
Abstract
Introduction
Two multicenter, double-blind, randomized controlled trials assessed the effect of Breathe Right Nasal Strips (BRNS) on sleep-related quality of life in otherwise healthy subjects with chronic nocturnal nasal congestion who reported trouble sleeping.
Methods
Subjects were randomized to BRNS or a placebo strip for approximately 8 h each night for 14 days. Efficacy was assessed in the clinic using the Nocturnal Rhinoconjunctivitis Quality of Life Questionnaire (NRQLQ).
Results
A total of 140 subjects were randomized in Study 1, and 130 in Study 2. There was no significant difference between BRNS and placebo on either the NRQLQ “Sleep Problems” domain or the “Feel Tired and Unrefreshed” item of the “Symptoms on Waking in the Morning” domain at day 7 or 14. There was, however, a significant change in the least squares mean difference from baseline to days 7 and 14 in both the BRNS and placebo arms for each of these endpoints. BRNS were well tolerated.
Conclusions
BRNS did not significantly improve subjective measures of sleep quality and nasal congestion compared with placebo strips in this population of chronic nocturnal congestion sufferers with self-reported sleep impairment, possibly due to a strong placebo effect.
Funding
GlaxoSmithKline Consumer Healthcare.
ClinicalTrials.gov Registration Numbers
Study 1: NCT03549117; Study 2: NCT03549130.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nasal congestion is thought to be a primary symptom underlying rhinitis-related sleep issues [1]. Congestion has been associated with issues of sleep, daytime somnolence, mood, and work and school productivity [2]. Nasal dilators such as Breathe Right® Nasal Strips (BRNS; GlaxoSmithKline Consumer Healthcare; Warren, NJ, USA) are medical devices used to provide temporary relief of breathing difficulties and nasal congestion. BRNS act by reducing nasal resistance mechanically, reducing the work of breathing, and increasing the supply of oxygen through stabilization of the lateral nasal vestibular wall [3,4,5,6,7]. Use of BRNS has been shown to increase the minimum cross-sectional area of the nasal valve (thereby decreasing nasal resistance), accompanied by improvement in symptoms of nasal congestion [8,9,10]. BRNS have also been shown to improve nasal patency in normal awake subjects [11, 12].
Although BRNS have demonstrated improvements in the ease of mechanical breathing and congestion, evidence of their effects on sleep outcomes has been mixed. In some studies, BRNS have demonstrated improvement in sleepiness symptoms in patients with obstructive sleep apnoea or snoring, including those with chronic congestion [13,14,15,16]. In a study of subjects who were heavy snorers, BRNS were associated with significant improvements in both snoring and sleepiness, as measured on the Epworth Sleepiness Scale [15]. However, a recent meta-analysis found that use of nasal dilators such as BRNS had no impact on sleep architecture or sleepiness [17]. The objective of the two studies described here, therefore, was to assess the effect of BRNS on sleep-related quality of life and the feeling of being refreshed upon waking in the morning relative to placebo in otherwise healthy subjects with leptorrhine noses suffering from chronic nocturnal nasal congestion who reported trouble sleeping.
Methods
Study Design
These were two nearly identical randomized, placebo-controlled, double-blind, two-arm, parallel-group, multicenter, pivotal phase 2 studies. Both studies were conducted at 2 study sites in the United States: Study 1 (NCT03549117) was conducted from October 2010 through January 2011 in Cincinnati, OH, and Verona, NJ, and Study 2 (NCT03549130) from November 2010 through February 2011 in Paramus, NJ, and Indianapolis, IN. The studies were performed in full concordance with good clinical practice (ICH 1996) and the Declaration of Helsinki and were approved by an institutional review board. All subjects gave written informed consent.
