not modify meaningful clinical outcomes when compared with the standard dose.
References
Gaunt ER, Harvala H, McIntyre C, Templeton KE, Simmonds P. Disease burden of the most commonly detected respiratory viruses in hospitalized patients calculated using the disability adjusted life year (DALY) model. J Clin Virol. 2011;52:215–21. https://doi.org/10.1016/j.jcv.2011.07.017.
Rath B, Conrad T, Myles P, et al. Influenza and other respiratory viruses: standardizing disease severity in surveillance and clinical trials. Expert Rev Anti Infect Ther. 2017;15:545–68. https://doi.org/10.1080/14787210.2017.1295847.
Saunders-Hastings PR, Krewski D. Reviewing the history of pandemic influenza: understanding patterns of emergence and transmission. Pathogens. 2016. https://doi.org/10.3390/pathogens5040066.
European Centre for Disease Prevention and Control (ECDC). Public health use of influenza antivirals during influenza pandemics. ECDC Interim guidance 2009. https://europa.eu/european-union/about-eu/agencies/ecdc_es. Accessed 16 Mar 2020
Uyeki TM, Bernstein HH, Bradley JS, et al. Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenzaa. Clin Infect Dis. 2019;68:895–902. https://doi.org/10.1093/cid/ciy874.
Kumar B, Asha K, Khanna M, Ronsard L, Meseko CA, Sanicas M. The emerging influenza virus threat: status and new prospects for its therapy and control. Arch Virol. 2018;163:831–44. https://doi.org/10.1007/s00705-018-3708-y.
Welch SC, Lam SW, Neuner EA, Bauer SR, Bass SN. High-dose versus standard dose oseltamivir for treatment of severe influenza in adult intensive care unit patients. Intensive Care Med. 2015;41:1365–6. https://doi.org/10.1007/s00134-015-3816-z.
Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62:e1–34. https://doi.org/10.1016/j.jclinepi.2009.06.006.
Jefferson T, Jones MA, Doshi P, et al. Neuraminidase inhibitors for preventing and treating influenza in adults and children. Cochrane Database Syst Rev. 2014. https://doi.org/10.1002/14651858.CD008965.pub4.
The Cochrane Collaboration. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. https://www.cochrane-handbook.org. Accessed June 21, 2019.
Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Methodol. 2005;5:13. https://doi.org/10.1186/1471-2288-5-13.
Lee N, Hui DSC, Zuo Z, et al. A prospective intervention study on higher-dose oseltamivir treatment in adults hospitalized with influenza A and B infections. Clin Infect Dis. 2013;57:1511–9. https://doi.org/10.1093/cid/cit597.
South East Asia Infectious Disease Clinical Research Network. Effect of double dose oseltamivir on clinical and virological outcomes in children and adults admitted to hospital with severe influenza: double blind randomised controlled trial. BMJ. 2013;346:f3039. https://doi.org/10.1136/bmj.f3039.
Marty FM, Vidal-Puigserver J, Clark C, et al. Intravenous zanamivir or oral oseltamivir for hospitalised patients with influenza: an international, randomised, double-blind, double-dummy, phase 3 trial. Lancet Respir Med. 2017;5:135–46. https://doi.org/10.1016/S2213-2600(16)30435-0.
Ison MG, Hui DS, Clezy K, et al. A clinical trial of intravenous peramivir compared with oral oseltamivir for the treatment of seasonal influenza in hospitalized adults. Antivir Ther. 2013;18:651–61. https://doi.org/10.3851/IMP2442.
Lee N, Chan PKS, Tam WWS, et al. Virological response to peramivir treatment in adults hospitalised for influenza-associated lower respiratory tract infections. Int J Antimicrob Agents. 2016;48:215–9. https://doi.org/10.1016/j.ijantimicag.2016.05.003.
Kohno S, Kida H, Mizuguchi M, et al. Intravenous peramivir for treatment of influenza A and B virus infection in high-risk patients. Antimicrob Agents Chemother. 2011;55:2803–12. https://doi.org/10.1128/AAC.01718-10.
Ison MG, Fraiz J, Heller B, et al. Intravenous peramivir for treatment of influenza in hospitalized patients. Antivir Ther. 2014;19:349–61. https://doi.org/10.3851/IMP2680.
Lee J, Park JH, Jwa H, Kim YH. Comparison of efficacy of intravenous peramivir and oral oseltamivir for the treatment of influenza: systematic review and meta-analysis. Yonsei Med J. 2017;58:778–85. https://doi.org/10.3349/ymj.2017.58.4.778.