The study designs included a screening phase, a 7-day baseline qualification phase, and a 14-day treatment phase. Clinic visits were scheduled at baseline, day 7, and day 14. Subjects were randomly assigned to receive either an asymmetric butterfly nasal dilator strip (BRNS) or an asymmetric butterfly placebo strip. Treatment allocation was conducted by dispensing staff using a randomization schedule provided by the sponsor. Aside from the dispensing staff, all study personnel at both the research centres and sponsor who could influence study outcomes were blinded to the treatment allocation.
BRNS or placebo strips were applied by the subjects to the outside of the nose, across the bridge from alar crease to alar crease, every night during the treatment phase. The strips were used for approximately 8 h per night but no more than 12 h per night.
Participants
Subjects were healthy adults who had a leptorrhine nose with a nasal tip protrusion index ≥ 45 and chronic nocturnal nasal congestion for at least the past year, while reporting trouble with sleep. They were also required to have recorded a score of ≤ 70 on the 100-point visual analogue scale (VAS) Nasal Openness Qualifying Question on at least 4 of 7 nights during the baseline qualification period prior to the baseline visit; on this scale, 0 corresponds to extremely blocked and 100 corresponds to extremely open. This instrument was administered at bedtime while the subject was in the supine position.
Exclusion criteria included allergy or intolerance to study materials (or to closely related materials such as adhesive bandages or latex), a history of skin cancer, the presence of a chronic skin condition, eczema of the face or nose, evidence of visible open sores, a diagnosis of sleep apnoea or any other major sleep disorder, a non-typical sleep schedule (e.g., shift work), regular/habitual consumption of more than five cups or glasses per day of xanthine-containing beverages (i.e., tea, coffee, cola), or current use of any product or medication that has any effect on nasal congestion or sleep within specified times of study entry (e.g., antihistamines, stimulants, antidepressants), severe nasal obstruction caused by a structural abnormality, or pregnancy or nursing.
Study Assessments
Efficacy was assessed in the clinic using the subjective, validated Nocturnal Rhinoconjunctivitis Quality of Life Questionnaire (NRQLQ) [18], which assesses specific problems in people with allergic rhinoconjunctivitis who experience symptoms predominantly at night, which in turn have an impact on how they feel and their ability to function the next day. The NRQLQ includes 16 items in 4 domains (i.e., composite variables): “Sleep Problems”, “Sleep Time Problems”, “Symptoms on Waking in the Morning”, and “Practical Problems”. Each NRQLQ item was scored using a scale ranging from 0 = not troubled to 6 = extremely troubled. The changes in score for each question and domain on the NRQLQ from baseline to the visits at days 7 and 14 were evaluated. The proportion of subjects showing improvement on each question and domain was also measured.
At-home measurements included questions that were answered by the subject and recorded in a daily diary. These questions related to the perception of nasal breathing and congestion. Subjects were instructed to record their responses while in the supine position before and after application of the strip at bedtime and before removal of the strip in the morning in Study 1, and before application of the strip at bedtime only in Study 2. In Study 1, the change from baseline to the visits at days 7 and 14 and the percentage of subjects showing improvement in the morning and evening ratings at days 1, 3, 7, and 14 were evaluated. In Study 2, mean daily scores were summarized for days 1 through 14. The incidence of adverse events, including severity and relationship to treatment (based on the investigator’s assessment), was evaluated throughout both studies.
Study Outcomes
The co-primary efficacy endpoints in these two studies were the mean change from baseline in the “Sleep Problems” domain of the NRQLQ and the “Feel Tired and Unrefreshed” item on the “Symptoms on Waking in the Morning” (during the first hour after waking) domain of the NRQLQ. The criteria for success for these endpoints required that the mean total score on the “Sleep Problems” domain of the NRQLQ was statistically significant and clinically relevant versus placebo, and there was a statistically significant difference versus placebo on the “Feel Tired and Unrefreshed” item on the “Symptoms on Waking in the Morning” domain.
In Study 1, secondary efficacy endpoints included the daily diary items, “How Easy It Is to Breathe Through Your Nose” and “How Open Your Nose Felt” after applying the strip at night. In order for these secondary endpoints to be met, there must have been a statistically significant difference in the change from baseline and an increase in the percentage of subjects showing improvement on these items in the BRNS group compared with the placebo group. There were no prespecified secondary endpoints for Study 2.
Statistical Methods
Sample size calculations were based on the results of a previous study (ClinicalTrials.gov identifier NCT01122849). To provide 80% power to detect a significant difference between BRNS and placebo (alpha = 0.05), it was estimated that a sample size of 57 subjects per arm would be required for an expected treatment difference of 2.30 units with a standard deviation of 4.31 units on the “Sleep Problems” domain of the NRQLQ on day 7, and a sample size of 64 subjects per arm would be required for an expected treatment difference of 0.62 units, with a standard deviation of 1.24 units on the “Feel Tired or Unrefreshed” item in the “Symptoms on Waking in the Morning” domain of the NRQLQ on day 7. In order to meet both of these primary endpoints, a sufficient number of subjects was enrolled to achieve a target sample size of 64 subjects per arm.
All subjects who were randomized, received study treatment, and had at least one follow-up visit were included in the safety population. Efficacy analyses were performed on the intent-to-treat population, which included subjects who were randomized and had at least one post-baseline efficacy assessment.
For the co-primary efficacy analyses (both studies), analysis of covariance (ANCOVA) was used to compare BRNS and placebo on subjective measures of congestion and sleep quality at days 7 and 14. In both studies, the model included treatment and site as factors and baseline score as a covariate; in Study 2, the model also included the following as covariates: baseline score, age, level of congestion symptoms at baseline, Berlin Questionnaire (risk of sleep apnoea) score, and Epworth Sleepiness Scale score. The adjusted mean changes from baseline and 95% confidence intervals were provided. Chi square tests were used to compare the proportion of subjects showing improvements in selected items on the NRQLQ, with all tests performed at the 5% significance level.
In Study 1, ANCOVA was used to analyze changes in the degree of perception of nasal breathing and nasal congestion recorded for the daily diary items. A secondary analysis was also conducted using age, level of congestion symptoms at baseline, Berlin Questionnaire score, and Epworth Sleepiness Scale score as covariates. Analyses were carried out for days 1, 3, 7, and 14; summary statistics were presented for other days. Chi square tests were used to compare the proportion of subjects showing improvements in selected diary items.
Separate analyses were performed in a number of subgroup categories, including age group (18−24 years, 25−54 years, and ≥ 55 years), Berlin Questionnaire score [≥ 2 (subjects at risk for sleep apnoea) and < 2 (not at risk)], and Epworth Sleepiness Scale score (≤ 10 and > 10). Subgroup analyses were conducted for daily diary items in Study 1, and for NRQLQ domains in Study 2.
Results
Study Subjects
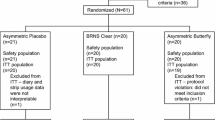
Subject dispositions for the two studies are shown in Fig. 1a, b, and baseline characteristics for the two study populations are summarized in Table 1. Overall, subject characteristics were similar between the two studies, with the mean age being approximately 47 years, and approximately two-thirds were women. The majority of subjects in the two studies were white, although a higher percentage of other races (specifically African Americans and those indicating multiple races) were represented in Study 2 compared with Study 1. Subject characteristics in the active treatment and placebo arms were also well balanced in both studies.