Doll MK, Winters N, Boikos C, Kraicer-Melamed H, Gore G, Quach C. Safety and effectiveness of neuraminidase inhibitors for influenza treatment, prophylaxis, and outbreak control: a systematic review of systematic reviews and/or meta-analyses. J Antimicrob Chemother. 2017;72:2990–3007. https://doi.org/10.1093/jac/dkx271.
Boikos C, Caya C, Doll MK, et al. Safety and effectiveness of neuraminidase inhibitors in situations of pandemic and/or novel/variant influenza: a systematic review of the literature, 2009–15. J Antimicrob Chemother. 2017;72:1556–733. https://doi.org/10.1093/jac/dkx013.
Muthuri SG, Myles PR, Venkatesan S, Leonardi-Bee J, Nguyen-Van-Tam JS. Impact of neuraminidase inhibitor treatment on outcomes of public health importance during the 2009–2010 influenza A(H1N1) pandemic: a systematic review and meta-analysis in hospitalized patients. J Infect Dis. 2013;207:553–63. https://doi.org/10.1093/infdis/jis726.
Muthuri SG, Venkatesan S, Myles PR, et al. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: a meta-analysis of individual participant data. Lancet Respir Med. 2014;2:395–404. https://doi.org/10.1016/S2213-2600(14)70041-4.
Birnkrant D, Cox E. The emergency use authorization of peramivir for treatment of 2009 H1N1 influenza. N Engl J Med. 2009;361:2204–7. https://doi.org/10.1056/NEJMp0910479.
Ariano RE, Sitar DS, Zelenitsky SA, et al. Enteric absorption and pharmacokinetics of oseltamivir in critically ill patients with pandemic (H1N1) influenza. CMAJ. 2010;182:357–63. https://doi.org/10.1503/cmaj.092127.
Lemaitre F, Luyt C-E, Roullet-Renoleau F, et al. Impact of extracorporeal membrane oxygenation and continuous venovenous hemodiafiltration on the pharmacokinetics of oseltamivir carboxylate in critically ill patients with pandemic (H1N1) influenza. Ther Drug Monit. 2012;34:171–5. https://doi.org/10.1097/FTD.0b013e318248672c.
Torti C, Mazzitelli M, Longhini F, et al. Clinical outcomes of patients treated with intravenous zanamivir for severe influenza A(H1N1)pdm09 infection: a case report series. BMC Infect Dis. 2019. https://doi.org/10.1186/s12879-019-4530-1.
Mazzitelli M, Garofalo E, Bruni A, et al. Severe myocarditis due to influenza A(H1N1)pdm09 viral infection in a young woman successfully treated with intravenous zanamivir: a case report. Clin Case Rep. 2019;7:2336–400. https://doi.org/10.1002/ccr3.2499.
Yamashita M. Laninamivir and its prodrug, CS-8958: long-acting neuraminidase inhibitors for the treatment of influenza. Antivir Chem Chemother. 2010;21:71–84. https://doi.org/10.3851/IMP1688.
Hayden FG, Shindo N. Influenza virus polymerase inhibitors in clinical development. Curr Opin Infect Dis. 2019;32:176–86. https://doi.org/10.1097/QCO.0000000000000532.
Ison M, Portsmouth S, Yoshida Y, Shishido T, Hayden F, Uehara T. Phase 3 trial of baloxavir marboxil in high-risk influenza patients (CAPSTONE-2 study). Open Forum Infect Dis. 2018;5:S764–S765765.
Acknowledgements
This study was part of the doctoral thesis from ST at the Medicine Department, University of Barcelona.
Funding
This work was funded by CIBERES, Instituto Salud Carlos III, Madrid, Spain (Fondos FEDER) (CB06-06–036). No funding or sponsorship was received for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Jordi Rello served as a consultant and received grant support from Genentech and Roche. Jordi Rello is a member of the journal’s Editorial Board. Sofia Tejada, Laura Campogiani and Candela Solé-Lleonart have nothing to declare.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
All data and materials used in this research are freely available. References have been provided.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12098139.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tejada, S., Campogiani, L., Solé-Lleonart, C. et al. Alternative Regimens of Neuraminidase Inhibitors for Therapy of Hospitalized Adults with Influenza: A Systematic Review of Randomized Controlled Trials. Adv Ther 37, 2646–2666 (2020). https://doi.org/10.1007/s12325-020-01347-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01347-5