Subject dispositions. Study 1 (a); Study 2 (b). BRNS Breathe Right Nasal Strip
Primary Efficacy Outcomes
Baseline scores in the “Sleep Problems” domain were similar for BRNS and placebo in both studies, with scores ranging from 13 to 14. Figure 2a, b shows the least square mean difference from baseline to days 7 and 14 for the “Sleep Problems” domain of the NRQLQ over the course of the two studies. There was no significant difference between BRNS and placebo at either time point in either study. However, there was a significant change in the least square mean difference from baseline to days 7 and 14 in both the BRNS and placebo arms of each study (P < 0.0001). In Study 1, 91.4% and 92.9% of subjects in the BRNS group showed improvement on any item in the “Sleep Problems” domain at days 7 and 14, respectively, compared with 97.1% in the placebo group at both time points. In Study 2, 92.2% of subjects in the BRNS group showed improvement on any item in the “Sleep Problems” domain at both days 7 and 14 compared with 95.3% and 93.8% in the placebo group at days 7 and 14, respectively.

Least square mean (95% CI) change from baseline on the “Sleep Problems” domain of the NRQLQ for Study 1 (a) and Study 2 (b), ITT populations. CI confidence interval, ITT intent-to-treat, LS least square, NRQLQ Nocturnal Rhinoconjunctivitis Quality of Life Questionnaire
Baseline scores in the “Feel Tired and Unrefreshed” item of the “Symptoms on Waking in the Morning” domain of NRQLQ, the other primary efficacy endpoint, were similar for BRNS and placebo in both studies, with scores ranging from 3.4 to 3.6. Figure 3a, b shows the least square mean difference from baseline to days 7 and 14 for this item over the course of the two studies. Again, there was no significant difference between BRNS and placebo at either time point in either study. Similar to the “Sleep Problems” domain, there was a significant change in the least square mean difference from baseline to days 7 and 14 in both the BRNS and placebo arms of each study (P < 0.0001).

Least square mean (95% CI) change from baseline on the “Feel Tired and Unrefreshed” item of the “Symptoms on Waking in the Morning” domain of the NRQLQ for Study 1 (a) and Study 2 (b), ITT populations. CI confidence interval, ITT intent-to-treat, LS least square, NRQLQ Nocturnal Rhinoconjunctivitis Quality of Life Questionnaire
Secondary Efficacy Outcomes (Study 1)
Secondary endpoints were prespecified in Study 1 only. There was no statistically significant treatment difference observed for the daily diary item scores for “How Easy It Is to Breathe Through Your Nose”, although both BRNS and placebo showed a statistically significant improvement from baseline (P < 0.0001). For daily diary item 4 regarding “How Breathing Felt after the Nasal Strip Was Applied”, BRNS had numerically higher mean scores than placebo at day 7 (2.03 vs. 1.75, respectively) and day 14 (2.01 vs. 1.74, respectively) over the 14-day study period.
Subgroup Analyses
A number of responses to various questions on the daily diary items and NRQLQ were marginally significant in different prespecified subgroups, stratified by age (18–24 years, 25–54 years, and ≥ 55 years), Berlin Questionnaire score [≥ 2 (subjects at risk for sleep apnoea) and < 2 (not at risk)], and Epworth Sleepiness Scale score (≤ 10 and > 10). However, treatment differences in any of these subgroups were not found to be significant for the same question on more than 1 day in either study. Only the daily diary question regarding nasal stuffiness on day 3 of Study 1 was highly significant (P < 0.01).
Other Efficacy Assessments
Responses for other NRQLQ domains and individual NRQLQ items were also recorded. In Study 1, no statistically significant treatment differences were observed for any domain of the NRQLQ at any time point (all P > 0.05). In Study 2, however, there were significant differences between BRNS and placebo for the “Sleep Time Problems” and “Symptoms on Waking in the Morning” domains of the NRQLQ at day 7 (both P < 0.05). There were also significant treatment differences for the individual items “Congestion in Sinuses” and “Takes Time to Clear Nighttime Drainage after Waking Up” at day 7 (both P < 0.05).
Safety
Treatment-emergent adverse events, all of which were mild or moderate in intensity, are summarized in Table 2. There were two treatment-emergent adverse events that were regarded by investigators as treatment related in Study 1 (mild nasal discomfort occurring in two subjects in the BRNS group), and five events in three subjects in Study 2 (two subjects in the BRNS group and one in the placebo group), all of which occurred at the application site [irritation (one event), pruritus (two events), discoloration (one event), and erythema (one event)].
Summary
Because there was no significant difference between BRNS and placebo on the “Sleep Problems” domain and the “Feel Tired and Unrefreshed” item at day 7 or 14, neither study met its primary endpoint.
Discussion
Both of these studies failed to meet their primary endpoint, indicating that BRNS had no significant benefit relative to placebo with regard to subjective sleep outcomes and next-day symptoms in subjects with chronic nocturnal nasal congestion and associated sleep impairment. Moreover, there was no reliable indication that BRNS had any differential effect in any of the subgroups analyzed, regardless of age, Berlin Questionnaire score, and Epworth Sleepiness Scale score. Although a number of subgroup analyses showed statistically significant differences between BRNS and placebo, these results are of limited value due to the increased potential for type 1 error (false positive findings) from the multiple comparisons. Furthermore, there appeared to be no particular pattern to these improvements, further suggesting that these may have been spurious findings.
The failure of these studies to demonstrate any clinical benefit was at least partly due to a placebo effect, with both BRNS and placebo displaying substantial and statistically significant improvements versus baseline in the “Sleep Problems”, “Sleep Time Problems”, “Symptoms on Waking in the Morning”, and “Practical Problems” domains of the NRQLQ. A strong placebo effect has been identified previously in studies of allergic rhinitis that relied on subjective measures, such as patient perceptions of symptoms (e.g., nasal obstruction, mucous secretion, etc.) [19]. A substantial placebo effect has also been seen with quality-of-life measures, where statistically significant and clinically relevant improvements in quality of life have been observed with both treatment and placebo, but with no difference between the two, despite significant treatment differences in total symptom scores [20]. For example, in a study by van Cauwenberge et al., loratadine was found to be significantly superior to placebo in terms of total symptom scores but showed no statistically significant effect on quality of life as measured with the Rhinoconjunctivitis Quality of Life Questionnaire [20]. It is also plausible that the popularity of BRNS contributed to the high placebo effect, as this product has been commercially available for decades [15]; subjects who received placebo strips may have simply assumed they were receiving BRNS based on their familiarity with the product and the fact that both strips were drug-free and had a similar asymmetrical design and directions for application.
Few of the previously published studies reporting the efficacy of BRNS were randomized, placebo-controlled trials [8, 14]. The majority of studies have measured mechanical improvement only or subjective responses relative to baseline only [3,4,5,6,7, 9,10,11,12]. Given the substantial placebo effect observed here, the positive results of some of these previous uncontrolled trials of BRNS are perhaps to be expected, and their results should be interpreted cautiously given the lack of a control group. One randomized controlled study did demonstrate a reduction in symptoms of congestion with BRNS compared with placebo using both a VAS and an ordinal scale for measuring treatment response [8]. This result parallels that of Study 2 reported here, which found significant improvements relative to placebo with regard to endpoints of congestion, including questions on the “Symptoms on Waking in the Morning” domain of the NRQLQ at day 7 (P < 0.05), and specific questions regarding “Congestion in Sinuses” and “Takes Time to Clear Nighttime Drainage after Waking Up” at day 7 (both P < 0.05). However, the validity of these comparisons is limited given the failure of the study to meet its primary endpoint. A second randomized-controlled study, while demonstrating a significant reduction in snoring frequency compared with placebo, found no significant change in sleep quality, arousal-index, apnoea–hypopnea index, or snoring loudness [14].
Another possible reason for the lack of improvement in these subjective measures, despite earlier positive results in patients with congestion and in healthy volunteers, as well as in other settings such as snoring and obstructive sleep apnoea [8, 9, 11,12,13,14,15,16], may be the size of the actual treatment effect. A study by Cohen suggested that a change in nasal resistance of 17.5–20.0% was required for patients to register clinical relief on subjective scales, such as the NRQLQ used here [21]. In two prior studies of BRNS that measured nasal resistance in patients with congestion, nasal resistance was improved by only 16.7–17.0% [4, 6]. As a result, the improvement in nasal resistance seen with BRNS in patients with congestion may be insufficient to reliably register improvement in subjective measures.
One possible limitation of this study was the inclusion of subjects with leptorrhine noses only. Such noses, due to their narrower airway, are generally thought to have greater resistance to airflow [22]. Although it is not clear whether this would have a positive or negative effect on the results of our two studies, one study found no correlation between narrow leptorrhine noses or broad platyrrhine noses and airway resistance or response to topical nasal decongestants as measured by nasal index [22]. Additional potential limitations of this study are the short follow-up time and the subjective assessment of sleep quality using questionnaires as opposed to or in addition to polysomnography. However, while polysomnography data may be less subjective than various scales and questionnaires, user variability across the four study sites may not be easily accounted for by existing software [23].
Conclusions
Over 14 nights of use, the BRNS did not significantly improve subjective measures of sleep quality and nasal congestion compared with placebo strips in this population of chronic nocturnal congestion sufferers with self-reported sleep impairment. Further research, including studies correlating changes in airway resistance with subjective quality-of-life measures and somnographic data over a longer follow-up interval, is needed to determine the role of BRNS in the management of impaired sleep caused by nocturnal congestion.
Change history
04 September 2019
The article “Sleep Quality and Congestion with Breathe Right Nasal Strips: Two Randomized Controlled Trials”, written by Michael J. Noss, Renee Ciesla, and Gilbert Shanga, was originally published electronically on the publisher’s internet portal (currently SpringerLink) on June 17, 2019 without Open Access. The article has now been made Open Access.
References
Storms W. Allergic rhinitis-induced nasal congestion: its impact on sleep quality. Prim Care Respir J. 2008;17(1):7–18.
Stull DE, Roberts L, Frank L, Heithoff K. Relationship of nasal congestion with sleep, mood, and productivity. Curr Med Res Opin. 2007;23(4):811–9.
Gehring JM, Garlick SR, Wheatley JR, Amis TC. Nasal resistance and flow resistive work of nasal breathing during exercise: effects of a nasal dilator strip. J Appl Physiol (1985). 2000;89(3):1114–22.
Wong LS, Johnson AT. Decrease of resistance to air flow with nasal strips as measured with the airflow perturbation device. Biomed Eng Online. 2004;3(1):38.
Di Somma EM, West SN, Wheatley JR, Amis TC. Nasal dilator strips increase maximum inspiratory flow via nasal wall stabilization. Laryngoscope. 1999;109(5):780–4.
Gosepath J, Mann WJ, Amedee RG. Effects of the Breathe Right nasal strips on nasal ventilation. Am J Rhinol. 1997;11(5):399–402.
Seto-Poon M, Amis TC, Kirkness JP, Wheatley JR. Nasal dilator strips delay the onset of oral route breathing during exercise. Can J Appl Physiol. 1999;24(6):538–47.
Latte J, Taverner D. Opening the nasal valve with external dilators reduces congestive symptoms in normal subjects. Am J Rhinol. 2005;19(2):215–9.
Hoyvoll LR, Lunde K, Li HS, Dahle S, Wentzel-Larsen T, Steinsvag SK. Effects of an external nasal dilator strip (ENDS) compared to xylometazolin nasal spray. Eur Arch Otorhinolaryngol. 2007;264(11):1289–94.
Griffin JW, Hunter G, Ferguson D, Sillers MJ. Physiologic effects of an external nasal dilator. Laryngoscope. 1997;107(9):1235–8.
Roithmann R, Chapnik J, Cole P, Szalai J, Zamel N. Role of the external nasal dilator in the management of nasal obstruction. Laryngoscope. 1998;108(5):712–5.
Burres SA. Acoustic rhinometry of the oriental nose. Am J Rhinol. 1999;13(5):407–10.
Gosepath J, Amedee RG, Romantschuck S, Mann WJ. Breathe Right nasal strips and the respiratory disturbance index in sleep related breathing disorders. Am J Rhinol. 1999;13(5):385–9.
Pevernagie D, Hamans E, Van Cauwenberge P, Pauwels R. External nasal dilation reduces snoring in chronic rhinitis patients: a randomized controlled trial. Eur Respir J. 2000;15(6):996–1000.
Ulfberg J, Fenton G. Effect of Breathe Right nasal strip on snoring. Rhinology. 1997;35(2):50–2.
Scharf MB, Brannen DE, McDannold M. A subjective evaluation of a nasal dilator on sleep & snoring. Ear Nose Throat J. 1994;73(6):395–401.
Camacho M, Malu OO, Kram YA, Nigam G, Riaz M, Song SA, et al. Nasal dilators (Breathe Right strips and NoZovent) for snoring and OSA: a systematic review and meta-analysis. Pulm Med. 2016;2016:4841310.
Juniper EF, Rohrbaugh T, Meltzer EO. A questionnaire to measure quality of life in adults with nocturnal allergic rhinoconjunctivitis. J Allergy Clin Immunol. 2003;111(3):484–90.
del Cuvillo A, Sastre J, Bartra J, Mullol J, DaVila I, Montoro J, et al. Placebo effect in clinical trials involving patients with allergic rhinitis. J Investig Allergol Clin Immunol. 2011;21(Suppl 3):40–5.
Van Cauwenberge P, Juniper EF. Comparison of the efficacy, safety and quality of life provided by fexofenadine hydrochloride 120 mg, loratadine 10 mg and placebo administered once daily for the treatment of seasonal allergic rhinitis. Clin Exp Allergy. 2000;30(6):891–9.
Cohen BM. Clinical correlants of changes in nasal flow/resistance (Rn) measurements. Allergol Immunopathol (Madr). 1978;6(3):217–23.
Doddi NM, Eccles R. The relationship between nasal index and nasal airway resistance, and response to a topical decongestant. Rhinology. 2011;49(5):583–6.
Allocca G, Ma S, Martelli D, Cerri M, Del Vecchio F, Bastianini S, et al. Validation of ‘Somnivore’, a machine learning algorithm for automated scoring and analysis of polysomnography data. Front Neurosci. 2019;13:207.
Acknowledgements
GlaxoSmithKline Consumer Healthcare provided a full review of the article. The authors would like to thank the participants of the studies.
Funding
This study and the article processing charges were sponsored by GlaxoSmithKline Consumer Healthcare.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing and other Editorial Assistance
Medical writing assistance was provided by James Street and Diane Sloan, PharmD, of Peloton Advantage, LLC, an OPEN Health Company, Parsippany NJ, USA, and was funded by GlaxoSmithKline Consumer Healthcare.
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were in accordance with the ethical standards of the research committee Allendale Investigational Review Board and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors. Informed consent was obtained from all individual participants included in the study.
Disclosures
Michael J. Noss is an independent contractor who conducts clinical trials for Synexus (formerly Radiant Research) and has nothing further to declare. Renee Cisla is an employee of GlaxoSmithKline Consumer Healthcare, Warren, NJ, USA. Gilbert Shanga is an employee of GlaxoSmithKline Consumer Healthcare, Warren, NJ, USA.
Data Availability Statement
Anonymized individual participant data and study documents can be requested for further research from http://www.clinicalstudydatarequest.com.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised due to retrospective open access.
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.8174621.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Noss, M.J., Ciesla, R. & Shanga, G. Sleep Quality and Congestion with Breathe Right Nasal Strips: Two Randomized Controlled Trials. Adv Ther 36, 1975–1985 (2019). https://doi.org/10.1007/s12325-019-01005-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-019-01005-5